

 This work is licensed under a
This work is licensed under a Abstract
Large prospective studies established a link between obesity and breast cancer (BC) development. Yet, the mechanisms underlying this association are not fully understood. Among the diverse adipocytokine secreted by hypertrophic adipose tissue, leptin is emerging as a key candidate molecule linking obesity and cancer, since it promotes proliferation and invasiveness of tumors. However, the potential implication of leptin on tumor escape mechanisms remains unknown. This study aims to explore the effect of leptin on tumor resistance to NK lysis and the underlying mechanism. We found that leptin promotes both BC resistance to NK92-mediated lysis and β oxidation on MCF-7, by the up-regulation of a master regulator of mitochondrial biogenesis, the peroxisome proliferator activated receptor coactivator-1 α (PGC1A). Using adenoviral approaches, we show that acute elevation of PGC1A enhances the fatty acid oxidation pathway and decreases the susceptibility of BC cells to NK92-mediated lysis. Importantly, we identified the involvement of PGC1A and leptin in the regulation of hypoxia inducible factor-1 alpha (HIF1A) expression by tumor cells. We further demonstrate that basal BC cells MDA-MB-231 and BT-20 exhibit an increased PGC1A mRNA level and an enhanced oxidative phosphorylation activity; in comparison with luminal BC cells MCF7 and MDA-361, which are associated with more resistance NK92 lysis. Altogether, our results demonstrate for the first time how leptin could promote tumor resistance to immune attacks. Reagents blocking leptin or PGC1A activity might aid in developing new therapeutic strategies to limit tumor development in obese BC patients.
Key Words: leptin, breast cancer, PGC-1α, tumor immune escape, β oxidation
Introduction
The prevalence of obesity in the world has reached epidemic proportions in recent years. Epidemiological studies have established a positive association between obesity and the risk of developing different types of cancer including breast cancer (BC) (1, 2, 3). Obese BC patients exhibit a higher risk for lymph node metastasis, aggressive tumor, and mortality when compared with non-obese BC patients. Yet, the cellular and molecular mechanisms underlying this association remain poorly understood.
As it is known, adipocytes hypertrophy leads to deregulated secretions of pro-inflammatory chemokines, cytokines and adipokines. Among various adipocytokines, leptin, a 16-kDa circulating hormone, is emerging as a key candidate mediator linking obesity with BC. Accumulated evidence supports this hypothesis: (1.) Hyperleptinemia and intratumoral leptin mRNA levels have been linked to a poor prognosis in BC patients (4), (2.) the leptin receptor (ObR) is highly expressed in primary and metastatic mammary carcinoma in comparison with normal tissues (5), and (3.) tumors transplanted into obese leptin receptor-deficient mice (db/db) grow much faster than tumors transplanted into lean (WT) mice (6).
The main role of leptin is to regulate energy homeostasis through the regulation of cellular metabolism. This adipokine has important additional effects, as in fact it is able to trigger a series of downstream signaling pathways, including both canonical (JAK2/STATs, MAPK/ERK 1/2, PI-3K/AKT) and non-canonical signaling pathways (JNK, p38 MAPK, PKC and AMPK), which promote proliferation, migration, and angiogenesis in BC (7). Yet, few studies have investigated the involvement of leptin on tumor/immune cells conflict. The main objective of this study was to explore the impact of leptin on tumor cell susceptibility to NK lysis. A more comprehensive assessment of the influence of this adipokine on tumor escape mechanisms may shed new light on the association between obesity and the poor prognosis for BC patients.
Materials and methods
Cell line and transient transfection experiments
MCF7, MDA-MB 361, BT20 and MDA-MB231 breast cancer cell lines were grown in RPMI 1640 medium with glucose (1 mg/mL) supplemented with 10% FCS, 100 U/mL penicillin and 100 mg/mL streptomycin (Life Technologies). The NK92 cell line was cultured in RPMI 1640/GlutaMAX supplemented with 10% of FCS and 300 U/mL recombinant human IL-2. Recombinant adenoviruses expressing PGC-1α were kindly provided by Dr Pere Puigserver, Dana Farber Cancer institute (Boston, MA, USA). For adenoviral transduction experiments, adenoviruses were used as described (8).
Western blot analysis
Cells were cultured in six-well plates in serum-free medium, then treated with leptin and incubated for 48 h. For extraction of total soluble proteins from the cultured cells, the Mammalian Cell - PE LB™ kit (Genotech, St. Louis, MO) containing EDTA, DTT and protease inhibitor was used. Cell lysates were sonicated and subsequently centrifuged at 16,000 g for 10 min at 4°C, and clear supernatants were collected. For the Western blot analysis, 10% SDS-PAGE was used and proteins were transferred to Polyvinylidene difluoride membranes (Immobilon – P, Millipore). Primary antibodies against HIF1A and β-actin (ACTB) were purchased from Cell Signaling Technology. TFAM antibody was kindly provided by Dr Gustafsson, Department of Medical Biochemistry and Cell Biology, University of Gothenburg, Sweden. Suitable secondary IgG conjugated with HRP was used as secondary antibody. Immunodetection was performed using enhanced chemiluminescence (ECL system, Amersham Biosciences Inc.) according to the manufacturer’s instructions.
RNA isolation and Real-time RT-PCR
Total RNA was extracted using RNeasy Mini Kit according to the manufacturer’s protocol. cDNA was synthesized by using iScript cDNA Synthesis kit. Total RNA (1.0 μg) was used as template for cDNA synthesis. For quantitative comparisons, cDNA samples were analyzed by real-time PCR using the SYBR green mix. Data were analyzed using the comparative cycle threshold (CT) method. RPLP0 was used as the internal loading control.
Flow cytometry analysis
The following monoclonal antibodies were used for flow cytometry: anti-TRAILR1-PE, anti-TRAILR2-PE, anti-Fas-PE and anti MHC-I-FITC, isotypic controls mouse IgG1 antibody from BD Pharmingen (San Diego, CA, USA) and anti-MICA/B-PE from R&D System. Samples were analyzed using an Acurri C6 flow cytometer (BD Biosciences, Le Pont de Claix, France). Data for at least 1 × 105 cells/sample were acquired and analyzed using Accurri C6 software.
Chromium release assay
Cytotoxicity was measured by a 4-h 51Cr release. Different effector/target (E:T) ratios were used in the final volume of 200 µL in 96-well plates. Experiments were performed in triplicate. After 4 h of co-culture, 40 µL of the supernatants were transferred to LumaPlate-96 wells (PerkinElmer) and, after overnight drying, counted on a Packard Instrument TopCount NXT. Data were expressed as the percentage of specific 51Cr release from target cells, calculated as: (experimental release − spontaneous release)/(maximum release − spontaneous release) × 100.
Relative mtDNA content measurements
Mitochondria DNA (mtDNA) was measured by quantitative PCR analysis of total DNA extracted from MCF7 cells by assessing the relative levels of cytochrome C oxydase 2 and the nuclear App gene (9).
Statistical analyses
One-way ANOVA and Student t-tests were used from Prism 5 software. P < 0.05 was designated as a statistically significant difference. Statistical significance is represented in figures as follows: *P < 0.05, **P < 0.01, ***P < 0.001. All experiments were performed in triplicate and repeated at least three times, and all samples were analyzed in duplicate. The data were expressed as means ± s.d..
Results
Impairment of MCF-7 susceptibility to NK92 lysis by leptin
In order to explore the role of leptin in tumor immune escape, we investigated whether leptin alters NK92-mediated lysis of MCF-7 cells. As shown in Fig. 1A, leptin decreased significantly the susceptibility of MCF-7 to lysis by NK92 in a dose-dependent manner. Therefore, the molecular mechanism involved in the resistance of leptin treated MCF-7 to NK lysis was investigated. To this end, the expression of receptors involved in apoptosis Fas, TRAILs (TRAILR1 and TRAILR2) and ligands of NK activatory and inhibitory receptors (MHC-I and MICA/B) were analyzed after leptin treatment. In comparison to non-treated cells, we found that leptin significantly down-regulates TRAILR1 and TRAILR2 expression (mean fluorescence intensity and percentage of positive cells) on MCF-7 cells. However, no effect was found on Fas, MICA/B or MHC-I expression (Fig. 1B).
Figure 1.

Leptin decreases the susceptibility of MCF-7 to NK92 lysis. (A) The impact of leptin on MCF-7 tumor cells to NK92-mediated lysis was studied. MCF-7 were incubated with two concentrations of leptin for 48 h. Cytotoxicity was determined by a conventional 4-h Cr release assay at 20:1 and 2:1 ratios. Data shown represent the means ± s.d. *P < 0.05, **P < 0.01. (B) Analysis of surface expression of MHC-I, Fas, MICA/B, TRAILR1 and TRAILR2 was conducted on MCF-7 tumor cells treated for 48 h by two doses of leptin (10 ng: black line and 200 ng: red line). Isotypic control mAb was included; blue line.
Leptin stimulates fatty acid oxidation pathway in MCF-7 BC cells
Recent studies have indicated that the apoptotic network is incorporated into multiple cell physiological processes, including cell metabolism. Therefore, we investigated the effect of leptin on the metabolic proprieties of MCF-7 cells by checking the expression of several genes involved in glycolysis, lipogenesis and mitochondrial biogenesis. Our results revealed that, compared with basal conditions (10 ng/mL), leptin at 100 ng/mL up-regulates the transcription of three important genes involved in mitochondrial oxidation function, carnitine palmitoyltransferase 1 (CPT1), cyclo oxygenase-2 (COX2) and ATP synthase (Fig. 2A). However, no significant effects on the lipogenic genes expression such as fatty acid synthase (FASN), sterol regulatory element binding protein (SREBP) and acetyl COA carboxylase (ACC1) were found (Fig. 2B). The impact of leptin on expression of the glycolytic genes, GLUT1, GLUT4, PFK, HKI and ALD, was also tested. As shown in Fig. 2C, leptin was found to increase significantly the expression of GLUT1, with no effect noted on the other tested genes.
Figure 2.

Leptin stimulates fatty acid oxidation in MCF-7 cells. Analysis of leptin effect on expression of genes involved (A) in fatty acid oxidation, (B) in lipogenesis and (C) in glycolysis. MCF-7 cells were treated with 10 and 200 ng/mL of leptin for 48 h. Relative expression of mRNA was examined by real-time RT-PCR in leptin treated MCF-7 cells. RPLP0 was used as the internal loading control. Data shown represent the means ± s.d. *P < 0.05, **P < 0.01, ***P < 0.001.
Leptin up-regulate PGC1A gene expression
To further dissect the molecular effect of leptin on the mitochondrial oxidative and respiratory functions, we thought to examine the gene expression status of several coregulators known to impact mitochondrial biogenesis and that are considered as ‘master genes’ susceptible to represent the missing link between fatty acid oxidation and resistance to NK92 lysis. The potential perturbations by leptin of gene expression levels of coactivators and corepressors such as the peroxisome proliferator-activated receptor-γ (PPARG) coactivator 1α and 1β (PGC1A, PGC1B), estrogen receptor-a (ESR1), estrogen-related receptor alpha and gamma (ESRRA, ESRRG) were studied. In Fig. 3, we showed that leptin increased the gene expression of PGC1A but had no impact on the gene expression of PGC1B, ESR1, ESRRA and ESRRG. These results indicate that leptin may enhance mitochondrial oxidative phosphorylation through the up-regulation of PGC1A.
Figure 3.

Leptin up-regulates PGC1A gene expression. Analysis of leptin impact on expression of genes involved in mitochondrial biogenesis. MCF-7 cells were treated with 10 and 200 ng/mL of leptin for 48 h. Relative expression of mRNA of coactivators and nuclear receptors was examined by real-time RT-PCR in leptin treated MCF-7 cells. RPLP0 was used as the internal loading control. Data shown represent the means ± s.d. *P < 0.05.
PGC1A induces both oxidative phosphorylation and resistance of MCF7 to NK92-mediated lysis
The potential involvement of PGC1A on tumor resistance to NK92 lysis was then explored using adenoviral approaches. First, we evaluated the impact of PGC1A overexpression on mitochondrial oxidation. As shown in Fig. 4A, the overexpression of PGC1A markedly affected the gene expression of COX2, CPT1, and TFAM (transcription factor A of mitochondria) involved in the expression of mitochondrial genome. While, no impact on the gene expression of COX1, INOS and ACC1 was noticed. Besides, the relative abundance of mtDNA was consistently augmented in the PGC1A transfected cells in comparison with the control condition (Fig. 4B), suggesting therefore that overexpression of PGC1A enhances fatty acid oxidation in MCF-7 cells. Next, we evaluated the susceptibility of control or PGC1A transfected MCF-7 cells after co-culturing them with NK92 lysis. As shown in Fig. 4C, MCF-7 BC cells overexpressing PGC1A were found to be more resistant to NK92-mediated lysis. These results underline for the first time the implication of the coactivator PGC1A, a powerful transcriptional regulator of mitochondria and oxidative metabolic programs, in BC tumor resistance to NK-mediated apoptosis.
Figure 4.

PGC1A promotes oxidative phosphorylation and resistance to NK92-mediated lysis. (A) PGC1A mRNA levels were overexpressed by adenovirus infection (48 h) of MCF-7 cells. The relative expression level of genes involved in oxidative phosphorylation pathway was evaluated by real-time RT-PCR. (B) Quantification of mitochondrial DNA (mtDNA) was evaluated by real-time RT-PCR. (C) Cytotoxicity was determined by a conventional 4-h Cr release assay at 20:1 and 2:1 ratios. Data shown represent the means ± s.d. *P < 0.05, **P < 0.01, ***P < 0.001.
PGC1A and leptin control HIF1A expression in MCF-7 BC cells
Given the key roles of hypoxia in tumor progression and survival, we asked whether PGC1A can affect HIF1A expression. We found that overexpression of PGC1A using adenovirus increased significantly HIF1A protein levels in MCF-7 cell lines (Fig. 5A). Real-time PCR analysis revealed, however, a slight induction of HIF1A mRNA levels upon elevation of PGC1A, further suggesting, therefore, that PGC1A mainly affects HIF-1α protein accumulation rather than exerting a direct action on transcription of HIF1A gene message (Fig. 5B). In line with our results showing a positive association between leptin and PGC1A expression, we then analyzed the effect of leptin on HIF1A expression and found that leptin was able to increase HIF1A protein levels in a dose-dependent manner (Fig. 5C), without affecting its mRNA levels (Fig. 5D).
Figure 5.

PGC1A and leptin control HIF1A expression. (A) PGC1A mRNA levels were overexpressed by adenovirus infection (48 h) of MCF-7 cells. Whole-cell lysates were subjected to SDS-PAGE, blotted, and probed with anti-HIF1A, as indicated. β-actin (ACTB) was used as the loading control. (B) Relative expression of HIF1A mRNA was examined by real-time RT-PCR in PGC1A-infected MCF-7 cells. RPLP0 was used as the internal loading control. (C) After 48 h of incubation of MCF7 with different concentrations of leptin, whole-cell lysates were subjected to SDS-PAGE, blotted, and probed with anti-HIF1A. β-actin (ACTB) was used as the loading control. (D) Relative expression of HIF1A mRNA was examined by real-time RT-PCR in MCF-7 cells (48 h) treated with different concentrations of leptin. RPLP0 was used as the internal loading control. Data shown represent the means ± s.d. **P < 0.01.
Basal BC cells exhibit enhanced PGC1A expression levels and are more resistant to NK92 lysis
To provide further evidence for the role of PGC1A in tumor development, we explore the possible relationship between PGC1A with tumor aggressiveness of BC cells. PGC1A gene expression was assessed in two basal cell lines with a high grade of invasiveness, BT20 and MDA-MB231, in comparison with two luminal BC cells with modest tumorigentic capacity in vivo, MDA-MB361 and MCF-7. Interestingly, our results revealed PGC1A mRNA levels as increased in the two basal cells compared to the luminal BC cells (Fig. 6A). Consistently, the direct PGC1A target gene, TFAM, was induced in the basal cells BT20 and MDA-MB231 (Fig. 6B), clearly confirming the functionality of PGC1A. Finally, to draw a link between high levels of PGC1A, increased oxidative phosphorylation status and a resistance to NK-mediated lysis, the four BC cells were tested for susceptibility to NK92 lysis. Our results revealed a clear association between increased PGC1A gene expression and resistance to NK92 killing (Fig. 6C).
Figure 6.

TNBC/basal BC cells exhibit enhanced PGC1A expression levels and are more resistant to NK lysis. (A) Relative expression of PGC1A mRNA was examined by real-time RT-PCR in in two TNBC BC, BT20 and MDA-MB231, in comparison with two luminal BC cells, MDA-MB361 and MCF-7. RPLP0 was used as the internal loading control. (B) Whole-cell lysates were subjected to SDS-PAGE, blotted, and probed with anti-TFAM. β-actin (ACTB) was used as the loading control. (C) Susceptibility to NK92-mediated lysis of luminal and TNBC BC tumor cells was determined by a conventional 4-h Cr release assay at 20:1 and 2:1 ratios. Data shown represent the means ± s.d.
Altogether, these results establish a relationship between leptin, PGC1A gene activity, mitochondrial oxidative phosphorylation and tumor resistance to NK lysis (Fg. 7).
Figure 7.
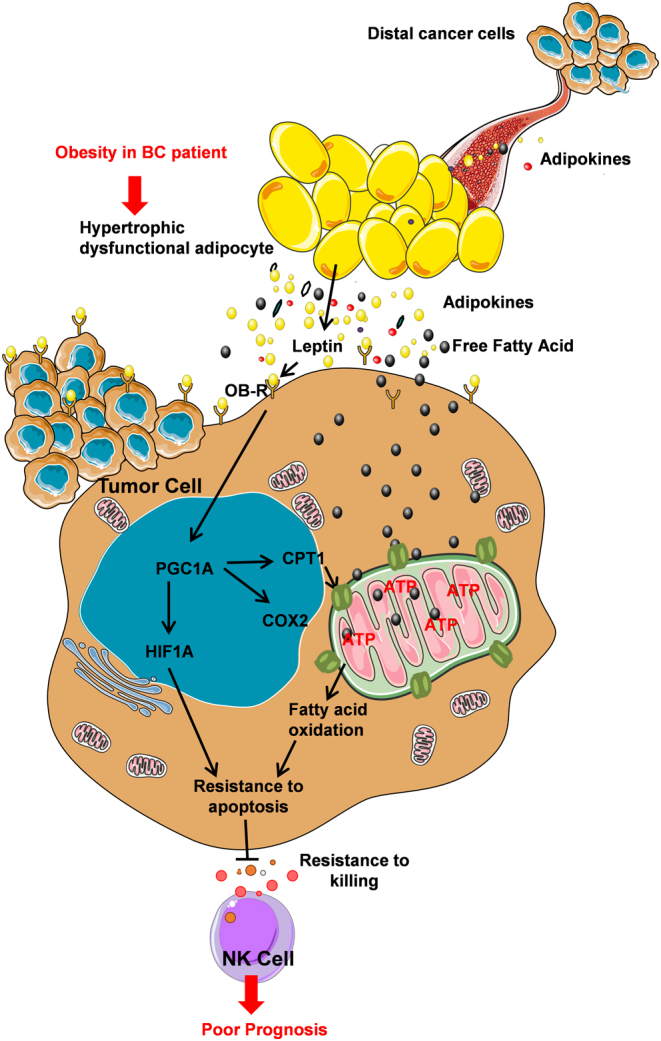
Working model depicting how leptin may promote MCF-7 resistance to apoptosis induced by NK cells while enhancing fatty acid oxidation through up-regulation of PGC1A.
Discussion
The excessive accumulation of adipose tissue in obesity has been linked to poorer prognosis in BC (10). The stromal cells in a breast tumor microenvironment are mainly composed of adipocytes, and these cells can supply cancer cells with energy and can secrete many obesity-associated mediators, such as leptin, that can play a role in tumor development. Leptin can act in an endocrine, paracrine and autocrine manner, to promote BC proliferation and survival (11, 12). Several carcinogenic actions have been attributed to leptin, including the modulation of cell cycle regulator proteins and secretion of angiogenic proteins and migration (13, 14, 15). However, few studies focused on the effects of leptin in the conflict of tumor with immune cells.
This work showed that leptin, by enhancing fatty acid oxidation pathway, could promote tumor resistance to NK lysis. Multiple reports have suggested the involvement of this pathway in the promotion of survival and growth of malignant cells (16, 20). A recent report from Le bleu et al. has shown that tumor cells are able to up-regulate oxidative phosphorylation by enhancing mitochondrial respiration in order to generate the ATP required for primary tumor escape, relocation and metastases (21). The relevance of the involvement of the fatty acid oxidation on cell survival was further supported by the use of inhibitors of this pathway. Treatment with etomoxir or ranolazine inhibits the proliferation and sensitizes human leukemia cells to apoptosis (22). Similarly, the inhibition of lipid synthesis and oxidation through the inhibition of lipogenic enzymes, such as FASN, acetyl-coenzyme A (ACACA), ACC or CPT1, results in decreased proliferation and increased apoptosis of cancer cells (23, 24).
The activation of PGC1A by leptin could represent an important molecular strategy for cancer cells to escape immune cells. The overexpression of this transcriptional coregulatory using adenovirus promoted the accumulation of HIF1A, a key protein that is involved in angiogenesis and resistance of tumor cells to apoptosis. Increasing evidence indicates that HIF1A may modify the balance between pro- and anti-apoptotic factors, promoting, therefore, tumor resistance to immune cells. In this context, a study from Noman et al. showed that the induction of HIF1A is associated with alteration of non-small-cell lung carcinoma target susceptibility to CTL-mediated killing (25). An association of overexpression of HIF1A with poor treatment response and outcome has also been demonstrated in several human tumors (26, 27, 28). Although some studies reported the involvement of HIF1A in the regulation of anti-apoptotic proteins expression such as BCL2, inhibitor of apoptosis proteins IAP2 and survivin (29, 30), one should consider that the precise mechanism by which HIF1A inhibits apoptosis is still far from being fully understood, and it may be dependent of various cellular contexts. In addition to its impact on anti-apoptotic proteins expression, HIF1A seems to be a major factor regulating leptin mRNA and protein expression. A report from Cascio et al. demonstrated that HIF1A binds multiple HRE motifs in the leptin promoter and induces expression of leptin in MCF-7 cells (31). Therefore, the overexpression of HIF1A by PGC1A may conduct to the maintenance of high leptin levels on obese patients. Nonetheless, we should mention that using adenovirus approach in our study we strongly increase PGC1A (150-fold higher), whereas leptin treatment increased PGC1A mRNA level by two fold.
Additionally, our data showed that basal BC cells, lacking ER, PR, and HER2 (32), express higher level of PGC1A and are more resistant to NK mediated lysis in comparison to the luminal BC cells. The role of PGC1A on the regulation of mitochondrial function and metabolism is well established; however, its impact on cancer metabolism and proliferation is only emerging. In BC, PGC1A has been previously shown to promote growth of tumors, regulate mitochondrial metabolism and endow angiogenic properties (33, 34). A recent study from Cai et al. reports that HIF1A and PGC1A may be promising biomarkers to identify subgroups of patients with poorer BC prognosis and to predict early and subclinical metastasis (35).
Overall, the present data demonstrate a key role of leptin in promoting tumor escape from anti-tumor immunity by enhancing fatty acid oxidation and tumor resistance to immune cells lysis. The potential use of leptin and PGC1A as plausible targets in the treatment of neoplastic progression of BC tumors deserves consideration, as it may constitute a promising therapeutic option for obese BC cancer patients.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This work was financially supported by the ‘PHC Utique’ program of the French Ministry of Foreign Affairs and Ministry of Higher Education and Research and the Tunisian Ministry of Higher Education and Scientific Research in the CMCU project number 13G0819.
Author contribution statement
A G designed research and revised the manuscript. H B performed research experiments and drafted the manuscript. A G and S A assisted with the research experiments and revised the manuscript. S C, H B, J F L helped to design research and revised the manuscript. All authors approved the final manuscript.
Acknowledgements
The authors thank Dr Pere Puigserver, Dana Farber Cancer institute (Boston, MA, USA), for providing PGC-1α adenovirus and Dr Gustafsson, Department of Medical Biochemistry and Cell Biology, University of Gothenburg, Sweden, for providing TFAM antibody.
References
- 1.Barone I, Giordano C, Bonofiglio D, Ando S, Catalano S. Leptin, obesity and breast cancer: progress to understanding the molecular connections. Current Opinion in Pharmacology 2016. 31 83–89. ( 10.1016/j.coph.2016.10.003 [DOI] [PubMed] [Google Scholar]
- 2.Renehan AG, Tyson M, Egger M, Heller RF, Zwahlen M. Body-mass index and incidence of cancer: a systematic review and meta-analysis of prospective observational studies. Lancet 2008. 371 569–578. ( 10.1016/S0140-6736(08)60269-X [DOI] [PubMed] [Google Scholar]
- 3.Dawood S, Broglio K, Gonzalez-Angulo AM, Kau SW, Islam R, Hortobagyi GN, Cristofanilli M. Prognostic value of body mass index in locally advanced breast cancer. Clinical Cancer Research 2008. 14 1718–1725. ( 10.1158/1078-0432.CCR-07-1479 [DOI] [PubMed] [Google Scholar]
- 4.Miyoshi Y, Funahashi T, Tanaka S, Taguchi T, Tamaki Y, Shimomura I, Noguchi S. High expression of leptin receptor mRNA in breast cancer tissue predicts poor prognosis for patients with high, but not low, serum leptin levels. International Journal of Cancer 2006. 118 1414–1419. ( 10.1002/ijc.21543 [DOI] [PubMed] [Google Scholar]
- 5.Garofalo C, Koda M, Cascio S, Sulkowska M, Kanczuga-Koda L, Golaszewska J, Russo A, Sulkowski S, Surmacz E. Increased expression of leptin and the leptin receptor as a marker of breast cancer progression: possible role of obesity-related stimuli. Clinical Cancer Research 2006. 12 1447–1453. ( 10.1158/1078-0432.CCR-05-1913 [DOI] [PubMed] [Google Scholar]
- 6.Zheng Q, Dunlap SM, Zhu J, Downs-Kelly E, Rich J, Hursting SD, Berger NA, Reizes O. Leptin deficiency suppresses MMTV-Wnt-1 mammary tumor growth in obese mice and abrogates tumor initiating cell survival. Endocrine-Related Cancer 2011. 18 491–503. ( 10.1530/ERC-11-0102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guo S, Liu M, Wang G, Torroella-Kouri M, Gonzalez-Perez RR. Oncogenic role and therapeutic target of leptin signaling in breast cancer and cancer stem cells. Biochimica et Biophysica Acta 2012. 1825 207–222. ( 10.1016/j.bbcan.2012.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Louet JF, Chopra AR, Sagen JV, An J, York B, Tannour-Louet M, Saha PK, Stevens RD, Wenner BR, Ilkayeva OR, et al. The coactivator SRC-1 is an essential coordinator of hepatic glucose production. Cell Metabolism 2010. 12 606–618. ( 10.1016/j.cmet.2010.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tyynismaa H, Sembongi H, Bokori-Brown M, Granycome C, Ashley N, Poulton J, Jalanko A, Spelbrink JN, Holt IJ, Suomalainen A. Twinkle helicase is essential for mtDNA maintenance and regulates mtDNA copy number. Human Molecular Genetics 2004. 13 3219–3227. ( 10.1093/hmg/ddh342 [DOI] [PubMed] [Google Scholar]
- 10.Bouguerra H, Guissouma H, Labidi S, Stambouli N, Marrakchi R, Chouaib S, Ben Ammar Elgaaied A, Boussen H, Gati A. Breast cancer in Tunisia: association of body mass index with histopathological aspects of tumors. Asian Pacific Journal of Cancer Prevention 2014. 15 6805–6810. ( 10.7314/apjcp.2014.15.16.6805 [DOI] [PubMed] [Google Scholar]
- 11.Sanchez-Jimenez F, Perez-Perez A, de la Cruz-Merino L, Sanchez-Margalet V. Obesity and breast cancer: role of leptin. Frontiers in Oncology 2019. 9 596 ( 10.3389/fonc.2019.00596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ando S, Gelsomino L, Panza S, Giordano C, Bonofiglio D, Barone I, Catalano S. Obesity, leptin and breast cancer: epidemiological evidence and proposed mechanisms. Cancers 2019. 11 62 ( 10.3390/cancers11010062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gati A, Kouidhi S, Marrakchi R, El Gaaied A, Kourda N, Derouiche A, Chebil M, Caignard A, Perier A. Obesity and renal cancer: role of adipokines in the tumor-immune system conflict. Oncoimmunology 2014. 3 e27810 ( 10.4161/onci.27810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fan Y, Gan Y, Shen Y, Cai X, Song Y, Zhao F, Yao M, Gu J, Tu H. Leptin signaling enhances cell invasion and promotes the metastasis of human pancreatic cancer via increasing MMP-13 production. Oncotarget 2015. 6 16120–16134. ( 10.18632/oncotarget.3878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saxena NK, Vertino PM, Anania FA, Sharma D. Leptin-induced growth stimulation of breast cancer cells involves recruitment of histone acetyltransferases and mediator complex to cyclin D1 promoter via activation of Stat3. Journal of Biological Chemistry 2007. 282 13316–13325. ( 10.1074/jbc.M609798200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blanquer-Rosselló MDM, Oliver J, Sastre-Serra J, Valle A, Roca P. Leptin regulates energy metabolism in MCF-7 breast cancer cells. International Journal of Biochemistry and Cell Biology 2016. 72 18–26. ( 10.1016/j.biocel.2016.01.002 [DOI] [PubMed] [Google Scholar]
- 17.Birsoy K, Possemato R, Lorbeer FK, Bayraktar EC, Thiru P, Yucel B, Wang T, Chen WW, Clish CB, Sabatini DM. Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature 2014. 508 108–112. ( 10.1038/nature13110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beloribi-Djefaflia S, Vasseur S, Guillaumond F. Lipid metabolic reprogramming in cancer cells. Oncogenesis 2016. 5 e189 ( 10.1038/oncsis.2015.49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ackerman D, Simon MC. Hypoxia, lipids, and cancer: surviving the harsh tumor microenvironment. Trends in Cell Biology 2014. 24 472–478. ( 10.1016/j.tcb.2014.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baenke F, Peck B, Miess H, Schulze A. Hooked on fat: the role of lipid synthesis in cancer metabolism and tumour development. Disease Models and Mechanisms 2013. 6 1353–1363. ( 10.1242/dmm.011338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.LeBleu VS, O'Connell JT, Gonzalez Herrera KN, Wikman H, Pantel K, Haigis MC, de Carvalho FM, Damascena A, Domingos Chinen LT, Rocha RM, et al. PGC-1alpha mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nature Cell Biology 2014. 16 992–1003, 1001–1015. ( 10.1038/ncb3039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Samudio I, Harmancey R, Fiegl M, Kantarjian H, Konopleva M, Korchin B, Kaluarachchi K, Bornmann W, Duvvuri S, Taegtmeyer H, et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. Journal of Clinical Investigation 2010. 120 142–156. ( 10.1172/JCI38942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brusselmans K, Vrolix R, Verhoeven G, Swinnen JV. Induction of cancer cell apoptosis by flavonoids is associated with their ability to inhibit fatty acid synthase activity. Journal of Biological Chemistry 2005. 280 5636–5645. ( 10.1074/jbc.M408177200 [DOI] [PubMed] [Google Scholar]
- 24.Kuhajda FP. Fatty-acid synthase and human cancer: new perspectives on its role in tumor biology. Nutrition 2000. 16 202–208. ( 10.1016/s0899-9007(99)00266-x [DOI] [PubMed] [Google Scholar]
- 25.Noman MZ, Janji B, Kaminska B, Van Moer K, Pierson S, Przanowski P, Buart S, Berchem G, Romero P, Mami-Chouaib F, et al. Blocking hypoxia-induced autophagy in tumors restores cytotoxic T-cell activity and promotes regression. Cancer Research 2011. 71 5976–5986. ( 10.1158/0008-5472.CAN-11-1094 [DOI] [PubMed] [Google Scholar]
- 26.Wilson WR, Hay MP. Targeting hypoxia in cancer therapy. Nature Reviews: Cancer 2011. 11 393–410. ( 10.1038/nrc3064 [DOI] [PubMed] [Google Scholar]
- 27.Ai Z, Lu Y, Qiu S, Fan Z. Overcoming cisplatin resistance of ovarian cancer cells by targeting HIF-1-regulated cancer metabolism. Cancer Letters 2016. 373 36–44. ( 10.1016/j.canlet.2016.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moschetta MG, Maschio LB, Jardim-Perassi BV, Gelaleti GB, Lopes JR, Leonel C, Goncalves Ndo N, Ferreira LC, Martins GR, Borin TF, et al. Prognostic value of vascular endothelial growth factor and hypoxia-inducible factor 1alpha in canine malignant mammary tumors. Oncology Reports 2015. 33 2345–2353. ( 10.3892/or.2015.3856 [DOI] [PubMed] [Google Scholar]
- 29.Chen YQ, Zhao CL, Li W. Effect of hypoxia-inducible factor-1alpha on transcription of survivin in non-small cell lung cancer. Journal of Experimental and Clinical Cancer Research 2009. 28 29 ( 10.1186/1756-9966-28-29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kilic M, Kasperczyk H, Fulda S, Debatin KM. Role of hypoxia inducible factor-1 alpha in modulation of apoptosis resistance. Oncogene 2007. 26 2027–2038. ( 10.1038/sj.onc.1210008 [DOI] [PubMed] [Google Scholar]
- 31.Cascio S, Bartella V, Auriemma A, Johannes GJ, Russo A, Giordano A, Surmacz E. Mechanism of leptin expression in breast cancer cells: role of hypoxia-inducible factor-1alpha. Oncogene 2008. 27 540–547. ( 10.1038/sj.onc.1210660 [DOI] [PubMed] [Google Scholar]
- 32.Badve S, Dabbs DJ, Schnitt SJ, Baehner FL, Decker T, Eusebi V, Fox SB, Ichihara S, Jacquemier J, Lakhani SR, et al. Basal-like and triple-negative breast cancers: a critical review with an emphasis on the implications for pathologists and oncologists. Modern Pathology 2011. 24 157–167. ( 10.1038/modpathol.2010.200 [DOI] [PubMed] [Google Scholar]
- 33.Deblois G, St-Pierre J, Giguere V. The PGC-1/ERR signaling axis in cancer. Oncogene 2013. 32 3483–3490. ( 10.1038/onc.2012.529 [DOI] [PubMed] [Google Scholar]
- 34.Klimcakova E, Chenard V, McGuirk S, Germain D, Avizonis D, Muller WJ, St-Pierre J. PGC-1alpha promotes the growth of ErbB2/Neu-induced mammary tumors by regulating nutrient supply. Cancer Research 2012. 72 1538–1546. ( 10.1158/0008-5472.CAN-11-2967 [DOI] [PubMed] [Google Scholar]
- 35.Cai FF, Xu C, Pan X, Cai L, Lin XY, Chen S, Biskup E. Prognostic value of plasma levels of HIF-1a and PGC-1a in breast cancer. Oncotarget 2016. 7 77793–77806. ( 10.18632/oncotarget.12796 [DOI] [PMC free article] [PubMed] [Google Scholar]