

Abstract
Background
Emerging evidence has suggested that dysbiosis of the lung microbiota may be associated with the development of lung diseases. However, the interplay between the lung microbiome and lung cancer remains unclear. The aim of the present study was to evaluate and compare differences in taxonomic and derived functional profiles in the lung microbiota between lung cancer and benign pulmonary diseases.
Methods
Bronchoalveolar lavage fluid (BALF) samples were collected from 32 patients with lung cancer and 22 patients with benign pulmonary diseases, and further analyzed by 16S rRNA amplicon sequencing. The obtained sequence data were deeply analyzed by bioinformatics methods.
Results
A significant differentiation trend was observed between the lung cancer and control groups based on principal coordinate analysis (PCoA), while richness and evenness in the lung microbiome of lung cancer patients generally resembled those of patients with benign pulmonary diseases. Phylum TM7 and six genera (c:TM7-3, Capnocytophaga, Sediminibacterium, Gemmiger, Blautia and Oscillospira) were enriched in the lung cancer group compared with the control group (adjust P<0.05). The area under the curve (AUC) combining the microbiome with clinical tumor markers to predict lung cancer was 84.52% (95% CI: 74.06–94.97%). In addition, predicted KEGG pathways showed that the functional differences in metabolic pathways of microbiome varied with groups.
Conclusions
The results indicated that differences existed in the lung microbiome of patients with lung cancer and those with benign pulmonary diseases, and some certain bacteria may have potential to predict lung cancer, though future larger-sample studies are required to validate this supposition.
Keywords: Lung cancer, 16S rRNA sequencing, lung microbiome, biomarkers
Introduction
Lung cancer is one of the most common malignancies worldwide, posing an enormous threat to human health due to high morbidity (1.82 million) and mortality (1.59 million) per year (1,2). The lung has historically been considered a sterile environment. However, high-throughput sequencing techniques have altered the previous cognition thoroughly and it is recognized that there are abundant and diverse bacterial communities, including Proteobacteria, Firmicutes and Bacteroidetes as the most prevalent phyla, even in the healthy human lungs (3,4). In addition, accumulating evidence has also suggested a link between altered pulmonary microbiota (dysbiosis) and lung diseases, such as asthma, chronic obstructive pulmonary disease (COPD), idiopathic pulmonary fibrosis (IPF), cystic fibrosis and lung cancer (5-9).
Furthermore, some specific microbes are known to be associated with cancer. For instance, Helicobacter pylori is recognized to be associated with the high risk of gastric cancer and the amount of Fusobacterium nucleatum is associated with the prognosis of colorectal carcinoma patients (10,11). However, compared with studies on the relationship between the microbiome and other cancers, the subtle association between the microbiome and lung cancer remains relatively unclear. Although some previous studies have noticed that changes of specific microbiota in bronchoalveolar lavage fluid (BALF), lung tissue, sputum, saliva and fecal samples may be related to lung cancer, these studies have yielded some conflicting specific results (9,12-16). Additionally, Gui et al. reported that Lactobacillus showed a promising trend in treating mice with lung cancer, implying that modulating the microbiome may impact the treatment of lung cancer (17). Therefore, we need to further explore the relationship between the microbiota and lung cancer so as to again a better understanding about the underlying mechanisms of their interaction.
A previous study demonstrated that the microbiota of an individual could keep long-term stable, which highlights the potential of the microbiota as a diagnostic method and therapeutic target (18). As saliva and sputum samples are susceptible to interference by the oral microbiota and it is usually difficult to obtain lung tissue specimens from patients with advanced lung cancer, and some experts believe that BALF is a viable option for sampling of the lung microbiome because BALF microbiome will not be confounded by oral contamination (19-21). To validate the previous research and provide a more specific understanding about how the lung microbiome affects lung cancer patients versus individuals with benign pulmonary diseases, the present study was intended to further apply 16S rRNA-based next-generation sequencing to investigate the differences in taxonomic and derived functional profiles in the lung microbiota in BALF samples.
Methods
Patients and sample collection
Enrolled in this study were patients who presented suspicious nodules on CT images and underwent clinical bronchoscopy at the Affiliated Hospital of Xuzhou Medical University (Xuzhou, China) between July 2018 and June 2019. Patients with acute pulmonary infection, second primary tumors, other lung comorbidities such as COPD, pulmonary fibrosis and bronchiectasis and other immune metabolic diseases or treated with antibiotics within a month were excluded from the study. Finally, a total of 54 patients were recruited in the research, including 32 patients who were pathologically diagnosed with lung cancer, and 22 patients who were diagnosed with benign pulmonary diseases as the control. Bronchoalveolar lavage (BAL) was performed on the side of the lung suspicious nodules following a standardized protocol developed to minimize oral contamination (22), and then 2 mL BALF was collected from each patient and stored at −80 °C within 30 min. In addition, prior to bronchoscopy, 10–20 mL sterile 0.9% saline was washed through the bronchoscope and divided into 5 aliquots in a sterile centrifuge tube to serve as the negative control. The demographic and clinical data of all participants including age, gender, body mass index (BMI), smoking history, smoking index, tumor markers, pathology type and tumor staging were recorded. The study (XYFY2019-KL110-01) was approved by the Medical Ethics Committee at the Affiliated Hospital of Xuzhou Medical University and written informed consent was obtained from the subjects.
Extraction of genome DNA
Total genome DNA from BALF samples was extracted using CTAB/SDS method. The DNA concentration and purity were monitored on 1% agarose gels using Qubit® dsDNA Assay Kit in Qubit® 2.0 Flurometer (Life Technologies, CA, USA) and NanoPhotometer® spectrophotometer (IMPLEN, CA, USA). The isolated DNA of BALF was diluted to 1 ng/µL using sterile water according to the concentration aimed to amplify 16S rRNA genes (V3–V4).
16S rRNA gene amplicon sequencing
16S rRNA genes of distinct regions (16SV3-V4) were amplified using specific primer with the barcode and Phusion® High-Fidelity PCR Master Mix (New England Biolabs). PCR products were electrophoresis on 2% agarose gel for detection, and samples with bright main strips between 400–450 bp were chosen to be purified with Qiagen Gel Extraction Kit (Qiagen, Germany). Sequencing libraries were generated usingTruSeq® DNA PCR-Free Sample Preparation Kit (Illumina, USA) following the manufacturer’s recommendations and index codes were added. The library quality was assessed on the Qubit@ 2.0Fluorometer (Thermo Scientific) and Agilent Bioanalyzer 2100 system and sequenced on an IlluminaHiSeq2500 platform and 250 bp paired-end reads were generated.
Data analysis
Samples were demultiplexed using an in-house script. Adaptors were trimmed, and paired end sequences were joined using FLASH (v1.2.7). Paired sequences were input into QIIME 2 (2019.7). Sequences were quality filtered and denoised using deblur (q2-deblur) with parameters on 400 bp amplicons to generate amplicon sequence variants (ASVs). A phylogenetic tree was built using fragment insertion into the gg-13-8-99 identity tree backbone with q2-phylogeny; taxonomic assignments were made with a naïve Bayesian classifier trained against the same reference (q2-feature-classifier). In cases where the classifier or reference database was unable to describe a taxonomic level (for instance, a missing genus), the taxonomy was described by inheriting the lowest defined level using a custom python script. Following sequencing and denoising, 24,285 high quality reads on average were retained. Alpha diversity was calculated as observed ASVs, Chao1, Shannon diversity, and Faith’s phylogenetic diversity using q2-diversity in QIIME 2 (2019.7). Beta diversity was measured using the Bray-Curtis metrics, unweighted UniFrac and weighted UniFrac on rarefied data (q2-diversity). PERMANOVA was applied to evaluate the diversity differences between groups. Principal coordinate analyses (PCoA) were performed to visualize distance of samples based on Bray-Curtis metrics, unweighted UniFrac and weighted UniFrac. Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt Version 2.2.0-b) was designed to predict KO abundance from ASVs data, and differentially enriched KEGG (Kyoto Encyclopedia of Genes and Genomes) pathways were estimated according to the LDA (Linear discriminant analysis) score by LEfSe (LDA Effect Size). The random forest model was performed to estimate the importance of each differential genus and predict lung cancer based on the receiver-operating characteristic curve (ROC) by the value of the AUC.
Statistical analysis
Wilcoxon rank sum test was used to perform hypothesis test on inter-group taxonomic abundance data (the occurrence rate >10% and the average abundance >0.5%) to obtain P value based on taxonomic abundance table of different levels and the P values were adjusted by the Benjamini-Hochberg correction for multiple tests when required. The relationships between the microbial data and clinical parameters were assessed using Spearman correlation test. All statistical data analyses were performed using R software (Version 3.6.1).
Results
Baseline clinical characteristics of the participants
The demographic and clinical data of the 54 participants including 32 patients with lung cancer and 22 patients with benign pulmonary diseases are shown in Table 1. No significant difference in age, gender, BMI, smoking status and smoking index was elicited between the two groups.
Table 1. The baseline clinical characteristics of the participants.
| Variables | Patients with lung cancer (N=32) | Patients with benign pulmonary diseases (N=22) | P value* |
|---|---|---|---|
| Age (years) | 64.3 (8.44) | 56.5 (14.30) | 0.0547 |
| Gender | 0.25 | ||
| Male | 23 (71.9) | 12 (54.5) | |
| Female | 9 (28.1) | 10 (45.5) | |
| BMI (kg/m2) | 23.1 (3.09) | 21.3 (2.95) | 0.0782 |
| Smoking status | 1 | ||
| Never smoking | 15 (46.9) | 13 (59.1) | |
| Ever smokers | 10 (31.2) | 5 (22.7) | |
| Current smokers | 7 (21.9) | 4 (18.2) | |
| Smoking index | 504 (627) | 252 (375) | 0.178 |
| Tumor markers | |||
| CEA | 78.05 (176.1) | 3.38 (2.79) | 0.00039* |
| NSE | 27.46 (30.59) | 14.43 (3.97) | 0.03104* |
| CYFRA21-1 | 11.3 (25.57) | 2.22 (0.92) | 0.00003* |
| Histology | |||
| LUAD | 16 (50.0) | – | |
| LUSC | 9 (28.1) | – | |
| SCLC | 7 (21.9) | – | |
| Tumor stage | |||
| I | 7 (21.9) | – | |
| II | 0 (0.0) | – | |
| III | 3 (9.4) | – | |
| IV | 15 (46.9) | – | |
| Limited stage | 3 (9.4) | – | |
| Extensive stage | 4 (12.5) | – |
The data are shown as the mean (standard deviation) for continuous variables and number (%) for categorical variables. *, P<0.05. BMI, body mass index; LUAD, adenocarcinoma; LUSC, squamous cell carcinoma; SCLC, small cell lung cancer; CEA, carcinoembryonic antigen; NSE, neuron specific enolase; CYFRA21-1, cytokeratin 19 fragment.
Alpha and beta diversity between lung cancer and control groups
Richness and evenness in the lung microbiome of the lung cancer patients generally resembled those of the patients with benign pulmonary diseases using observed ASVs (P=0.89), Chao1 (P=0.85), Shannon (P=0.086) and faith_pd index (P=0.17) to display the alpha diversity (Figure 1A). However, the beta diversity of the two groups was significantly different (P<0.05) (Figure 1B), and a trend of distinct separation in microbiota composition was also observed in the two groups using principal coordinate analysis (PCoA) plot based on the bray-curtis distance (PERMANOVA, P<0.01) (Figure 1C), weighted and unweighted UniFrac distance matrix (Figure S1).
Figure 1.

Comparison of alpha-diversity and beta-diversity of the lung microbiota between patients with lung cancer and those with benign pulmonary diseases. (A) Observed ASVs, Chao1, Shannon and faith_pd indices were used to evaluate the evenness and richness in the lung microbiome between lung cancer and controls. Each p-value was calculated by Wilcoxon rank sum test; (B) difference in microbiota composition between the groups based on Bray-Curtis metrics was displayed by box plot according to the Wilcoxon rank sum test; (C) principal coordinate analysis (PCoA) of Bray-Curtis metrics. The proportion of variance explained by each principal component was performed in the corresponding box plot. Red and blue dot represent cancer and nonmalignant control, respectively.
Figure S1.

Principal coordinate analysis (PCoA) based on weighted (A) and unweighted UniFrac distance matrix (B). The axes are labeled with the variation they explain. The proportion of variance explained by each principal component by groups was performed in the corresponding box plot. Red and blue dot represent cancer and nonmalignant control, respectively.
Taxonomic profiles of the lung microbiota composition
According to the relative abundance of the microbiota in the BALF samples of the two groups, classification and analysis were carried out on the phylum, class, order, family and genus and species levels. The phylum/genus taxonomic profiles of the lung microbiota composition between the two groups are presented in Figure 2A,B, and the microbiota composition profiles based on the family level are displayed in Figure S2A. At the phylum level, Proteobacteria, Firmicutes and Bacteroidetes were the most common in both the cancer and control groups. Besides, the mean abundance of Actinobacteria, Fusobacteria and TM7 in both groups was greater than 0.5%. At the genus level, Stenotrophomonas, Prevotella, Streptococcus, Haemophilus and Neisseria were the core genera in the study samples. The relative abundance of TM7 was significantly increased (adjust P=0.005634) and Proteobacteria was significantly depleted (adjust P=0.01388) in patients with lung cancer compared with benign pulmonary diseases. Additionally, at the family level, the relative frequency of Ruminococcaceae, c: TM7-3 and Chitinophagaceae were increased more significantly in lung cancer patients than the controls (adjust P<0.05) (Figure S2B). Furthermore, six genera (c:TM7-3, Capnocytophaga, Sediminibacterium, Gemmiger, Blautia and Oscillospira) were more enriched in lung cancer patients (adjust P<0.05) and the relative abundance of four genera (Microbacterium, Stenotrophomonas, Lautropia and f:Pseudomonadaceae) was lower than that in the control group (adjust P<0.05) (Figure 2C).
Figure 2.

Taxonomic profiles of the lung microbiota in patients with lung cancer and those with benign pulmonary diseases. (A) The relative frequency of lung microbiota in each BALF sample at the phylum level; (B) the relative frequency of lung microbiota in each BALF sample at the genus level. Only the mean abundance within each group >0.5% taxa is shown; (C) the two significantly different bacterial phyla and 10 genera were displayed by box plot (Wilcoxon rank sum test. P adjust <0.05).
Predicted functional profiles of the lung microbiota
The LEfSe analysis was used to find significantly imbalanced KEGG metabolic pathways between lung cancer patients and benign pulmonary diseases patients, and the differences between the lung cancer patients versus controls are displayed in Figure 3. In general, there were 46 and 57 discrepant pathways enriched in the lung cancer and control groups respectively. The microbiota in the BALF samples of lung cancer patients always showed obvious metabolic behaviors for the pathways of Ribosome (with the metabolic pathways most affected), Pyrimidine metabolism and Purine metabolism. Conversely, several metabolic pathways, such as Two component system, Flagellar assembly, and Bacterial secretion system were overrepresented significantly in patients with benign pulmonary diseases.
Figure 3.

Prediction of the lung microbiota function. The impact of differentially enriched KEGG pathways between lung cancer and benign pulmonary diseases was evaluated through the linear discriminant analysis (LDA) score. Only metabolic pathways meeting an LDA score >2.0 are shown. Red and blue histograms represent the cancer and control respectively.
Potential bacterial biomarkers for lung cancer patients
A random forest model was used to predict lung cancer combining ten bacterial genera with the significant difference between groups (f:Pseudomonadaceae, Capnocytophaga, Stenotrophomonas, Microbacterium, Gemmiger, c:TM7-3, Oscillospira, Blautia, Lautropia, Sediminibacterium) with three clinical tumor markers (CEA, NSE, CYFRA21-1). The receiver operating characteristic (ROC) analysis was performed to validate the diagnostic ability of these potential biomarkers for lung cancer based on the significantly different genera between groups and tumor markers, and the calculated area under the curve (AUC) was 84.52% (95% CI: 74.06–94.97%). The AUC generated by ten remarkable differential genera and commonly used clinical diagnostic markers were 79.12% (95% CI: 66.41–91.83%) and 78.27% (95% CI: 65.73–90.81%) to distinguish lung cancer patients and patients with benign pulmonary diseases (Figure 4A). The genera f:Pseudomonadaceae and Capnocytophaga were found to play a more important role among the classifier compared with the clinical tumor markers (Figure 4B). The heat map of the Spearman’s rank correlation between significantly different genera between groups and tumor markers illustrates that the microbiota, especially Capnocytophaga, Sediminibacterium and c:TM7-3, enriched in the BALF of lung cancer patients vs. patients with benign pulmonary diseases was also positively and significantly correlated with the markers CEA and CYFRA21-1 (Figure S3).
Figure 4.

The random forest model based on the microbiota and tumor markers (TMs) to distinguish lung cancer and benign pulmonary diseases patients. (A) Receiver operating characteristic (ROC) curves with the ten significant differential genera (AUC =79.12%), three tumor markers for lung cancer (AUC =78.27%) and combination of these genera and clinical tumor markers (AUC =84.52%) to predict lung cancer versus benign pulmonary diseases; (B) the importance of Gini coefficient was arranged from top to bottom.
Figure S3.
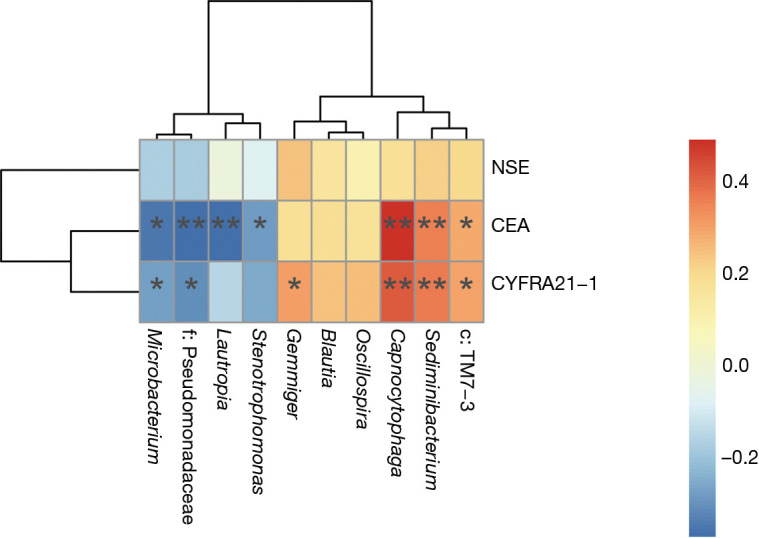
Heat map of Spearman’s rank correlation between significant differential bacterial genera and clinical tumor markers. Spearman correlation coefficient r on the right side of the heat map is between −1 and 1, with r<0 as negative correlation, represented by blue and r>0 as positive correlation, represented by red, respectively. *, P<0.05; **, P<0.01.
Discussion
The human microbiota is known to play a critical role in the development and progression of cancers by affecting host inflammation, immunity and metabolism (23,24). In this study, we examined and explored the taxonomic and derived functional profiles of the lung microbiome between patients with lung cancer versus benign pulmonary diseases, and found that the microbiota composition and metabolic activity differed significantly in BALF samples between the two groups.
The difference of patient clinical characteristics including age, gender, BMI and smoking may impact individual bacterial communities (16,25,26), so we ensured no statistically significant differences in these factors between the two groups, devoid of the interference with the final microbiota sequencing to the greatest extent.
The trend of differentiation was observed in the overall structure of the microbial communities between the two groups based on PCoA, which is consistent with the findings by Tsay et al. and Liu et al. (27,28), suggesting that there was a significant difference in the lung microbiome composition between lung cancer and nonmalignant diseases. The a-diversity analysis results showed that there was no significant difference in the richness and diversity of microbiota in the BALF samples from the patients with lung cancer versus non-malignant pulmonary diseases, which is similar to the report from Jin et al. (29). In contrast, Lee et al. reported that a-diversity of the microbiome was significantly different between the groups (9). We think that the heterogeneity may be due to the difference in the environmental and air particulates exposures, the geography and diet of enrolled patients, the depth of microbiota sequencing, and other factors (9,19,30,31).
Our study showed that the relative abundance of TM7 and c:TM7-3 was significantly elevated in BALF samples of lung cancer patients, which is in agreement with the finding of Lee et al. (9). A significant increase in TM7 was also observed in COPD patients compared with the healthy controls, indicating that TM7 may play an essential role in COPD and lung cancer patients (32). Moreover, Yan et al. demonstrated that Capnocytophaga and Veillonella could serve as potential biomarkers for the detection of lung cancer by quantitative PCR (33). In addition, Wang et al. showed that Veillonella and Capnocytophaga were enriched in the BALF samples of their 51 patients with lung cancer as compared with the healthy controls (16). It was found in our study that the level of Capnocytophaga in lung cancer patients was significantly higher than that in patients with benign pulmonary diseases, which is in concordance with previous studies. The stimulation of chronic inflammation by more Capnocytophaga may be one of the carcinogenic mechanisms of lung cancer, which is similar to the intrinsic relationship between Capnocytophaga gingivalis and oral squamous cell carcinoma (OSCC) (34). However, there was no significant increase in Veillonella in the BALF samples of lung cancer patients as compared with the controls. In addition, certain significantly differential genera in our study also differed from previous reports, and therefore further research is needed to validate the role of Veillonella in the development of lung cancer and verify the effectiveness of these differential genera. In the BALF samples of lung cancer patients enrolled in this study, the genus Sediminibacterium and Gemmiger were significantly increased versus benign pulmonary diseases, which, to the best of our knowledge, has not been reported in previous related studies. However, the relative abundance of the genus Sediminibacterium was also much higher in participants with type 2 diabetes mellitus (T2DM) and the genus Gemmiger was enriched in early hepatocellular carcinoma (HCC) versus cirrhosis (35,36). The role of these genera in the development of lung cancer remains to be further investigated in more larger-sample cohorts.
We further analysed the inferred metabolic function of the lung microbiome between the two groups based on the KEGG pathways predication. Ribosome and Pyrimidine metabolism pathways were significantly enriched in lung cancer patients. The above pathways were also reported to be correlated with the progression of breast cancer and lung cancer (16,37). The abnormal Ribosome biogenesis was reported to increase tumor cell proliferation and be negatively correlated with patient survival, and the nucleotide metabolism imbalance was closely related to tumor cell growth and proliferation (38,39). It was also found in our study that methane metabolism was enriched in lung cancer patients, which is consistent with the previous finding in colorectal cancer (40). Additionally, further research based on large clinical samples needs to focus on metabolic pathways that may be involved in the interaction between lung cancer and the microbiome.
In our study, all 10 distinct differential bacterial genera were applied to distinguish lung cancer and benign pulmonary diseases via building a random forest classifier and the AUC was 79.12%, indicating that these bacterial genera are closely linked to the development of lung cancer and in the classification of the two groups is moderately valuable. The combination of the bacteria and the clinical tumor markers showed a higher ROC value (AUC =84.52%) than that of the bacteria alone, suggesting that the joint multi-dimensional data could better predict lung cancer to some extent, but this model requires a larger sample cohort for exploration and validation. The f:Pseudomonadaceae and Capnocytophaga were found to play a more important role among the classifier compared with the clinical tumor markers, indicating that the lung microbiome may have the potential as bacterial biomarkers and new targets for the treatment of lung cancer, which is worth further exploration. Jin et al. also built a diagnostic model based on age, pack year of smoking and 11 types of bacteria to predict lung cancer and obtained a higher AUC (29). However, there were some differences between previous studies on the bacterial genus involved in classifying and identifying patients with lung cancer. Therefore, future larger-sample and dynamic longitudinal studies are required to verify the association between the microbiome and different pathological types of lung cancer based on different regions and populations.
Additionally, there are some limitations to our study. First, the number of patients enrolled in this study is not large enough and lung cancer patients were not classified by histological subtypes or different stages, and there may be heterogeneity. Second, the use of specific bacteria to distinguish lung cancer from benign lung diseases without a validation cohort may result in the false positive value and unreliability. Third, the study is a cross-sectional study and only illustrates the phenomenon from microbiology. Although the metabolic pathways that may be involved are initially predicted based on the microbiome results, the mechanism of their interaction is not further explored.
Conclusions
In the present study, we provided new insights into changes in the composition of the lung microbiome and predicted the metabolic function of the lung microbiome in patients with lung cancer. The differential genera discovered in this study may prove to be potential bacterial biomarkers and new targets for the diagnosis and treatment of lung cancer. Further related larger-sample studies are needed to validate the potential of these genera as bacterial biomarkers. Animal research is also needed to understand whether these differential genera affect the development of lung cancer by exploring possible mechanisms or whether lung cancer results in changes in the microbiome.
Figure S2.

Taxonomic profiles of the lung microbiota of lung cancer patients and patients with benign pulmonary diseases at the family level. Only the mean abundance within one group >0.5% taxa is shown. The significant different eight bacterial families were displayed by box plot (Wilcoxon rank sum test. P adjust <0.05).
Supplementary
The article’s supplementary files as
Acknowledgments
Funding: This research was supported by the National Natural Science Foundation of China (81871734, 81600044, 81700463), Jiangsu Provincial Medical Talent (ZDRCA2016053), Six Talent Peaks Project of Jiangsu Province (WSN-135, WSN-081), Advanced Health Talent of Six-One Project of Jiangsu Province (LGY2016042).
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. The study was conducted in accordance with the Declaration of Helsinki (as revised in 2013). The study (XYFY2019-KL110-01) was approved by the Medical Ethics Committee at the Affiliated Hospital of Xuzhou Medical University and written informed consent was obtained from the subjects.
Footnotes
Conflicts of Interest: All authors have completed the ICMJE uniform disclosure form (available at http://dx.doi.org/10.21037/tlcr-19-590). The authors have no conflicts of interest to declare.
References
- 1.Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer 2015;136:E359-86. 10.1002/ijc.29210 [DOI] [PubMed] [Google Scholar]
- 2.Arenberg D. Update on screening for lung cancer. Transl Lung Cancer Res 2019;8:S77-87. 10.21037/tlcr.2019.03.01 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ubags NDJ, Marsland BJ. Mechanistic insight into the function of the microbiome in lung diseases. Eur Respir J 2017;50:1602467. 10.1183/13993003.02467-2016 [DOI] [PubMed] [Google Scholar]
- 4.Maddi A, Sabharwal A, Violante T, et al. The microbiome and lung cancer. J Thorac Dis 2019;11:280-91. 10.21037/jtd.2018.12.88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hilty M, Burke C, Pedro H, et al. Disordered microbial communities in asthmatic airways. PLoS One 2010;5:e8578. 10.1371/journal.pone.0008578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Millares L, Ferrari R, Gallego M, et al. Bronchial microbiome of severe COPD patients colonised by Pseudomonas aeruginosa. Eur J Clin Microbiol Infect Dis 2014;33:1101-11. 10.1007/s10096-013-2044-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takahashi Y, Saito A, Chiba H, et al. Impaired diversity of the lung microbiome predicts progression of idiopathic pulmonary fibrosis. Respir Res 2018;19:34. 10.1186/s12931-018-0736-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coburn B, Wang PW, Diaz Caballero J, et al. Lung microbiota across age and disease stage in cystic fibrosis. Sci Rep 2015;5:10241. 10.1038/srep10241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee SH, Sung JY, Yong D, et al. Characterization of microbiome in bronchoalveolar lavage fluid of patients with lung cancer comparing with benign mass like lesions. Lung Cancer 2016;102:89-95. 10.1016/j.lungcan.2016.10.016 [DOI] [PubMed] [Google Scholar]
- 10.Polk DB, Peek RM. Helicobacter pylori: gastric cancer and beyond. Nat Rev Cancer 2010;10:403-14. 10.1038/nrc2857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mima K, Nishihara R, Qian ZR, et al. Fusobacterium nucleatum in colorectal carcinoma tissue and patient prognosis. Gut 2016;65:1973-80. 10.1136/gutjnl-2015-310101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peters BA, Hayes RB, Goparaju C, et al. The microbiome in lung cancer tissue and recurrence-free survival. Cancer Epidemiol Biomarkers Prev 2019;28:731-40. 10.1158/1055-9965.EPI-18-0966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cameron SJS, Lewis KE, Huws SA, et al. A pilot study using metagenomic sequencing of the sputum microbiome suggests potential bacterial biomarkers for lung cancer. PLoS One 2017;12:e0177062. 10.1371/journal.pone.0177062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang J, Mu X, Wang Y, et al. Dysbiosis of the salivary microbiome is associated with non-smoking female lung cancer and correlated with immunocytochemistry markers. Front Oncol 2018;8:520. 10.3389/fonc.2018.00520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang WQ, Zhao SK, Luo JW, et al. Alterations of fecal bacterial communities in patients with lung cancer. Am J Transl Res 2018;10:3171-85. [PMC free article] [PubMed] [Google Scholar]
- 16.Wang K, Huang Y, Zhang Z, et al. A preliminary study of microbiota diversity in saliva and bronchoalveolar lavage fluid from patients with primary bronchogenic carcinoma. Med Sci Monit 2019;25:2819-34. 10.12659/MSM.915332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gui QF, Lu HF, Zhang CX, et al. Well-balanced commensal microbiota contributes to anti-cancer response in a lung cancer mouse model. Genet Mol Res 2015;14:5642-51. 10.4238/2015.May.25.16 [DOI] [PubMed] [Google Scholar]
- 18.Zheng Y, Wang T, Tu X, et al. Gut microbiome affects the response to anti-PD-1 immunotherapy in patients with hepatocellular carcinoma. J Immunother Cancer 2019;7:193. 10.1186/s40425-019-0650-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu G, Gail MH, Consonni D, et al. Characterizing human lung tissue microbiota and its relationship to epidemiological and clinical features. Genome Biol 2016;17:163. 10.1186/s13059-016-1021-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bassis CM, Erb-Downward JR, Dickson RP, et al. Analysis of the upper respiratory tract microbiotas as the source of the lung and gastric microbiotas in healthy individuals. mBio 2015;6:e00037. 10.1128/mBio.00037-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang D, Su X, Yuan M, et al. The characterization of lung microbiome in lung cancer patients with different clinicopathology. Am J Cancer Res. 2019;9:2047-63. [PMC free article] [PubMed] [Google Scholar]
- 22.Segal LN, Alekseyenko AV, Clemente JC, et al. Enrichment of lung microbiome with supraglottic taxa is associated with increased pulmonary inflammation. Microbiome 2013;1:19. 10.1186/2049-2618-1-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garrett WS. Cancer and the microbiota. Science 2015;348:80-6. 10.1126/science.aaa4972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen J, Domingue JC, Sears CL. Microbiota dysbiosis in select human cancers: Evidence of association and causality. Semin Immunol 2017;32:25-34. 10.1016/j.smim.2017.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fang X, Wei J, He X, et al. Quantitative association between body mass index and the risk of cancer: A global Meta-analysis of prospective cohort studies. Int J Cancer 2018;143:1595-603. 10.1002/ijc.31553 [DOI] [PubMed] [Google Scholar]
- 26.Budden KF, Gellatly SL, Wood DL, et al. Emerging pathogenic links between microbiota and the gut-lung axis. Nat Rev Microbiol 2017;15:55-63. 10.1038/nrmicro.2016.142 [DOI] [PubMed] [Google Scholar]
- 27.Tsay JJ, Wu BG, Badri MH, et al. Airway microbiota is associated with upregulation of the PI3K pathway in lung cancer. Am J Respir Crit Care Med 2018;198:1188-98. 10.1164/rccm.201710-2118OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu Y, O'Brien JL, Ajami NJ, et al. Lung tissue microbial profile in lung cancer is distinct from emphysema. Am J Cancer Res 2018;8:1775-87. [PMC free article] [PubMed] [Google Scholar]
- 29.Jin J, Gan Y, Liu H, et al. Diminishing microbiome richness and distinction in the lower respiratory tract of lung cancer patients: A multiple comparative study design with independent validation. Lung Cancer 2019;136:129-35. 10.1016/j.lungcan.2019.08.022 [DOI] [PubMed] [Google Scholar]
- 30.Anand S, Mande SS. Diet, Microbiota and Gut-Lung Connection. Front Microbiol 2018;9:2147. 10.3389/fmicb.2018.02147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.He Y, Wu W, Zheng HM, et al. Regional variation limits applications of healthy gut microbiome reference ranges and disease models. Nat Med 2018;24:1532-5. 10.1038/s41591-018-0164-x [DOI] [PubMed] [Google Scholar]
- 32.Pragman AA, Kim HB, Reilly CS, et al. The lung microbiome in moderate and severe chronic obstructive pulmonary disease. PLoS One 2012;7:e47305. 10.1371/journal.pone.0047305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yan X, Yang M, Liu J, et al. Discovery and validation of potential bacterial biomarkers for lung cancer. Am J Cancer Res 2015;5:3111-22. [PMC free article] [PubMed] [Google Scholar]
- 34.Mager DL, Haffajee AD, Devlin PM, et al. The salivary microbiota as a diagnostic indicator of oral cancer: a descriptive, non-randomized study of cancer-free and oral squamous cell carcinoma subjects. J Transl Med 2005;3:27. 10.1186/1479-5876-3-27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Qiu J, Zhou H, Jing Y, et al. Association between blood microbiome and type 2 diabetes mellitus: A nested case-control study. J Clin Lab Anal 2019;33:e22842. 10.1002/jcla.22842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ren Z, Li A, Jiang J, et al. Gut microbiome analysis as a tool towards targeted non-invasive biomarkers for early hepatocellular carcinoma. Gut 2019;68:1014-23. 10.1136/gutjnl-2017-315084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meng S, Chen B, Yang J, et al. Study of microbiomes in aseptically collected samples of human breast tissue using needle biopsy and the potential role of in situ tissue microbiomes for promoting malignancy. Front Oncol 2018;8:318. 10.3389/fonc.2018.00318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Prakash V, Carson BB, Feenstra JM, et al. Ribosome biogenesis during cell cycle arrest fuels EMT in development and disease. Nat Commun 2019;10:2110. 10.1038/s41467-019-10100-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fan TWM, Bruntz RC, Yang Y, et al. Synthesis of serine and glycine fuels purine nucleotide biosynthesis in human lung cancer tissues. J Biol Chem 2019;294:13464-77. 10.1074/jbc.RA119.008743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sereme Y, Mezouar S, Grine G, et al. Methanogenic Archaea: Emerging Partners in the Field of Allergic Diseases. Clin Rev Allergy Immunol 2019;57:456-66. 10.1007/s12016-019-08766-5 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The article’s supplementary files as