

Abstract
Staphylococcus aureus is notoriously known for its rapid development of resistance to conventional antibiotics. S. aureus can alter its membrane composition to reduce the killing effect of antibiotics and antimicrobial peptides (AMPs). To obtain a more complete picture, this study identified the resistance genes of S. aureus in response to human cathelicidin LL-37 peptides by screening the Nebraska Transposon Mutant Library. In total, 24 resistant genes were identified. Among them, six mutants, including the one with the known membrane-modifying gene (mprF) disabled, became more membrane permeable to the LL-37 engineered peptide 17BIPHE2 than the wild type. Mass spectrometry analysis detected minimal lysyl-phosphatidylglycerol (lysylPG) from the mprF mutant of S. aureus JE2, confirming loss-of-function of this gene. Moreover, multiple mutants showed reduced surface adhesion and biofilm formation. In addition, four S. aureus mutants were unable to infect wax moth Galleria mellonella. There appears to be a connection between the ability of bacterial attachment/biofilm formation and infection. These results underscore the multiple functional roles of the identified peptide-response genes in bacterial growth, infection, and biofilm formation. Therefore, S. aureus utilizes a set of resistant genes to weave a complex molecular network to handle the danger posed by cationic LL-37. It appears that different genes are involved depending on the nature of antimicrobials. These resistant genes may offer a novel avenue to designing more potent antibiotics that target the Achilles heels of S. aureus USA300, a community-associated pathogen of great threat.
Keywords: Biofilms, Galleria mellonella, LL-37, Nebraska transposon mutant library, Staphylococcal resistance
Introduction
Staphylococcus aureus is a highly adaptable pathogen and could rapidly develop resistance to nearly all antimicrobials. It is documented that this organism became resistant to penicillin only two years after its large-scale production in 19421. Likewise, methicillin was introduced in 1960 and methicillin-resistant Staphylococcus aureus (MRSA) strains were observed in 19622. This led to the introduction of vancomycin in 1972. In 1988, vancomycin-resistant enterococci (VRE) emerged3. It appears that S. aureus is highly capable of reprograming its genome to diminish the effect of antibiotics. Nowadays, MRSA and VRE are detected in both clinical and community-associated settings, making it necessary to identify new antibiotics to fight such pathogens.
Antimicrobial peptides (AMPs), usually cationic and less than 50 amino acids (http://aps.unmc.edu/AP), are potent against resistant bacteria. The importance of AMPs is frequently attributed to rapid killing of invading pathogens. The classic amphipathic helix is the most popular model to understand the selective effect of cationic AMPs on anionic bacterial membranes rather than zwitterionic host membranes. A large-scale membrane damage makes it difficult for bacteria to repair4–6. Human cathelicidin LL-37 is a typical example in this class. It is a linear peptide with 37 amino acids and starting with a pair of leucines. LL-37 has 13 hydrophobic amino acids, 5 acidic and 11 basic amino acids, leading to a net charge of +6 at pH 7. LL-37 is capable of killing numerous pathogens, including bacteria, fungi, viruses, and parasites7–9. The structural basis for bacterial killing has been elucidated. LL-37 utilizes a long amphipathic helix spanning residues 2–31 to attack bacterial membranes10. In particular, four phenylalanines (F5, F6, F17 and F27) and arginines can directly interact with bacterial anionic phosphatidylglycerol (PG). Interestingly, LL-37 also regulates the immune system and plays a role in wound healing and cancer metastasis11–13. The multiple mechanisms of action of LL-37 are one possible reason for its lasting antimicrobial potency.
LL-37 is relatively long and thereby costly to synthesize by the solid-phase method. Multiple fragments of LL-37 are antimicrobial14,15. Using NMR spectroscopy, we identified the major antimicrobial peptide GF-17, corresponding to residues 17–32 of LL-3716. This protease-susceptible peptide formed the template for subsequent design of a protease-resistant peptide 17BIPHE2, which can kill the ESKAPE resistant pathogens, including Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species17. Both GF-17 and 17BIPHE2 are superior to LL-37 in disrupting the S. aureus biofilms18. Among the five basic amino acids in GF-17 and 17BIPHE2, R23 appears to be critical for membrane permeation and bacterial killing19,20. Interestingly, chemical modification of hydrophobic phenylalanines F17 and F27 with aliphatic or aromatic moieties modulates the ability of the 17BIPHE2 analogs to eliminate the ESKAPE pathogens21.
This study investigates how S. aureus JE2 responds to human cathelicidin LL-37, including its derived peptides GF-17 and 17BIPHE2. We took advantage of the Nebraska Transposon Mutant Library (NTML), which contains a collection of 1920 S. aureus mutants, each with a single non-essential gene disabled22. Our library screen relative to the wild type (WT) JE2 strain of S. aureus led to the identification of 24 susceptible mutant strains for us to study their growth, membrane permeation, biofilm formation, and fitness in an invertebrate animal model. We discuss these genes in terms of resistance mechanisms of S. aureus JE2, which lacks two plasmids compared to the community-isolated methicillin-resistant S. aureus USA300 LAC22.
RESULTS
Identification of susceptible genes based on the Nebraska Transposon Mutant Library of S. aureus JE2
Because human cathelicidin derived peptides are potent against MRSA and VRE, they have potential as new topical antimicrobials. We then identified the bacterial genes in response to the action of these LL-37 peptides based on reduced minimal inhibitory concentration (MIC) values of S. aureus mutants relative to the WT JE2 in 96-well plates. We hypothesized that bacterial strains became more susceptible to killing in the absence of resistance genes. The MIC values for these peptides are listed in supporting Table S1 as this study focuses on the MIC differences between the WT and mutants. The MIC values of LL-37 and its derived peptides against S. aureus JE2 are 3.1 μM. The use of a sub-MIC in the screening allowed the identification of susceptible S. aureus mutants that could be inhibited at reduced MIC values. Different media conditions were tested and found 50% tryptic soy broth (12.5% TSB for LL-37) was useful for screening. For comparison, both polystyrene and polypropylene plates were utilized. The polypropylene plates were used for the search of the susceptible mutants in response to LL-37. In total, we identified 24 susceptible S. aureus mutants by using LL-37 and its peptides (Table 1). These 24 mutants were more susceptible to at least one of the three LL-37 peptides. Among them, we observed MIC decreases for 12 mutants when treated with human LL-37. We also identified 11 mutants using GF-17 and 17 mutants in response to 17BIPHE2. Table 2 lists the possible roles of these S. aureus genes based on their corresponding gene ID. Each of these genes is disabled by the transposon insertion and harbored in an S. aureus mutant we identified. The insertion of the transposon for each was validated by designing primers (Table S2) followed by PCR. Each generated a band at the anticipated PCR product size (Figure S1), confirming the successful insertion of the transposon.
Table 1.
Fold decrease of MIC values of antimicrobial peptides and antibiotics against S. aureus JE2 and its mutants identified from the Nebraska Transposon Mutant Library1
| SAUSA300 ID | LL-37 | GF-17 | 17BIPHE2 | DFTamP1 | Dap | Linez | Tige | Tobra | Van |
|---|---|---|---|---|---|---|---|---|---|
| 0186 | 2 | 2 | 2 | ||||||
| 0394 | 2 | ||||||||
| 0645 | 2 | ||||||||
| 0646 | 2 | 2 | 2 | 2 | 2 | 2–4 | |||
| 0647 | 2–10 | 2 | 2–6 | 2 | 6 | 2 | 2 | 2–4 | |
| 0829 | 2 | 2 | 2 | 2 | |||||
| 0988 | 2 | 2 | 2 | 2 | |||||
| 1036 | 2 | 2 | |||||||
| 1037 | 2 | 2 | 2–4 | 8 | 2 | 2–4 | 4 | 4 | |
| 1089 | 2 | ||||||||
| 1097 | 2 | ||||||||
| 1228 | 2 | ||||||||
| 1255 | 2 | 2 | 2 | 2 | 8 | 2 | 4 | 2 | |
| 1308 | 2 | ||||||||
| 1336 | 2 | 2 | 2 | ||||||
| 1465 | 10 | 10 | 8 | 6 | 10 | 6 | 6 | 4 | 4 |
| 1503 | 2 | 2 | 2 | 2 | |||||
| 1515 | 2 | ||||||||
| 1962 | 8 | 2 | 2 | 4 | 2 | 2–4 | 4 | 2 | 2 |
| 1865 | 2 | 2 | |||||||
| 1867 | 2 | 2 | 2 | 2 | 2 | 2 | |||
| 1967 | 6 | 2 | |||||||
| 1898 | 2 | 2 | 2 |
Values in the table represent the MIC ratios of the antimicrobials against the WT and each mutant of S. aureus JE2. Abbreviations used are: Dap, daptomycin; Linez, linezolid; Tige, tigecycline; Tobra, tobramycin; Van, vancomycin.
Table 2.
Summary of the transposon mutants of S. aureus JE2 and their possible roles
| Gene ID | Gene name | Possible roles |
|---|---|---|
| SAUSA300_0186 | argC | N-acetyl-gamma-glutamyl-phosphate reductase |
| SAUSA300_0394 | FAD/NAD(P)-binding Rossmann fold superfamily protein | |
| SAUSA300_0645 | graR | DNA-binding response regulator |
| SAUSA300_0646 | graS | Sensor histidine kinase |
| SAUSA300_0647 | vraF | ABC transporter ATP-binding protein |
| SAUSA300_0829 | lipA | Lipoyl synthase |
| SAUSA300_0988 | trkA | Potassium uptake protein |
| SAUSA300_1036 | RNA methyltransferase | |
| SAUSA300_1037 | pheS | Phenylalanyl-tRNA synthetase subunit alpha |
| SAUSA300_1089 | lspA | lipoprotein signal peptidase |
| SAUSA300_1097 | pyrF | orotidine 5’-phosphate decarboxylase |
| SAUSA300_1228 | thrB | homoserine kinase |
| SAUSA300_1255 | mprF | Oxacillin resistance-related FmtC protein |
| SAUSA300_1308 | arlR | DNA-binding response regulator |
| SAUSA300_1336 | Hypothetical protein | |
| SAUSA300_1465 | 2-oxoisovalerate dehydrogenase, E1 component, beta subunit | |
| SAUSA300_1469 | argR | arginine repressor |
| SAUSA300_1503 | Putative competence protein ComGB | |
| SAUSA300_1515 | ABC transporter permease | |
| SAUSA300_1865 | vraR | DNA-binding response regulator |
| SAUSA300_1867 | Conserved hypothetical protein | |
| SAUSA300_1898 | Conserved hypothetical protein | |
| SAUSA300_1962 | phiPVL ORF39-like protein | |
| SAUSA300_1967 | conserved hypothetical phage protein |
Next, we compared the susceptibility of the identified S. aureus mutants relative to the WT in the presence of database-designed peptide DFTamP1, daptomycin, and conventional antibiotics. Unlike 17BIPHE2, less mutants were more susceptible to DFTamP123 with MIC reduced by two fold or more (Table 1). Likewise, we only found a few mutants that showed decreased MIC values when daptomycin, linezolid, tobramycin, and vancomycin were utilized in the antimicrobial assays. However, nearly all the mutants were more susceptible to tigecycline (Table 1).
Mass Spectrometry Analysis of S. aureus JE2 and Its Mutants.
To validate the mutants, we also analyzed the lipid composition of the wild type JE2 and two known S. aureus mutants: mprF (SAUSA300_1255) and lspA (SAUSA300_1089) using a previously established method24. We selected the mprF mutant as a positive control since it has a defined function of modifying anionic PG with lysine (Figure 1). LspA is a membrane enzyme that cleaves the signal peptide from lipoproteins. As anticipated, a minimal amount of lysylphosphatidylglycerols (lysylPGs) was identified for the mprF mutant (Figure 1B), confirming a lack of function of this gene due to the transposon insertion. For the lspA mutant, there is a small decrease in the level of free fatty acids (Figure 1E), while changes for other lipids, including PG, cardiolipin, lysylPG, were insignificant. For the mprF mutant, there is a significant increase in digalactosyldiacylglycerol (DGDG, Figure 1A).
Fig. 1.
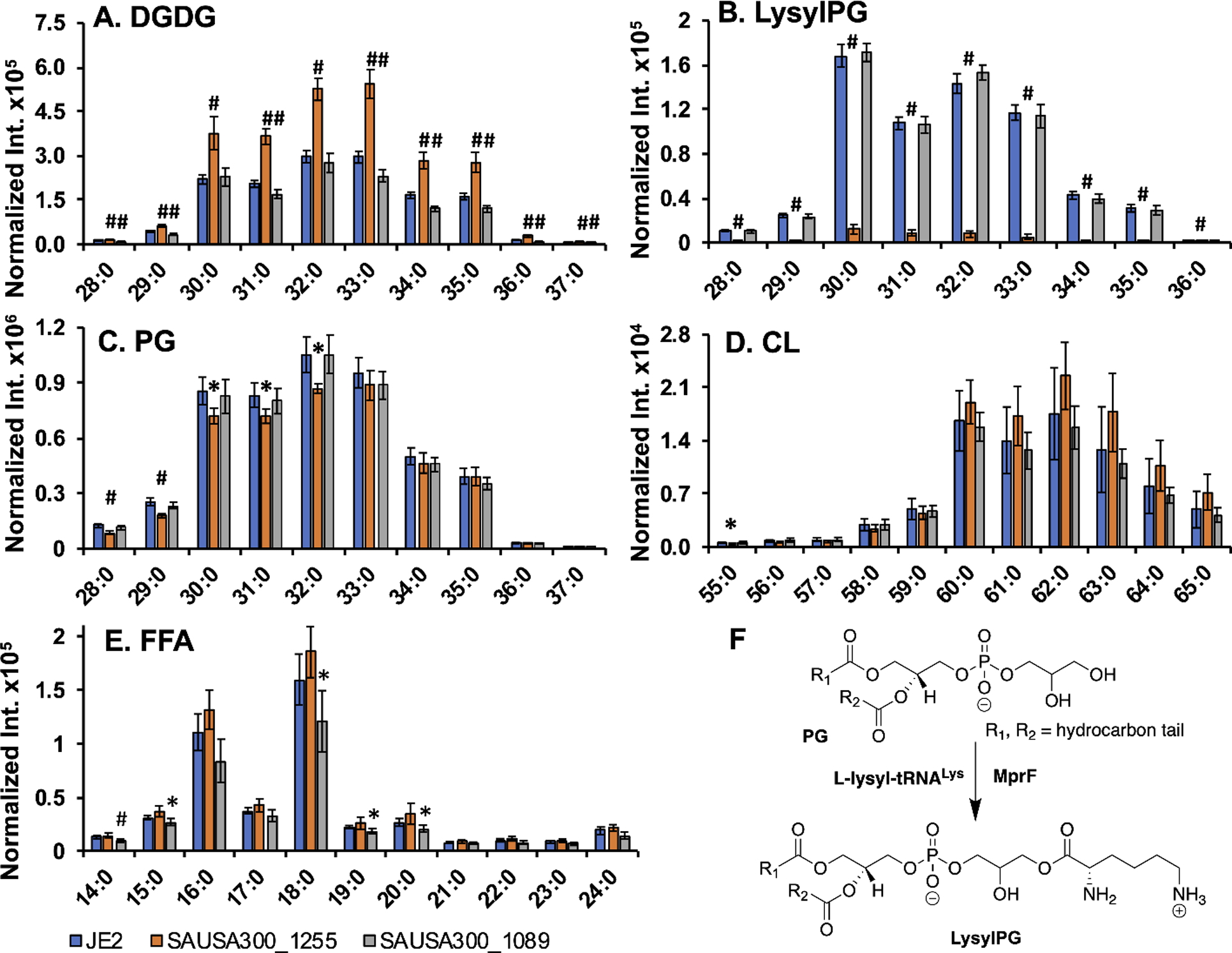
(A-E) HILIC-IM-MS lipidomics analysis of JE2, SAUSA300_1255, and SAUSA300_1089 strains. Digalactosyldiacylglycerol (DGDG; A) and lysyl-phosphatidylglycerol (LysylPG; B) species were observed in positive ionization mode as [M+NH4]+ and [M+H]+ adducts, respectively. Phosphatidylglycerol (PG; C), cardiolipin (CL; D) species, and free fatty acids (FFA; E) were observed in negative ionization mode as [M-H]−, [M-2H]2−, and [M-H]− adducts, respectively. Intensities were normalized by dry bacterial pellet weight and internal standard ion abundance. Statistical significance of differences in intensity compared to JE2 were determined for SAUSA300_1255 and SAUSA300_1089 by Student’s t-test. *, p < 0.05; #, p < 0.005. X axis numbers represent [total number of carbon atoms in the lipid acyl chains]:[total number of double bonds in acyl chains] of a specific lipid molecular species. All lipids shown are unsaturated and shown in the ascending order of chain length. (F) Scheme illustrating synthesis of lysylPG from PG via MprF.
Growth curves
We first checked whether any of the susceptible strains identified from the NTML resulted from growth defect. Relative to the wild type JE2, nearly all the mutants were found to grow well in the TSB medium and only the SAUSA300_1465 (Figure 2A) and SAUSA300_1097 (Figure 2C) mutants showed slightly reduced growth. Considering the growth differences are small and TSB is an artificial condition, we did not remove these two mutants from other assays below.
Fig. 2.
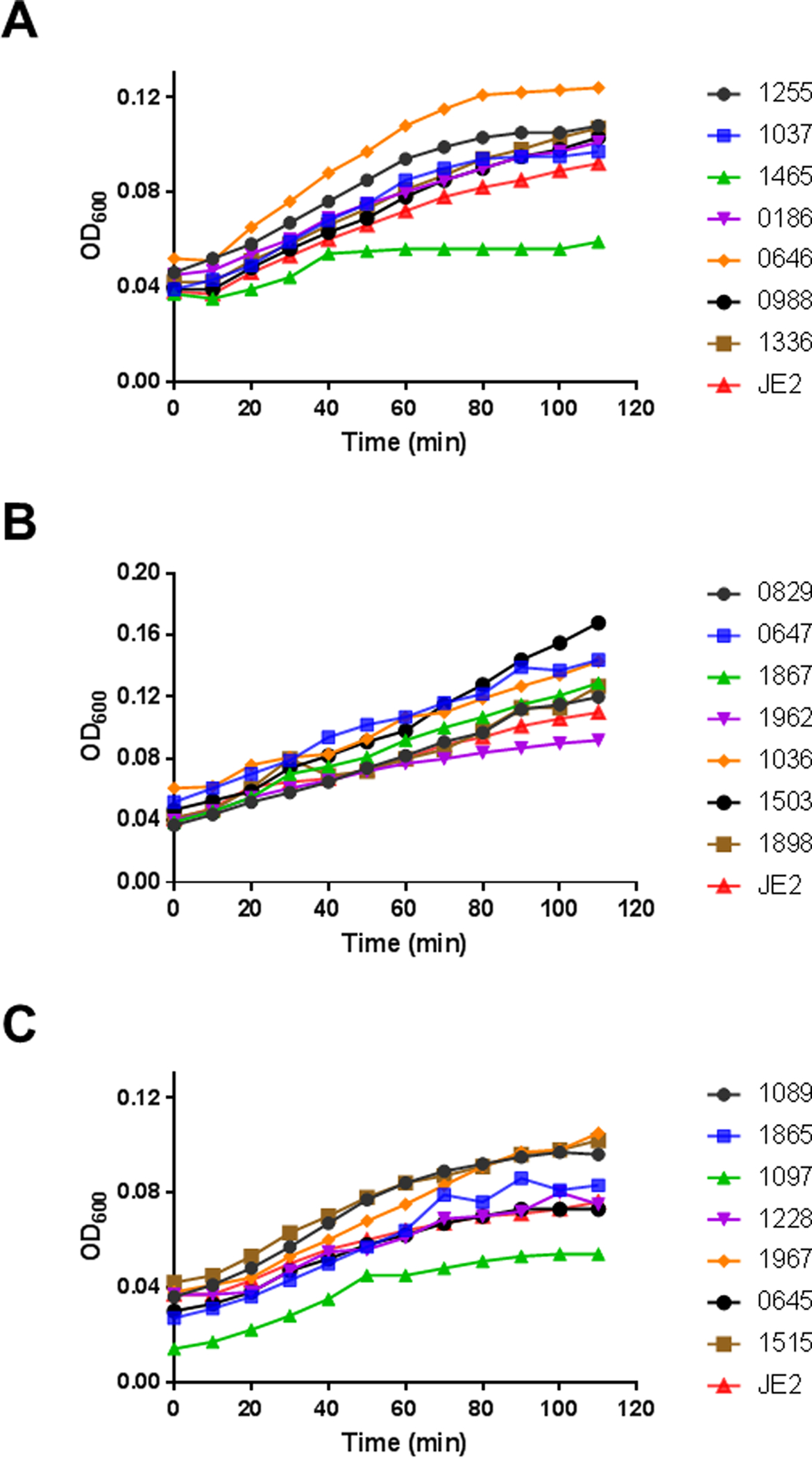
Growth curves of the S. aureus transposon mutants identified from library screening. For clarity, the mutants are presented in three panels: A, B, and C. The wild type JE2 was used as a control and presented in all the panels. The experiment was conducted in rich TSB media at 37°C with a constant shaking speed at 100 rpm and the absorbance was monitored at 600 nm.
Membrane permeation
S. aureus is a Gram-positive pathogen with a cell wall outside the membranes. Disruption of certain genes may compromise the integrity of cell wall/membranes, making such mutants more susceptible to antimicrobials. To gauge the membrane integrity, we made use of fluorescence spectroscopy in the presence of propidium iodide. This membrane non-permeable dye is unable to enter bacteria to bind DNA and emit fluorescence when the bacterial membrane is intact. We compared the effects of 17BIPHE2 and LL-37 treated at 1.56 μM (Figure 3, three panels). At this concentration, 17BIPHE2 was more potent in membrane permeation than LL-37 (Figure 3, right bottom). We then compared the fluorescence spectroscopy of the wild type JE2 with each mutant identified above. When treated with LL-37, mutants SAUSA300_1036, 1037, and 1255 became clearly more permeable than JE2, while mutants SAUSA300_0645, 0647, and 1503 were slightly more permeable. In the case of 17BIPHE2, nearly all the mutants became more permeable, although the magnitudes for SAUSA300_1097, 1228, and 1465 were smaller. Thus, 17BIPHE2 appeared to be more powerful than LL-37 in membrane permeation in most of the cases. The increased permeation of the mprF mutant (SAUSA300_1255) to both LL-37 and 17BIPHE2 is attributed to its lack of positively charged lysylPGs (Figure 1B)25. In the case of LL-37, there were essentially no differences between the WT and numerous mutants, including SAUSA300_0186, 0829, 1089, 1097, 1220, 1228, 1515, 1865, 1867, 1962, and 1967 suggesting that these genes might not be involved in membrane modifications. Rarely, mutant SAUSA300_0646 became less permeable to LL-37 than JE2, although the engineered peptide 17BIPHE2 remained effective (Figure 3, panel 1B).
Fig. 3.
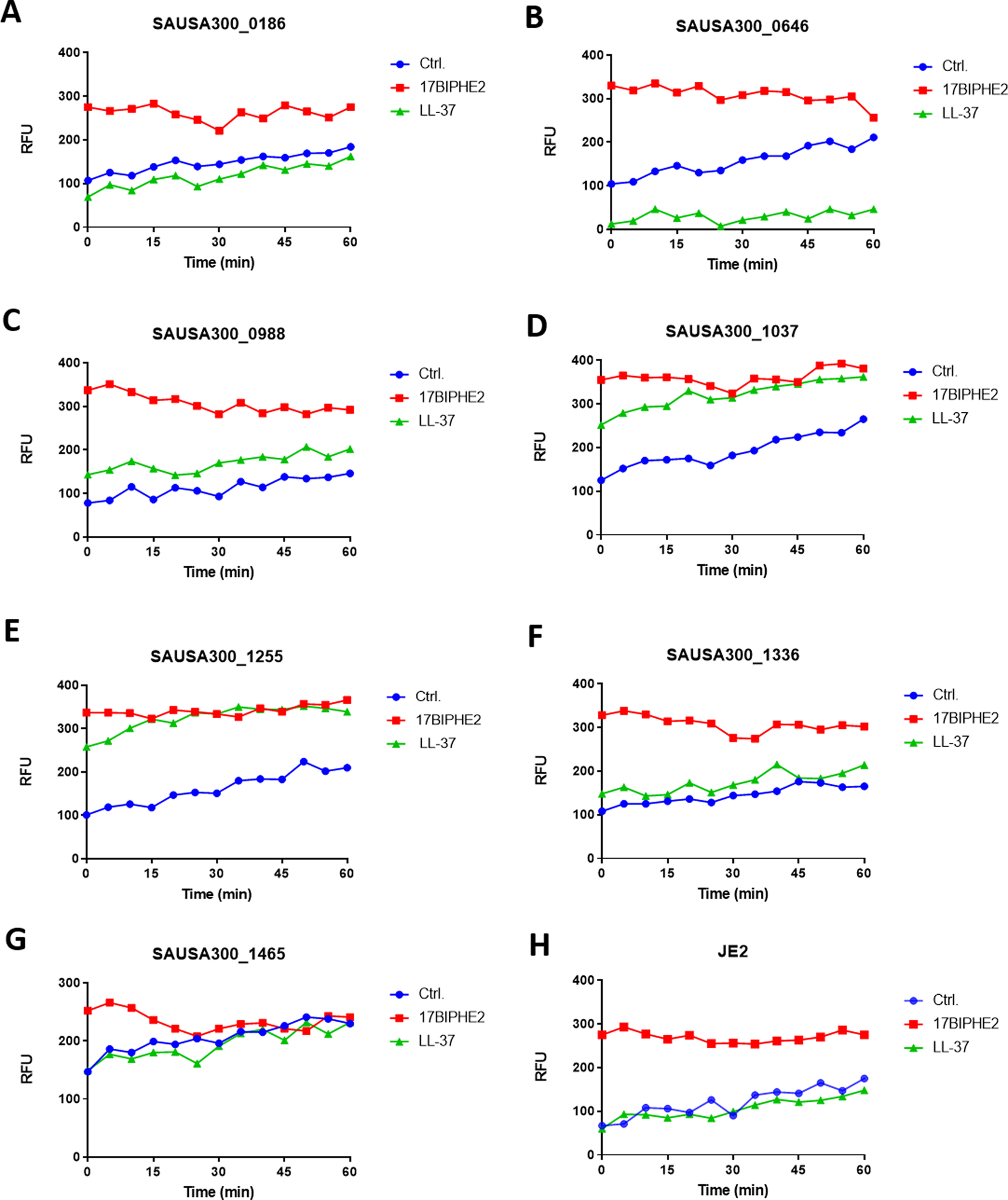
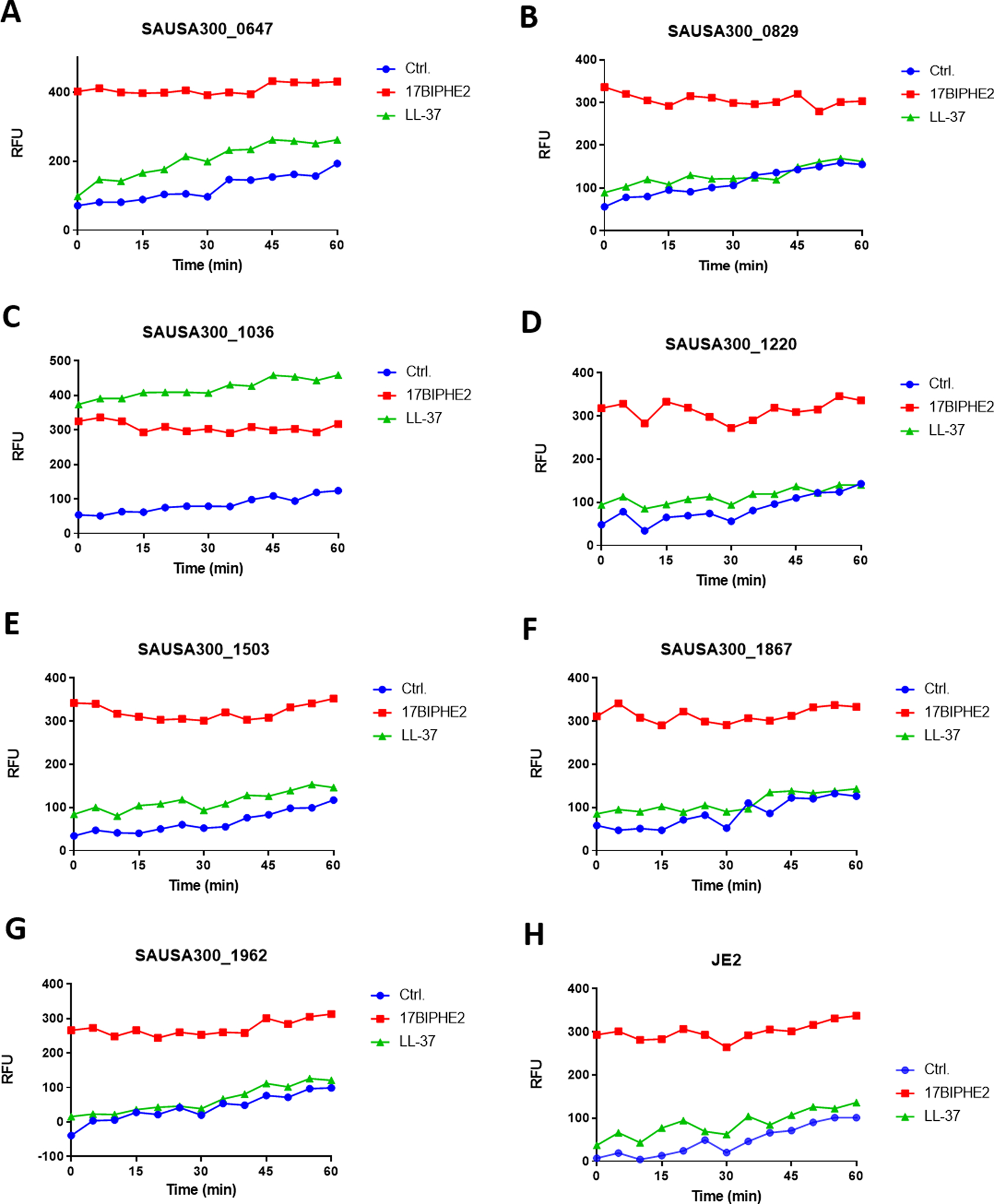
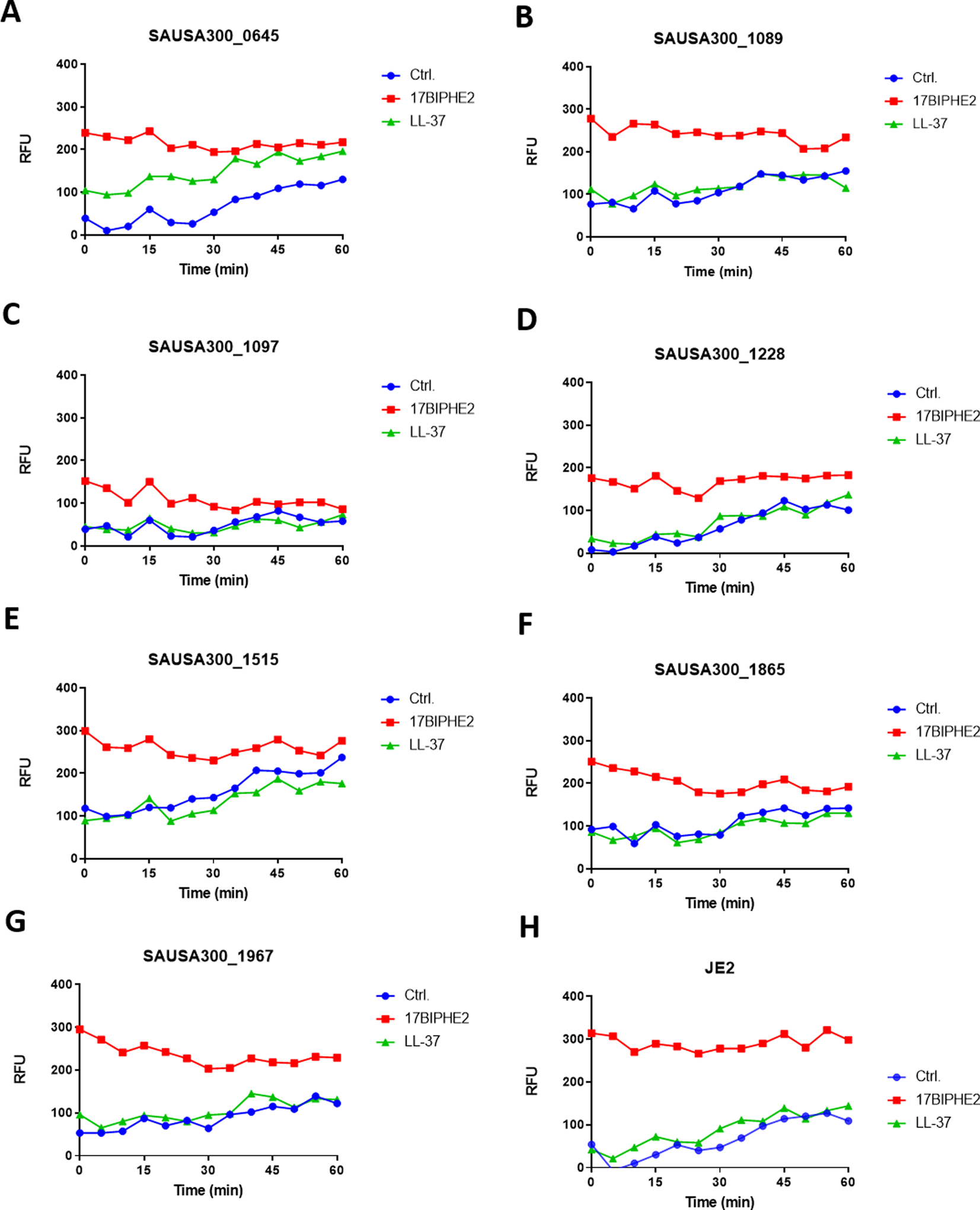
Membrane permeabilization assay of the S. aureus transposon mutants identified from library screening. For clarity, the mutants are presented in three panels: 1, 2, and 3. In each sub-panel, a single transposon mutant was treated with water, LL-37 or 17BIPHE2 at a sub-MIC concentration (1.56 μM) and compared to the wild type JE2 strain in a separate subpanel at the right bottom corner. Propidium iodide fluorescence increment is plotted as a measure of relative fluorescence intensities (RFU).
Biofilms of the susceptible transposon mutants of S. aureus
Biofilm formation constitutes one important mechanism for pathogen survival in the host to evade the host immune system as well as conventional antibiotics. Our identification of a set of S. aureus mutants here in response to human defense peptide LL-37 offered a great opportunity for us to evaluate the role of these genes in bacterial attachment and biofilm formation. Bacterial attachment to the substratum is usually the first step for biofilm formation. To better understand the biofilm forming process, we combined the use of two dyes26. While crystal violet (CV) stains all the biomass (e.g., live and dead cells) in bacterial biofilms, XTT, a tetrazolium salt, only stains live cells. During this initial stage of biofilms, it was found that most of the net biomass (Figure 4A, stained with CV) and the live cells (Figure 4B, stained with XTT) of the identified mutants are in correspondence, meaning the higher the biomass the more live cells in biofilms. The biomasses of SAUSA300_1465 and 1967 appeared to be composed of more non-cell substances than live cells. For mutants SAUSA300_0186, 0646, 0647, 0829, 0988, 1036, 1867, 1898, and 1962, however, the major contributor to the biomass was live cells (Figure 4B). Mutants like SAUSA300_1037, 1255, 1465, and 1503 had a biomass comparable to the wild type JE2. However, the remaining mutants had a reduced biomass than the wild type. For those mutants with reduced biomass, the biofilms mostly comprised live cells. These genes may influence the initial stage of bacterial biofilm formation. The attached mutants with more biomasses (CV stained) could better be viewed in the ratio plot between CV and XTT (Figure 4C). These include SAUSA300_0394, 1037, 1089, 1255, 1465, 1865, and 1967.
Fig. 4.
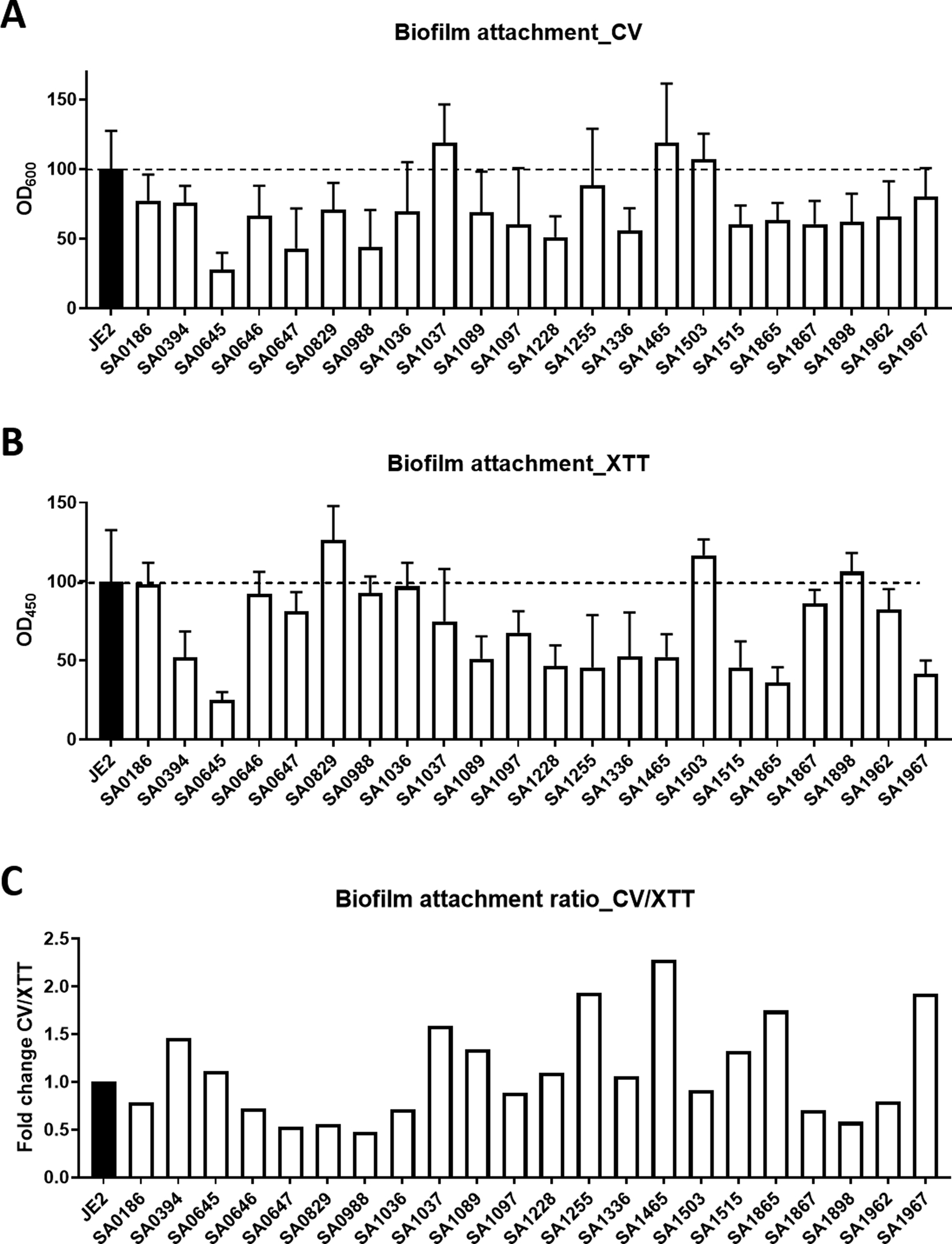
Bacterial attachment to polypropylene microtiter plate surfaces. S. aureus biofilms were quantitated by using both the crystal violet (CV) and the XTT staining methods. The CV for total biomass and XTT for live cell contents in the biofilms.
We also compared the biomasses in the mature biofilms of these S. aureus mutants, again stained with both CV (Figure 5A) and XTT (Figure 5B). Relative to the JE2 wild type, multiple mutant strains, including SAUSA300_0394, 0645, 1089, 1228, 1336, 1503, 1515, 1962, 1865, 1867, and 1967 were found to have a reduced biomass. In particular, mutants SAUSA300_1089, 1336, and 1865 had the least biomass. However, some S. aureus mutant strains (e.g., SAUSA300_0186, 1037, and 1465) accumulated a biomass similar to the WT, suggesting a minor role of these S. aureus genes in biofilm formation. Overall, we also found a proportional amount of biomass (Figure 5A) and live cell counts (Figure 5B) in the biofilms of these mutants, indicating the live S. aureus cells are the major component in mature biofilms. In most of the cases, including SAUSA300_0645, 0646, 0647, 0829, 0988, 1036, 1228, 1503, 1898, and 1967, the biomasses were higher than live cell counts, probably implying more secreted materials in the extracellular matrix. However, the biofilms of SAUSA300_0186, 1037, 1255, 1336, 1465, 1515, and 1865 were composed of mostly live bacteria. The dominance of either biomass (higher than WT) or live cells (lower than WT) in 24-h formed biofilms of S. aureus JE2 and its mutants could be viewed more clearly in the CV/XTT ratio plot (Figure 5C).
Fig. 5.
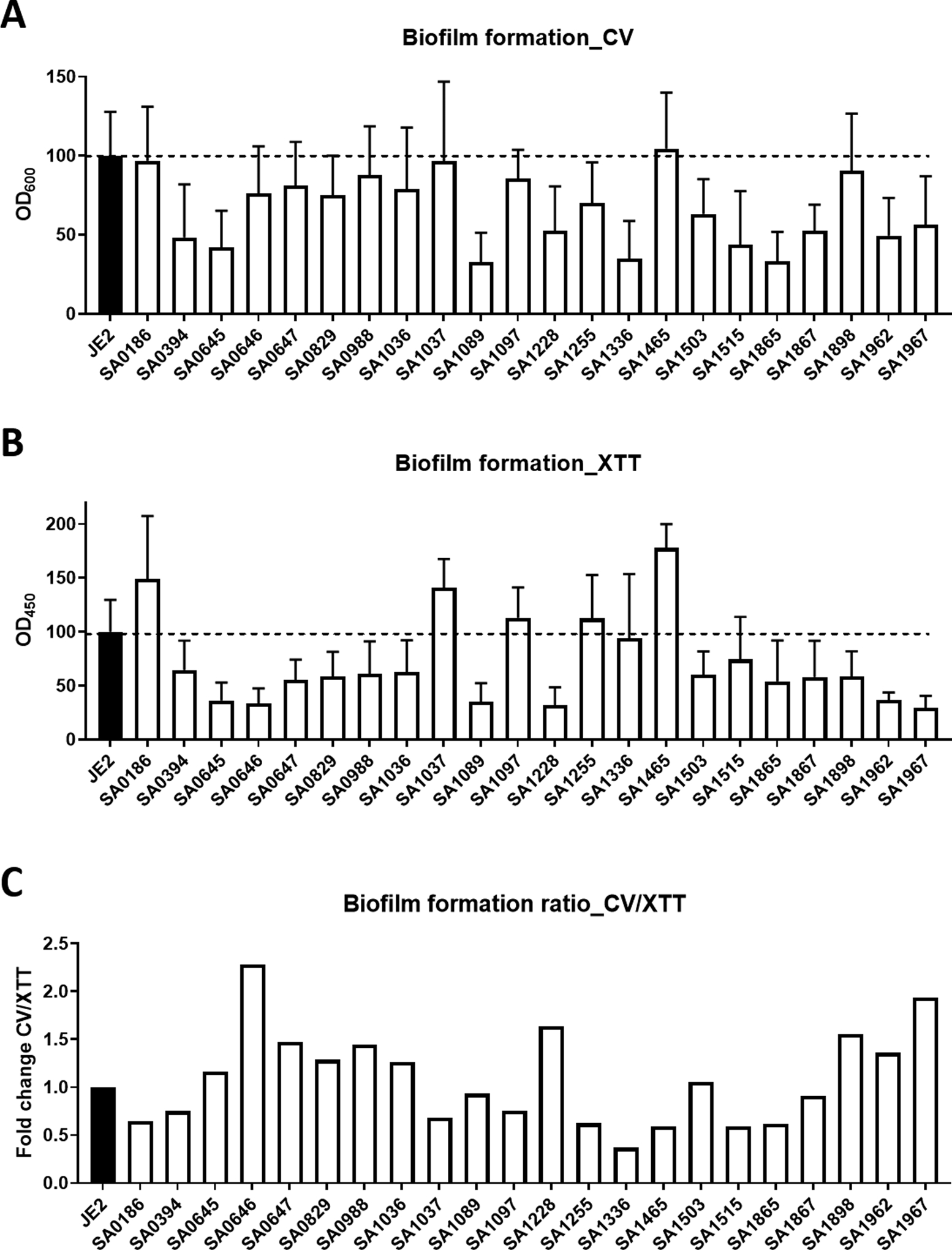
Biofilm formation of S. aureus transposon mutants on polypropylene microtiter plate surfaces. Biofilms were quantitated by using both the CV and the XTT staining methods.
It is also interesting to compare the trend of biomass in the attachment experiment with that in the mature biofilm. For the S. aureus mutants poor in biofilm formation in Figure 5 (e.g., SAUSA300_0645, 1089, and 1515), the corresponding less attached cells in Figure 4 would suggest an important role of initial attachment in biofilm formation of S. aureus. While the graR gene (SAUSA300_0645) of S. aureus regulates the mprF gene, the lipoprotein signal peptidase LspA (SAUSA300_1089) is required for lipoprotein processing.
Fitness of the S. aureus mutants in a G. mellonella wax moth model
Natural S. aureus strains may differ in their ability to infect the host. Those frequently identified clinically from patients are infectious pathogens. S. aureus USA300 LAC is one such pathogen isolated from communities. Compared to the S. aureus USA300 LAC, JE2 lacks two plasmids22. Here we compared the infectivity of the JE2-based mutant strains using a wax moth model we used previously20. S. aureus mutants suspended in phosphate buffer (PBS) were injected into the lumen of the worms at 1 × 106 colony forming units (CFU) per animal and the survival of these insects was recorded for 5 days. Non-responsive and morphologically distinct animals were considered dead and discarded daily during the study. For clarity, these survival curves were displayed in multiple panels in Figure 6. As a control, the PBS treated animals were all fine during the 5-day observation (panel A). In contrast, the S. aureus wild type JE2 appeared to be the most infectious with only 40% survival at the end of day 5. Surprisingly, the JE2 strain was more infectious than S. aureus USA300, where 60% insects survived (Figure 6H). Compared to JE2, all mutant strains became less infectious with few insect deaths. In the extreme cases, mutants SAUSA300_0988, 1089, 1336, and 1515 behaved like the PBS control and no animal death was observed. These genes might be essential for S. aureus infection. Moreover, there are multiple mutants that retained more than 90% insect survival. These include SAUSA300_0394, 0647, 0829, 1036, 1037, 1097, 1255, 1308, 1465, 1503, and 1962 mutants. Although both SAUSA300_1097 and 1465 showed slightly reduced growth in vitro, other mutants growed well (Figure 2), making it difficult to correlate mutant growth in vitro with animal survival. In addition, we observed 80% survival when the wax moths were infected with SAUSA300_1228, 1865, and 1967. The remaining mutants, including SAUSA300_0186, 0645 (graR), 0646 (graS), 1867, and 1898, were responsible for 30–40% death (60–70% survival). These mutants retained the degree of infectivity similar to S. aureus USA300, implying that these genes, including the graRS two component system (Table 2), play a smaller role in bacterial fitness in this insect model. We observed a larger MIC decrease for SAUSA300_0647, 1037, 1465, and 1962 (Table 1). However, these mutants showed poor infectivity (Figure 6). Thus, the magnitude of MIC decreases had no correlation with insect survival. Future studies are needed to elucidate how the known 20 endogenous AMPs from Galleria mellonella collected in the antimicrobial peptide database (http://aps.unmc.edu/AP) influence the infectivity of S. aureus JE2 and its mutants.
Fig. 6.
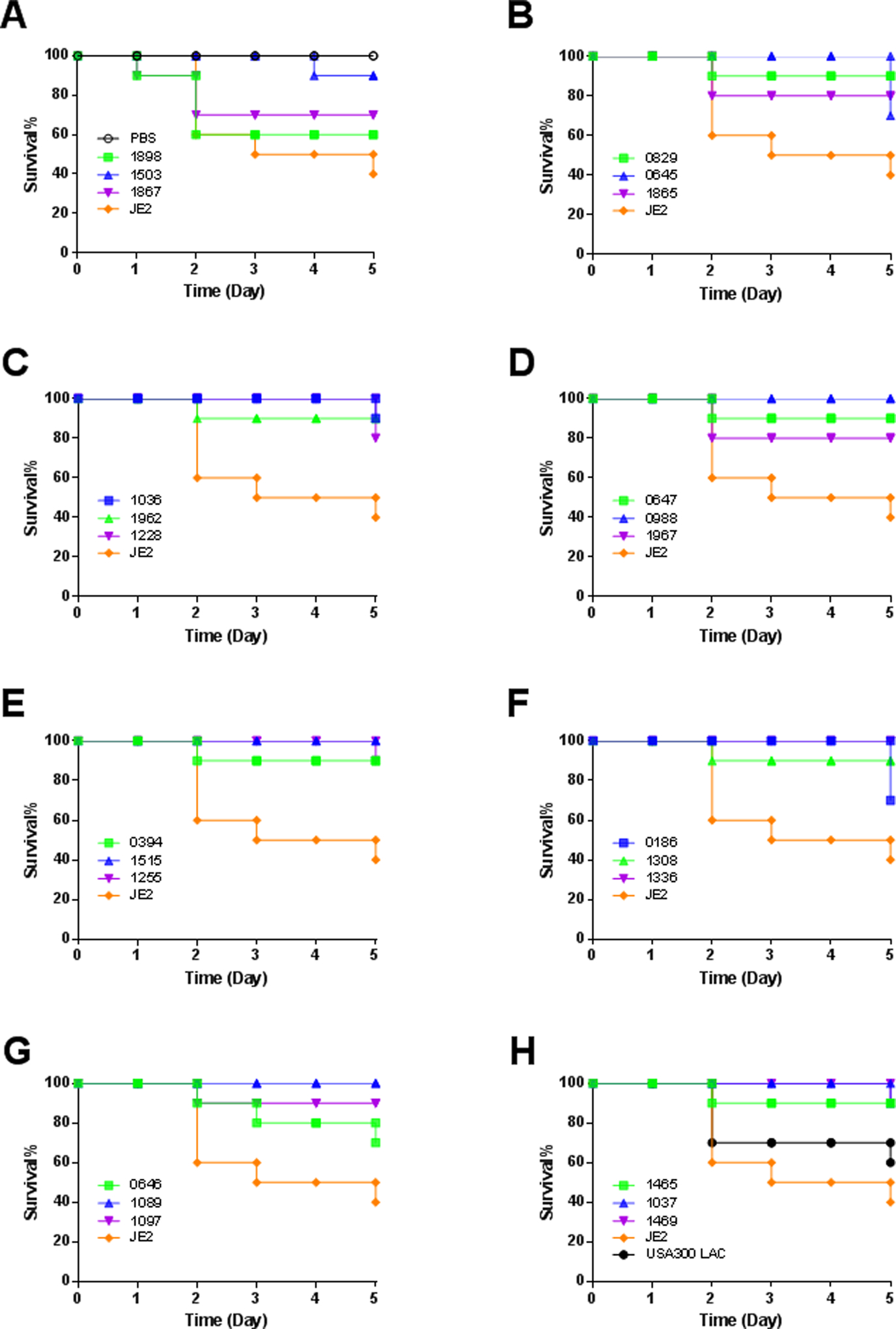
Fitness test of select S. aureus library mutants on a wax moth infection model. Shown are panels A-H with survival rates of Galleria mellonella determined with n=10 animals per group for five days. For clarity, the PBS vehicle is included only in panel A, S. aureus USA300 LAC only in panel H and wild type (JE2) in all panels.
DISCUSSION
S. aureus is highly effective in reducing the impact of antibiotics, including innate immune peptides such as LL-37. A few mechanisms are known. S. aureus secretes a variety of proteases (aureolysin, SepA, SpeB, and V8) to degrade LL-37. The cleavage sites tend to be located within the major antimicrobial region of LL-3714. S. aureus can also reduce the effect of basic AMPs by making the cell surface less negatively charged27,28. S. aureus also forms biofilms by secreting exopolymers as biofilm matrix molecules (exopolysaccharides, capsular polysaccharides, polysaccharide intercellular adhesin (PIA)). However, a more complete picture for S. aureus to evade killing by human cathelicidin LL-37 is lacking. Our unbiased screening of the S. aureus transposon mutant library22 using LL-37, its major antimicrobial peptide GF-17, and a chymotrypsin-resistant peptide 17BIPHE216,17 led to the identification of two dozen susceptible S. aureus mutant strains. It may be projected that the presence of these genes contributes to S. aureus resistance to cationic AMPs, thereby forming the “resistome” of S. aureus JE2. Based on possible roles, we may classify these resistant genes into eight groups: (1) AMP major resistance pathway (graR-0645, graS-0646, vraF-0647, and mprF-1255), (2) two-component system (vraR-1865 and SAUSA300_1867), (3) transporters (trkA-0988 and znuB-1515), (4) lipoprotein export (lspA-1089), (5) RNA biogenesis (SAUSA300_1036 and 1336), (6) biosynthesis and metabolism (argC-0186, lipA-0829, pheS-1037, pyrF-1097, and thrB-1228, SAUSA300_1465, and SAUSA300_1898), (7) bacterial secretion system (SAUSA300_1503), and (8) unknown function (phiPVL ORF39-like protein-1962 and conserved hypothetical phage protein SAUSA300_1967).
The five-component system GraRSX-VraGF of S. aureus is known to play a major role in AMP sensing and resistance. It regulates the expression of genes involved in major AMP resistance mechanisms, such as the dlt operon for D-alanylation of lipoteichoic acid, the mprF gene for PG lysinylation and the vraFG ABC transporter29,30. Our identification of multiple genes for this known system underscores the effectiveness of our manual screening method and validates the essential role of this system for the survival of this commensal bacterium31. Deletion of the graS, graR, mprF, or varF gene resulted in a two or more-fold decrease in MIC for cationic LL-37 peptides compared to the wild type JE2 (Table 1). Our findings support the role of this system by placing positively charged moieties on bacterial surface, as validated by our MS characterization (Figure 1), rendering S. aureus less susceptible to cationic LL-37 peptides. This S. aureus system appears to be powerful and shows a common resistance to different cationic peptides27. In addition, similar surface modification mechanisms are also utilized by other bacterial pathogens, including Gram-negative bacteria31.
S. aureus possesses multiple two-component systems (TCS), including the above upgraded five component system. TCS are a predominant means by which bacteria sense and respond to the environment32. Two-component systems have been implicated in antibiotic resistance33. The vraSR system appears to be involved in cell wall synthesis and thickening34. This system consists of two conserved modular proteins: a histidine protein kinase (vraS) and a response regulator protein (vraR)33. Inactivation of vraSR of MRSA resulted in increased susceptibility to daptomycin (DAP) and plasmid complementation restored DAP resistance to the WT level35. Likewise, the S. aureus SAUSA300_1865 (vraR) mutant identified here is more susceptible to LL-37 and its major antimicrobial peptide GF-17. Moreover, the vraSR mutant was more susceptible to phagocytosis by human polymorphonuclear leukocytes (PMNs) and had greater sensitivity to oxidation and lysozyme than wild-type Streptococcus suis36. We also showed here that the vraR mutant of S. aureus has a reduced ability to attach to the plastic surface for biofilm formation or infect wax moth larva.
LiaFSR orchestrates the cell envelope response to stress. The Lia system comprises the LiaRS TCS and the accessory protein LiaF. The latter acts as a negative regulator of LiaR-mediated gene regulation37. Mutants deficient in one or all of the liaFSR genes were susceptible to Lipid II cycle interfering antibiotics and to chemicals that perturbed the cell membrane integrity38. Mutations in liaFSR also have been linked with the development of DAP resistance in both Enterococcus faecalis and E. faecium39. A deletion of isoleucine in position 177 of LiaF, is likely to negatively affect bacterial response to DAP therapy and abolishes daptomycin bactericidal activity against vancomycin-resistant E. faecalis40. We found that, although the SAUSA300_1867 mutant is less effective in biofilm formation, it remained infective in the wax moth model similar to S. aureus USA300 LAC.
S. aureus also contains multiple K+ uptake systems, and four K+ transport systems have been identified, which include the proteins Trk, Kdp, Kup, and Kch41. Four nonlinked genes (trkA, trkE, trkG, and trkH) are key components of the constitutive Trk system42. The K+ uptake protein (TrkA) was found to be required for protamine and polymyxin B resistance in Vibrio vulnificus. The trkA mutant was much more easily killed by protamine and polymyxin B than the WT42. Moreover, this gene also influences the membrane potential of S. aureus related to antimicrobial susceptibility43. Biofilm formation in limited K+ conditions requires the Trk2 system in Streptococcus mutans41. When supplemented with 5 mM K+, the trk2 mutant strain formed a fragile and thin biofilm compared to the parent strain. Our results suggest that the SAUSA300_0988 mutant is more susceptible to membrane permeation by both LL-37 and its engineered peptide 17BIPHE2. Also, this gene is required for initial attachment of S. aureus for biofilm formation (Figure 3). This initial attachment appears to be significant for S. aureus infection because this transposon mutant is unable to infect wax moths (Figure 6D).
Zinc is an abundant transition metal that serves catalytic, structural, redox-modulatory roles44. In enterobacteria, zinc uptake is mediated by two major types of transporters: high-affinity ZnuABC (ABC transporter permease) and low-affinity ZupT45. Accumulated evidence revealed that mutants of the Znu permease could result in decreased zinc uptake and a distinctive growth defect45. In the case of S. aureus, two transporters related to zinc utilization and resistance are known46–48. We found the disruption of the ABC transporter (SAUSA300_1515) of S. aureus JE2 did not show growth defect. Like the WT JE2, it showed similar membrane permeation to LL-37, but increased permeation to its engineered peptide 17BIPHE2. However, this mutant showed reduced attachment and subsequent biofilm formation, probably due to a limited Zn2+ supply. Additional studies are required to understand the role of SAUSA300_1515 in Zn2+ resistance, infection and immunity.
Bacterial lipoproteins have many important functions, including substrate binding and transport, adherence, protein export and folding, cell signaling, sporulation, and antibiotic resistance49. Previous studies have identified lipoproteins as a virulence factor in bacteria50. Lipoprotein precursors undergo lipid modification by lipoprotein diacylglycerol transferase followed by removal of the signal sequence by lipoprotein type II signal peptidase (LspA)51. Type II signal peptidase has been identified in many Gram-negative and Gram-positive bacteria52. In Mycobacterium tuberculosis, the lspA mutant has a 104-fold reduction in CFU-forming efficiency with malachite green (a synthetic antimicrobial dye). Malachite green is decolorized faster in the presence of the lspA mutant than wild-type bacteria53. We found here that S. aureus, lacking the lspA (SAUSA300_1089) gene, had reduced biofilm-forming capability (Figure 5) and lost infectivity at least in the wax moth model (Figure 6G). Since there is no human protein homologous to bacterial LspA, this S. aureus protein is a potential target for structure-based design of novel inhibitors51.
RNA methyltransferases are a group of post-transcriptional RNA modification enzymes. Transfer RNA (tRNA) genes play an essential role in protein translation in all living cells54. To date, transfer RNA molecules are the most heavily methylated molecules55. Transfer RNA methyltransferases (TrmH or SpoU), which belong to Class IV, catalyze the transfer of a methyl group from S-adenosyl-L-methionine to the 2’-OH of guanosine 18 (Gm18) in tRNA56. It has been reported that the mutant lacking both Gm18 and ψ55 in E. coli will result in a decreased growth rate57. Prokaryotic rRNA modifications are mostly conducted by specific methyltransferases and pseudouridine synthases58. Some rRNA methyltransferases are important under stress conditions and their knockout affects antibiotic resistance and S. aureus virulence59. Methylation of rRNA in antibiotic-producing microorganisms confers resistance to their own toxic products60. Gene disruption of an rRNA methyltransferase gene hom12 results in the sensitivity of the mutant toward holomycin, the antibacterial compound produced by Yersinia ruckeri itself61. In order to avoid the toxicity of its own natural antibiotic ketolide, Streptomyces venezuelae expresses two paralogous rRNA methylase genes pikR1 and pikR262. In the case of S. aureus here, the SAUSA300_1036 mutant became highly membrane permeable to a low concentration of LL-37 and 17BIPHE2 (Figure 3, panel 2C) and poor in infecting the insect (Figure 6).
A further analysis of these mutants showed that those defective in biosynthesis and metabolism pathways had increased susceptibility to LL-37 compared to the wild type JE2 strain. These genes include pectate argC-0186, lipA-0829, pheS-1037, pyrF-1097, thrB-1228, acoB-1465, and lyase-1898. These mutants can compromise various functions of S. aureus. For instance, the SAUSA300_1097 and SAUSA300_1465 mutants displayed reduced growth in vitro. While SAUSA300_1037 became more membrane permeable to both LL-37 and 17BIPHE2, SAUSA300_1097 and 1228 are less permeable than the WT. In addition, the SAUSA300_1228 mutant has a reduced ability in biofilm formation. Most of these mutants became much less infective than the WT JE2. It appears that multiple genes are involved in Staphylococcal infection.
In natural transformation, exogenous DNA can be directly taken up from the environment by competent bacteria across the cell wall. In most bacteria, homologous type II secretion (T2S) systems and type IV pili (T4P) constitute the transformation machinery63. ComG, a locus with homology to T2S, is regulated by the competence state in naturally competent Gram-positive organisms. The comG locus consists of seven genes and a pseudogene64. The comGB gene encodes an integral membrane protein. A comGB mutant of Streptococcus mutans showed reduced biofilm formation and deficiency in DNA binding and uptake65. We found here that disruption of this gene (SAUSA300_1503) in S. aureus reduced biofilm formation and made the mutant strain much less infective (Figures 5 and 6).
Finally, it is useful to compare our findings with a recent screen of the NTML using polymyxin B66. Multiple genes, including SAUSA300_0645, 0646, 0647, 0988, 1089, 1254 (1255 in our case), and 1866, are also identified in our screen first completed in 2014. Because polymyxin is a cationic AMP from bacteria, this correspondence between the two independent screens indicates an overlap in the resistome of S. aureus in response to these two cationic AMPs.
Such a “resistome” overlap indicates that the set of the S. aureus mutants we identified from the NTML could be meaningful for us to compare the susceptibility of other AMPs as well as traditional antibiotics. Unlike LL-37 peptides, there are fewer S. aureus genes in response to DFTamP1, daptomycin, linezolid, tobramycin, and vancomycin (Table 1). A common feature of these peptides and antibiotics is that they all have a narrow-spectrum activity. While DFTamP1, daptomycin, linezolid, and vancomycin are only active against Gram-positive pathogens such as S. aureus, tobramycin is primarily used to treat the infection of Pseudomonas aeruginosa. In contrast, tigecycline has a broad-spectrum antibacterial activity and most of the S. aureus mutants we identified are susceptible to this antibiotics. Such a response pattern resembles LL-37 and its peptides, which kill both Gram-positive and Gram-negative pathogens. It appears that different resistant genes are involved depending on the nature of the antimicrobials. It is interesting that the five-component system (e.g., sensor graS, transporter vraF, and surface modifier mprF) is a general sensing and responsive system for all antimicrobials we tested in Table 1.
Conclusions
Human cathelicidin LL-37 is an innate immune peptide that plays an important role in host defense. This study identified a set of the resistant genes of S. aureus JE2 in response to human cathelicidin LL-37 and its engineered peptides. Gene disruption made the membranes of numerous mutants more permeable, reduced their capability in forming biofilms, and made most mutants less infectious. Our results suggest that antimicrobial susceptibility, membrane permeation, biofilm formation, and in vivo fitness of S. aureus are somehow connected. Such a connection reveals the importance of a bacterial gene-encoded network in mitigating toxins for survival. Our comparison of the S. aureus gene response patterns to nine antimicrobial peptides and antibiotics (Table 1) revealed that more genes might be involved in the network in facing broad-spectrum antimicrobials than narrow-spectrum ones. The major sensing and resistance system of S. aureus plays a general role in resistance to antimicrobial peptides and antibiotics, thereby offering an avenue to the development of a new generation of antibiotics or inhibitors that weaken this resistance mechanism. Indeed, crystal structures of the S. aureus lipoprotein signal peptidase LspA in complex with antibiotics have been determined, laying the foundation for designing a new generation of inhibitors.51 Such an inhibitor may be combined with structure-engineered peptide 17BIPHE2 to better combat MRSA. Alternatively, the antimicrobial peptide database67 can be utilized to design peptides with systemic efficacy against MRSA68.
METHODS
Peptide Synthesis and purification.
Peptides were purchased from Genemed Synthesis, Inc. (San Antonio, TX, USA). In brief, they were synthesized using the standard Fmoc Chemistry at 0.1 mmol scale. Peptides were then deprotected and cleaved from the Wang resin for LL-37 or rink amide resin for both GF-17 and 17BIPHE2 peptides using trifluoroacetic acid (TFA). After TFA removal on a rotary evaporator, the peptide was precipitated with dry ether and kept at 4°C. The ether was discarded and the peptide was solubilized and lyophilized. The peptide was finally purified via preparative HPLC. The quality of each peptide is indicated by Mass Spectroscopy (MS) and analytical HPLC (>95% pure). Fresh stock solutions were made by solubilizing peptides in autoclaved distilled water and their concentrations were determined by UV spectroscopy as described16.
Screen of the Nebraska Transposon Mutant Library of S. aureus.
S. aureus mutants were manually screened by using the NTML22. This mutant library, generated based on the S. aureus JE2 background, contains 1920 well-characterized strains, each with a single non-essential gene disabled due to the insertion of the transposon gene. Compared to S. aureus USA300 LAC, the JE2 strain lacks two plasmids deleted to facilitate the generation of the NTML22. The S. aureus mutant plates were duplicated and grown in 50% TSB in the presence of a sublethal concentration of 17BIPHE2, a protease-resistant peptide engineered based on human cathelicidin LL-3717. In each library plate, one strain was replaced with the wild type JE2 and another strain was replaced with TSB itself. These not-included strains were evaluated separately for potential susceptibility to peptides. Strains in the wells of each 96-well plate that showed no growth were validated by inoculating in 100% TSB media and grew overnight for antibacterial assays. Next day, bacteria were freshly inoculated in 100% TSB media to obtain logarithmic phase cultures (i.e., optical density at 600 nm ≈ 0.5). They were then diluted to OD600~0.001 in 50% TSB media and partitioned into a 96-well polystyrene microplate (Corning Costar Cat No. 3595) at 90 μL per well. After treatment with 10 μL of peptide solutions (two fold diluted to various concentrations), microplates were incubated at 37°C overnight and read on a ChroMate 4300 Microplate Reader at 630 nm (Awareness Technology, FL, USA). The minimal inhibitory concentration (MIC) is the lowest peptide concentration that fully inhibited bacterial growth. In this setup, susceptible strains have reduced MIC compared to JE2. A subset of potentially susceptible strains from the mutant library were also screened in the same manner as 17BIPHE2 by using human cathelicidin LL-37 or its major antimicrobial peptide GF-1716. Since LL-37 did not inhibit S. aureus in 100% TSB, we screened mutant strains susceptible to LL-37 in 12.5% TSB using polypropylene plates (Evergreen Scientific, CA, USA). For comparison, the inhibitory concentrations of select conventional antibiotics (Table 1) were also measured using the identified set of susceptible strains. We also evaluated the MIC values of this mutant set using DFTamP1, the first antimicrobial peptide designed based on the database filtering technology since this peptide is only active against Gram-positive pathogens23.
Validation of the Insertion of the Transposon Gene in the Identified S. aureus Mutants.
The insertion of the transposon gene to each S. aureus mutant was validated by PCR. PCR was performed on a Bio-Rad T100 Cycler (Bio-Rad, USA) using a mixture (25 μL) of reaction: 5 μL of 5 × Phusion HF Buffer (NEB, USA), 0.5 μL of dNTPs (10 mM), 0.5 μL of each primer (10 μM), 2 μL of DNA, 0.25 μL of Phusion DNA Polymerase (NEB, USA) and 8 μL of nuclease-free water. PCR cycling conditions were as follows: denaturation at 98℃ for 30 s, followed by 40 cycles of 98℃ for 10 s, 55℃ 30 s, and 72℃ for 30 s, final extension at 72℃ for 5 min. Primers (Supporting Table S1) were designed to amplify a targeted gene fused with the transposon. The designed primers were then paired with a transposon primer depending on the transcriptional direction of the mutated gene and the transposon gene. If correct, a DNA band with an anticipated size will be observed on the agarose gel (1%)69.
MS Characterization of Select S. aureus Mutants.
Lipid extraction was performed using a modified Bligh and Dyer method and untargeted lipidomics analysis was carried out by hydrophilic interaction liquid chromatography-ion mobility-mass spectrometry (HILIC-IM-MS) as described previously24,70. Briefly, dried bacteria pellets were suspended in 1 mL of water in 10 ml glass centrifuge tubes and sonicated in an ice bath for 30 min to dislodge the pellet. The internal standard was 1,2-dipentadecanoyl-sn-glycero-3-phosphoethanolamine (Avanti Polar Lipids, Inc., 850704) and the stock solution was prepared at 5 mM in chloroform. Four milliliters of chilled chloroform:methanol (1:2) solution and 4 μL of 5 mM internal standard were added to each tube (final concentration of internal standard in range of 1–5 μM). All of the samples were vortexed intermittently over 5 min. One milliliter of chilled chloroform and water were added to each tube and all of the samples were vortexed for 30 s. The samples were centrifuged at 1,000 × g for 10 min at 10°C to separate the organic and aqueous layers. The bottom organic layer was transferred to new 10 mL glass centrifuge tubes and dried in a SpeedVac concentrator. Dried extracts were reconstituted in 200 μL of chloroform:methanol (1:1) and stored at −80°C in glass vials until analysis. For analysis, 5 μL of sample was dried in a nitrogen atmosphere and reconstituted in 200 μl of acetonitrile:methanol (2:1).
Data acquisition was carried out on the Waters Synapt G2-Si. Post-acquisition data alignment, peak detection, and normalization were performed using the Progenesis QI software as described previously24,70. Data were normalized to dry bacterial pellet weight and internal standard ion abundance (positive mode [M+H]+ = 664.491 m/z; negative mode [M-H]− = 662.477 m/z). Lipid identification was based on retention time, m/z, and collision cross sections using an in-house database.
Simultaneous Bacterial Growth and Fluorescence-based Membrane Permeation Kinetics.
S. aureus wild type JE2 and its transposon mutants were inoculated and cultured overnight in 100% TSB media. Next day, the exponential phase cultures were made in 12.5% TSB media and were reconstituted in the same media conditions with a final OD600 ~0.11. Serially diluted 10× peptides (10 μL each well) were created in 96-well microtiter plates. Propidium iodide (2 μL) at a fixed concentration of 20 μM was added to each well followed by 88 μL of the bacterial culture. The plate was incubated at 37°C with continuous shaking at 100 rpm in a FLUOstar Omega (BMG LABTECH Inc, NC, USA) microplate reader. The fluorescence from the plate was read every 5 minutes for a total duration of 2 h with an excitation and emission wavelengths of 584 nm and 620 nm, respectively. Simultaneous absorbance measurements of bacterial growth of the transposon mutants were recorded at 600 nm. Wells without any peptide treatment were used as controls and compared with the wild type JE2. Data were analyzed using the MARS data analysis software provided by the manufacturer and representative plots were made using the Graph pad prism software. Since all the mutants treated at multiple peptide concentrations could not be accommodated in the same microtiter plate, we performed the experiments in multiple 96-well plates with a maximum of seven transposon mutants and one wild type JE2 in a plate.
Surface Adhesion of the S. aureus JE2 Mutants.
To understand the potential role of each gene in biofilm formation, we compared the ability of these S. aureus mutants in surface adhesion and biofilm formation. For bacterial attachment, transposon mutants were grown overnight in 100% TSB media. Two hundred microliters of this culture were added to each well of the microtiter plates. The plates were then incubated at 37 °C for 1 h. Media with planktonic bacteria were then pipetted out and chambers were washed with 1 × PBS to remove non-adherent cells. To provide additional insight, both CV and XTT staining methods were used to quantitate either the total biomass or live cells in the biofilms26. Live cells in the biofilms were quantitated using the XTT assay by following the manufacturer’s instructions with modifications. One hundred and eighty microliters of fresh TSB and 20 μL of XTT solution was added to each well and the plates were again incubated at 37°C for 2 h. Absorbance at 450 nm (media only wells stained with XTT served as the blank) was obtained using a ChroMate 4300 Microplate Reader. Percentages of biofilm growth was plotted by assuming 100% biofilm growth in the bacterial wells without peptide treatment. The total biomass content was quantitated using the standard CV staining method. In short, after removing the non-adherent cells, 200 μL of 99% methanol was added for fixation and the plates were allowed to sit for 15 min. The plates were aspirated and dried. Staining was done with 200 μL of 1% CV in water for 5 min. Excess stain was gently rinsed off with tap water, and plates were air-dried. Finally, stains were solubilized in 200 μL of 33% glacial acetic acid followed by colorimetric measurements at 600 nm.
Biofilm Formation of S. aureus Mutants.
In order to see the potential of the transposon mutants of S. aureus JE2 in forming biofilms, they were allowed for 24 h in polypropylene microtiter plates. Briefly, S. aureus cultures (106 CFU/ mL) in 100% TSB media were made from the exponential phase bacteria. Two hundred microliters of the above cultures were distributed to each well of the microtiter plate. The plates were incubated at 37°C for 24 h under static condition to form biofilms. Media were then pipetted out and the attached biofilms were washed with 1× PBS to remove the planktonic bacteria. To gain additional insight, biofilms for each mutant as well as the WT were formed in two different plates and stained with two different dyes: XTT and CV.
In Vivo Fitness Studies in an Invertebrate Model.
Wax moths Galleria mellonella are an established model to study pathogen infection71,72. Here we used this insect model to investigate the fitness of the S. aureus transposon mutants relative to the WT JE2. The animals (~250 mg/larva) were distributed by Timberline Live Pet Foods Marion, IL. Each transposon mutant of S. aureus JE2 was injected at a dose of 1×106 CFU/moth. Animals (n=10 per group) were kept at room temperature and observed daily for five days to record live and dead ones.
Statistics.
All experiments were replicated at least twice. For biofilms, plots represent the average values with standard deviation error bars. Membrane permeation experiments were duplicated for each condition and processed using the vendor’s software (MARS, BMG Labtech). For all the experiments, the level of significance was determined by performing paired Student’s t‐test with parameters of two‐tailed distribution. P values < 0.05 were considered significant (*).
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to Dr. Kenneth Bayles for his consistent support and helpful suggestions that made this study possible. We are indebted to the Bayles lab, especially Jennifer L. Endres who provided selected mutants for our pilot studies, duplicated the 21 plates of the S. aureus mutant library for this screen, and instructed us to validate the transposon insert in the mutants identified and reported herein before publication. We acknowledge the assistance of Dr. Tamara Lushnikova with the wax moth study.
Funding
This work was supported by the National Institute of Allergy and Infectious Diseases (NIAID) of the National Institutes of Health (NIH) under Award Number R01AI105147 to GW. The lipidomics research was supported by NIAID R01AI136979 to LX. XD is a UNMC visiting scholar who is also supported by the China Scholarship Council. The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
ABBREVIATIONS
- AMPs
antimicrobial peptides
- PBS
phosphate buffer
- CFU
colony forming units
- CL
cardiolipin
- DGDG
digalactosyldiacylglycerol
- CV
crystal violet
- HPLC
high performance liquid chromatography
- lysylPGs
lysylphosphatidylglycerols
- MRSA
methicillin-resistant Staphylococcus aureus
- MS
mass spectroscopy
- NTML
Nebraska Transposon Mutant Library
- PCR
polymerase chain reaction
- PG
Phosphatidylglycerol
- TCS
two-component system
- TSB
tryptic soy broth
- WT
wild type
- XTT
2,3-bis(2-methyloxy-4-nitro-5-sulfophenyl)-2H-tertazolium-5-carboxanilide
Footnotes
The authors declare no completing financial interest.
REFERENCES
- (1).Bayles KW (2000) The bactericidal action of penicillin: new clues to an unsolved mystery. Trends Microbiol. 8(6), 274–278. [DOI] [PubMed] [Google Scholar]
- (2).Zaffiri L, Gardner J, Toledo-Pereyra LH (2013) History of antibiotics: from fluoroquinolones to daptomycin (Part 2). J Invest Surg. 26(4), 167–179. [DOI] [PubMed] [Google Scholar]
- (3).Uttley AH, Collins CH, Naidoo J, George RC (1988) Vancomycin-resistant enterococci. Lancet. 331, 57–58. [DOI] [PubMed] [Google Scholar]
- (4).Boman HG (2003) Antibacterial peptides: basic facts and emerging concepts. J Intern Med. 254(3), 197–215. [DOI] [PubMed] [Google Scholar]
- (5).Lehrer RI (2007) Multispecific myeloid defensins. Curr Opin Hematol. 14(1), 16–21. [DOI] [PubMed] [Google Scholar]
- (6).Zasloff M (2002) Antimicrobial peptides of multicellular organisms. Nature 415(6870), 389–395. [DOI] [PubMed] [Google Scholar]
- (7).Frohm M, Agerberth B, Ahangari G, Stâhle-Bäckdahl M, Lidén S, Wigzell H, Gudmundsson GH (1997) The expression of the gene coding for the antibacterial peptide LL-37 is induced in human keratinocytes during inflammatory disorders. J Biol Chem. 272(24), 15258–15263. [DOI] [PubMed] [Google Scholar]
- (8).Sørensen O, Arnljots K, Cowland JB, Bainton DF, Borregaard N (1997) The human antibacterial cathelicidin, hCAP-18, is synthesized in myelocytes and metamyelocytes and localized to specific granules in neutrophils. Blood 90(7), 2796–803. [PubMed] [Google Scholar]
- (9).Bals R, Wang X, Zasloff M, Wilson JM (1998) The peptide antibiotic LL-37/hCAP-18 is expressed in epithelia of the human lung where it has broad antimicrobial activity at the airway surface. Proc Natl Acad Sci U S A. 95(16), 9541–9546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Wang G (2008) Structures of human host defense cathelicidin LL-37 and its smallest antimicrobial peptide KR-12 in lipid micelles. J Biol Chem. 283(47), 32637–32643. [DOI] [PubMed] [Google Scholar]
- (11).Scott MG, Davidson DJ, Gold MR, Bowdish D, Hancock RE (2002) The human antimicrobial peptide LL-37 is a multifunctional modulator of innate immune responses. J Immunol. 169(7), 3883–3891. [DOI] [PubMed] [Google Scholar]
- (12).Dorschner RA, Pestonjamasp VK, Tamakuwala S, Ohtake T, Rudisill J, Nizet V, Agerberth B, Gudmundsson GH, Gallo RL (2001) Cutaneous injury induces the release of cathelicidin anti-microbial peptides active against group A Streptococcus. J Invest Dermatol. 117(1), 91–97. [DOI] [PubMed] [Google Scholar]
- (13).Weber G, Chamorro CI, Granath F, Liljegren A, Zreika S, Saidak Z, Sandstedt B, Rotstein S, Mentaverri R, Sánchez F, Pivarcsi A, Ståhle M (2009) Human antimicrobial protein hCAP18/LL-37 promotes a metastatic phenotype in breast cancer. Breast Cancer Res. 11(1), R6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Wang G, Narayana JL, Mishra B, Zhang Y, Wang F, Wang C, Zarena D, Lushnikova T, Wang X (2019) Design of antimicrobial peptides: Progress made with human cathelicidin LL-37. Adv Exp Med Biol. 1117, 215–240. [DOI] [PubMed] [Google Scholar]
- (15).Vandamme D, Landuyt B, Luyten W, Schoofs L (2012) A comprehensive summary of LL-37, the factotum human cathelicidin peptide. Cell Immunol. 280(1), 22–35. [DOI] [PubMed] [Google Scholar]
- (16).Li X, Li Y, Han H, Miller DW, Wang G (2006) Solution structures of human LL-37 fragments and NMR-based identification of a minimal membrane-targeting antimicrobial and anticancer region. J Am Chem Soc. 128(17), 5776–85. [DOI] [PubMed] [Google Scholar]
- (17).Wang G, Hanke ML, Mishra B, Lushnikova T, Heim CE, Chittezham Thomas V, Bayles KW, Kielian T (2014) Transformation of human cathelicidin LL-37 into selective, stable, and potent antimicrobial compounds. ACS Chem Biol. 9(9), 1997–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Mishra B, Golla RM, Lau K, Lushnikova T, Wang G (2016) Anti-staphylococcal biofilm effects of human cathelicidin peptides. ACS Med Chem Lett. 7(1), 117–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Wang G, Epand RF, Mishra B, Lushnikova T, Thomas VC, Bayles KW, Epand RM (2012) Decoding the functional roles of cationic side chains of the major antimicrobial region of human cathelicidin LL-37. Antimicrob Agents Chemother. 56(2), 845–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Wang X, Mishra B, Lushnikova T, Narayana JL, Wang G (2018) Amino acid composition determines peptide activity spectrum and hot-spot-based design of merecidin. Adv Biosyst. 2(5), 1700259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Narayana JL, Mishra B, Lushinikova T, Golla RM, Wang G (2019) Modulation of antimicrobial potency of human cathelicidin peptides against the ESKAPE pathogens and in vivo efficacy in a murine catheter-associated biofilm model. Biochim. Biophys. Acta 1861, 1592–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Fey PD, Endres JL, Yajjala VK, Widhelm TJ, Boissy RJ, Bose JL, Bayles KW (2013) A genetic resource for rapid and comprehensive phenotype screening of nonessential Staphylococcus aureus genes. MBio. 4(1), e00537–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Mishra B, Wang G (2012) Ab initio design of potent anti-MRSA peptides based on database filtering technology. J. Am. Chem. Soc 134, 12426–12429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Hines KM, Waalkes A, Penewit K, Holmes EA, Salipante SJ, Werth BJ, Xu L (2017). Characterization of the mechanisms of daptomycin resistance among gram-positive bacterial pathogens by multidimensional lipidomics. mSphere 2(6), e00492–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Bayer AS, Mishra NN, Chen L, Kreiswirth BN, Rubio A, Yang SJ (2015). Frequency and Distribution of Single-Nucleotide Polymorphisms within mprF in Methicillin-Resistant Staphylococcus aureus Clinical Isolates and Their Role in Cross-Resistance to Daptomycin and Host Defense Antimicrobial Peptides. Antimicrobial Agents and Chemotherapy 59(8), 4930–4937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Zarena D, Mishra B, Lushnikova T, Wang F, Wang G (2017) The π configuration of the WWW motif of a short Trp-rich peptide is critical for targeting bacterial membranes, disrupting preformed biofilms and killing methicillin-resistant Staphylococcus aureus. Biochemistry 56, 4039–4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Peschel A, Sahl HG (2006) The co-evolution of host cationic antimicrobial peptides and microbial resistance. Nat Rev Microbiol. 4, 529–536. [DOI] [PubMed] [Google Scholar]
- (28).Rahman MM, Hunter HN, Prova S, Verma V, Qamar A, Golemi-Kotra D (2016) The Staphylococcus aureus Methicillin Resistance Factor FmtA Is a d-Amino Esterase That Acts on Teichoic Acids. mBio. 7(1), e02070–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Falord M, Karimova G, Hiron A, Msadek T (2012) GraXSR proteins interact with the VraFG ABC transporter to form a five-component system required for cationic antimicrobial peptide sensing and resistance in Staphylococcus aureus. Antimicrob Agents Chemother 56, 1047–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Falord M, Mäder U, Hiron A, Débarbouillé M, Msadek T (2011) Investigation of the Staphylococcus aureus GraSR regulon reveals novel links to virulence, stress response and cell wall signal transduction pathways. PLoS One 6, e21323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Wang G, Mishra B, Lau K, Lushnikova T, Golla R, Wang X (2015) Antimicrobial peptides in 2014. Pharmaceuticals 8, 123–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Capra EJ, Laub MT (2012) Evolution of two-component signal transduction systems. Annu Rev Microbiol. 66, 325–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Belcheva A, Golemi-Kotra D (2008) A close-up view of the VraSR two component system. A mediator of Staphylococcus aureus response to cell wall damage. J Biol Chem. 283, 12354–12364. [DOI] [PubMed] [Google Scholar]
- (34).Dai Y, Chang W, Zhao C, Peng J, Xu L, Lu H, Zhou S, Ma X (2017) VraR binding to the promoter region of agr inhibits its function in vancomycin-intermediate Staphylococcus aureus (VISA) and heterogeneous VISA. Antimicrob Agents Chemother. 61(5), e02740–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Mehta S, Cuirolo AX, Plata KB, Riosa S, Silverman JA, Rubio A, Rosato RR, Rosato AE (2012) VraSR two-component regulatory system contributes to mprF-mediated decreased susceptibility to daptomycin in in vivo-selected clinical strains of methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother. 56(1), 92–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Chang P, Li W, Shi G, Li H, Yang X, Xia Z, Ren Y, Li Z, Chen H, Bei W (2018) The VraSR regulatory system contributes to virulence in Streptococcus suis via resistance to innate immune defenses. Virulence 9(1), 771–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Kesel S, Mader A, Höfler C, Mascher T, Leisner M (2013) Immediate and heterogeneous response of the LiaFSR two-component system of Bacillus subtilis to the peptide antibiotic bacitracin. PLoS One. 8(1), e53457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Suntharalingam P, Senadheera MD, Mair RW, Lévesque CM, Cvitkovitch DG (2009) The liaFSR system regulates the cell envelope stress response in Streptococcus mutans. J Bacteriol. 191(9), 2973–2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Arias CA, Panesso D, McGrath DM, Qin X, Mojica MF, Miller C, Diaz L, Tran TT, Rincon S, Barbu EM, Reyes J, Roh JH, Lobos E, Sodergren E, Pasqualini R, Arap W, Quinn JP, Shamoo Y, Murray BE, Weinstock GM (2011) Genetic basis for in vivo daptomycin resistance in enterococci. N. Engl. J. Med 365, 892–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Munita JM, Tran TT, Diaz L, Panesso D, Reyes J, Murray BE, Arias CA (2013) A liaF codon deletion abolishes daptomycin bactericidal activity against vancomycin-resistant Enterococcus faecalis. Antimicrob Agents Chemother. 57(6), 2831–2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Binepal G, Gill K, Crowley P, Cordova M, Brady LJ, Senadheera DB, Cvitkovitch DG (2016) Trk2 potassium transport system in Streptococcus mutans and its role in potassium homeostasis, biofilm formation, and stress tolerance. J Bacteriol. 198(7), 1087–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Chen YC, Chuang YC, Chang CC, Jeang CL, Chang MC (2004) A K+ uptake protein, TrkA, is required for serum, protamine, and polymyxin B resistance in Vibrio vulnificus. Infect Immun. 72(2), 629–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Gries CM, Sadykov MR, Bulock LL, Bulock L, Chaudhari SS, Thomas VC, Bose JL, Bayles KW (2016) Potassium uptake modulates Staphylococcus aureus metabolism. mSphere 1(3), e00125–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Choi SH, Lee KL, Shin JH, Cho YB, Cha SS, Roe JH (2017) Zinc-dependent regulation of zinc import and export genes by Zur. Nat Commun. 8, 15812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Gabbianelli R, Scotti R, Ammendola S, Petrarca P, Nicolini L, Battistoni A (2011) Role of ZnuABC and ZinT in Escherichia coli O157:H7 zinc acquisition and interaction with epithelial cells. BMC Microbiol. 11, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Cassat JE, Skaar EP (2012) Metal ion acquisition in Staphylococcus aureus: overcoming nutritional immunity. Semin Immunopathol. 34, 215–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Yoon KP, Silver S (1991) A second gene in the Staphylococcus aureus cadA cadmium resistance determinant of plasmid pI258. J Bacteriol. 173(23), 7636–7642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Xiong A, Jayaswal RK (1998) Molecular characterization of a chromosomal determinant conferring resistance to zinc and cobalt ions in Staphylococcus aureus. J Bacteriol 180(16), 4024–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Kumari SR, Kadam K, Badwaik R, Jayaraman VK (2012) LIPOPREDICT: Bacterial lipoprotein prediction server. Bioinformation. 8(8), 394–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Das S, Kanamoto T, Ge X, Xu P, Unoki T, Munro CL, Kitten T (2009) Contribution of lipoproteins and lipoprotein processing to endocarditis virulence in Streptococcus sanguinis. J Bacteriol. 191(13), 4166–4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Olatunji S, Yu X, Bailey J, Huang CY, Zapotoczna M, Bowen K, Remškar M, Müller R, Scanlan EM, Geoghegan JA, Olieric V, Caffrey M (2020) Structures of lipoprotein signal peptidase II from Staphylococcus aureus complexed with antibiotics globomycin and myxovirescin. Nat. Comm 11, 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Rahman MS, Ceraul SM, Dreher-Lesnick SM, Beie MS, Azad AF (2007) The lspA gene, encoding the type II signal peptidase of Rickettsia typhi: transcriptional and functional analysis. J Bacteriol. 189(2), 336–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Banaei N, Kincaid EZ, Lin SY, Desmond E, Jacobs WR Jr., Ernst JD (2009) Lipoprotein processing is essential for resistance of Mycobacterium tuberculosis to malachite green. Antimicrob Agents Chemother. 53(9), 3799–3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Chan PP, Lowe TM (2009) GtRNAdb: a database of transfer RNA genes detected in genomic sequence. Nucleic Acids Res. 37, D93–D97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Swinehart WE, Jackman JE (2015) Diversity in mechanism and function of tRNA methyltransferases. RNA Biol. 12(4), 398–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Watanabe K, Nureki O, Fukai S, Ishii R, Okamoto H, Yokoyama S, Endo Y, Hori H (2005) Roles of conserved amino acid sequence motifs in the SpoU (TrmH) RNA methyltransferase family. J Biol Chem. 280(11), 10368–10377. [DOI] [PubMed] [Google Scholar]
- (57).Urbonavicius J, Durand JMB, Björk GR (2002) Three modifications in the D and T arms of tRNA influence translation in Escherichia coli and expression of virulence genes in Shigella flexneri. J Bacteriol. 184(19), 5348–5357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Wang KT, Ma L, Nan J, Su XD, Li L (2010) Purification, crystallization and preliminary X-ray crystallographic analysis of 23S RNA m2G2445 methyltransferase RlmL from Escherichia coli. Acta Crystallogr Sect F Struct Biol Cryst Commun. 66, 1484–1486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kyuma T, Kimura S, Hanada Y, Suzuki T, Sekimizu K, Kaito C (2015) Ribosomal RNA methyltransferases contribute to Staphylococcus aureus virulence. FEBS J. 282(13), 2570–2584. [DOI] [PubMed] [Google Scholar]
- (60).Morić I, Savić M, Ilić-Tomić T, Vojnović S, Bajkić S, Vasiljević B (2010) rRNA methyltransferases and their role in resistance to antibiotics. J Med Biochem. 29, 165–174. [Google Scholar]
- (61).Qin Z, Baker AT, Raab A, Huang S, Wang T, Yu Y, Jaspars M, Secombes CJ, Deng H (2013) The fish pathogen Yersinia ruckeri produces holomycin and uses an RNA methyltransferase for self-resistance. J Biol Chem. 288(21), 14688–14697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Almutairi MM, Park SR, Rose S, Hansen DA, Vázquez-Laslop N, Douthwaite S, Sherman DH, Mankin AS (2015) Resistance to ketolide antibiotics by coordinated expression of rRNA methyltransferases in a bacterial producer of natural ketolides. Proc Natl Acad Sci U S A 112(42), 12956–12961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Muschiol S, Balaban M, Normark S, Henriques-Normark B (2015) Uptake of extracellular DNA: Competence induced pili in natural transformation of Streptococcus pneumoniae. Bioessays. 37(4), 426–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Balaban M, Bättig P, Muschiol S, Tirier SM, Wartha F, Normark S, Henriques-Normark B (2011) Secretion of a pneumococcal type II secretion system pilus correlates with DNA uptake during transformation. Proc Natl Acad Sci U S A. 111(7), E758–E765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Petersen FC, Tao L, Scheie AA (2005) DNA binding-uptake system: a link between cell-to-cell communication and biofilm formation. J Bacteriol. 187(13), 4392–4400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Vestergaard M, Leng B, Haaber J, Bojer MS, Vegge CS, Ingmer H (2016) Genome-wide identification of antimicrobial intrinsic resistance determinants in Staphylococcus aureus. Front Microbiol. 7, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Wang G (2020). The antimicrobial peptide database provides a platform to decode the design principles of naturally occurring antimicrobial peptides. Protein Sci. 29, 8–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Mishra B, Narayana JL, Lushinikova T, Wang X, Wang G (2019). Low cationicity is important for systemic in vivo efficacy of database-derived peptides against drug-resistant Gram-positive pathogens. Proc Natl Acad Sci USA 116, 13517–13522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Endres JL, Yajjala VK, Fey PD, Bayles KW (2019) Construction of a Sequence-Defined Transposon Mutant Library in Staphylococcus aureus. Methods Mol Biol. 2016 29–37. [DOI] [PubMed] [Google Scholar]
- (70).Hines KM, Shen T, Ashford NK, Waalkes A, Penewit K, Holmes EA, McLean K, Salipante SJ, Werth BJ, Xu L (2020) Occurrence of cross-resistance and beta-lactam seesaw effect in glycopeptide, lipopeptide, and lipoglycopeptide-resistant MRSA correlates with membrane phosphatidylglycerol levels. J. Antimicrob. Chemother pii: dkz562. doi: 10.1093/jac/dkz562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Brennan M, Thomas DY, Whiteway M, Kavanagh K (2002) Correlation between virulence of Candida albicans mutants in mice and Galleria mellonella larvae. FEMS Immunol Med Microbiol. 34(2), 153–157. [DOI] [PubMed] [Google Scholar]
- (72).Jander G, Rahme LG, Ausubel FM (2000) Positive correlation between virulence of Pseudomonas aeruginosa mutants in mice and insects. J Bacteriol. 182(13), 3843–3845. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.