

Abstract
The current coronavirus disease 2019 (COVID‐19) pandemic presents a global challenge for managing acutely ill patients and complications from viral infection. Systemic inflammation accompanied by a “cytokine storm,” hemostasis alterations and severe vasculitis have all been reported to occur with COVID‐19, and emerging evidence suggests that dysregulation of lipid transport may contribute to some of these complications. Here, we aim to summarize the current understanding of the potential mechanisms related to COVID‐19 dyslipidemia and propose possible adjunctive type therapeutic approaches that modulate lipids and lipoproteins. Specifically, we hypothesize that changes in the quantity and composition of high‐density lipoprotein (HDL) that occurs with COVID‐19 can significantly decrease the anti‐inflammatory and anti‐oxidative functions of HDL and could contribute to pulmonary inflammation. Furthermore, we propose that lipoproteins with oxidized phospholipids and fatty acids could lead to virus‐associated organ damage via overactivation of innate immune scavenger receptors. Restoring lipoprotein function with ApoA‐I raising agents or blocking relevant scavenger receptors with neutralizing antibodies could, therefore, be of value in the treatment of COVID‐19. Finally, we discuss the role of omega‐3 fatty acids transported by lipoproteins in generating specialized proresolving mediators and how together with anti‐inflammatory drugs, they could decrease inflammation and thrombotic complications associated with COVID‐19.
Keywords: COVID‐19, dyslipidemia, inflammation, lipoproteins, oxidation
Abbreviations
- ACE2
angiotensin‐converting enzyme 2
- ACEi
angiotensin‐converting enzyme inhibitor
- ACS
acute coronary syndrome
- ALI
acute lung injury
- AMI
acute myocardial infarction
- ARB
angiotensin II receptor blocker
- ARDS
acute respiratory distress syndrome
- COVID‐19
coronavirus disease 2019
- CVD
cardiovascular disease
- HDL
high‐density lipoprotein
- LDL
low‐density lipoprotein
- LOX
lipoxygenase
- LOX‐1
lectin‐like oxLDL receptor
- MERS
Middle East respiratory syndrome
- OxLDL
oxidized LDL
- OxPLs
oxidized phospholipids
- PON1
paraoxonase 1
- RAAS
renin‐Angiotensin‐Aldosterone System
- ROS
reactive oxygen species
- SAA
serum amyloid protein A
- SARS‐CoV‐2
severe acute respiratory syndrome coronavirus 2
- SPMs
specialized proresolving lipid mediators
1. INTRODUCTION
Severe acute respiratory syndrome (SARS) coronavirus 2 (SARS‐CoV‐2) is one of the 36 coronaviruses in the family Coronaviridae, within the order Nidovirales that are known to cause respiratory or intestinal infections in humans and other animals. 1 In humans, SARS‐CoV‐2 infection causes acute lung injury (ALI) with rapid progression to acute respiratory distress syndrome (ARDS), leading to COVID‐19 disease. 2 The American‐European Consensus Conference recognized ARDS as an acute condition characterized by diffuse alveolar injury, bilateral pulmonary infiltrates, and severe hypoxemia in the absence of evidence for cardiogenic pulmonary edema. 3 It has long been recognized that ARDS due to viral infections is associated with alterations in the host immunological status, including decreases in circulating neutrophils, dendritic cell subsets, natural killer cells, CD4+ and CD8+ T lymphocytes, and B lymphocytes, and increased levels of pro‐inflammatory cytokines. 1 Some of the locally produced chemokines, such as monocyte chemoattractant protein‐1 (MCP1) and macrophage inflammatory proteins (MIPs) attract monocytes, macrophages, and T cells to the site of inflammation. 4 More recently, it has been suggested that overactivation of the immune system due to SARS‐CoV‐2 infection causes a surge of pro‐inflammatory factors, referred to as “cytokine storm,” resulting in host organ damage, such as lung damage, increased lung endothelial and epithelial permeability, impaired gas exchange and severe respiratory failure with a high mortality rate. 5 , 6
Severe acute respiratory syndrome coronavirus 2 is an enveloped, nonsegmented, positive‐sense, single‐stranded RNA virus that enters the host cells through endocytosis or membrane fusion. The first step in viral entry of SARS‐CoV‐2 into host cells is binding of a viral envelope transmembrane glycoprotein, known as the SPIKE (S) protein, to a cell host cell membrane receptor‐enzyme protein, namely the angiotensin‐converting enzyme 2 (ACE2) protein. 7 Subsequently, host cell infection by SARS‐CoV‐2 results in ACE2 internalization and reduction of cell surface ACE2 enzymatic activity. It has been reported that ACE2 membrane protein is highly expressed on the mucosa of oral cavity 8 and lung epithelial cells, 9 which determines the initial tissue entry site of the virus. Of note, endothelial cells, 10 along with human heart pericytes 11 also have high expression of ACE2, which may explain the pathophysiological effects related to the cardio‐pulmonary system. ACE2 is an important enzyme involved in modulation of the renin‐angiotensin‐aldosterone system (RAAS). ACE2 converts Angiotensin II (ANGII) to ANG1‐7, which binds to MAS, a receptor that signals to reduce the activity of the ANGII receptor AT1. 12 Decreasing cell surface ACE2 results in both increased ANGII and overactivation of AT1 and reduction of ANG1‐7, both of which could contribute to SARS‐CoV‐2 infection cardiopulmonary complications. Of note, because both classes of antihypertensive medicines, angiotensin‐converting enzyme inhibitor (ACEi) and angiotensin II receptor blocker (ARB), are known to increase ACE2 levels, there has been some concern that the use of these medications may increase susceptibility to SARS‐CoV‐2 infection, 13 although more data are needed to clarify this issue. 14
The clinical presentation for COVID‐19 varies from asymptomatic cases to individuals with severe pneumonia associated with ARDS and cardiogenic shock, particularly in older patients with chronic comorbidities. 2 , 15 Adaptive immune dysregulation associated with aging or with chronic systemic dysmetabolic conditions may result in lower tolerability to viral infections. 16 Hyperlipidemia and diabetes are known to compromise the immune response, and can lead to a sustained chronic inflammatory state, which leads to high cardiovascular (CVD) risk. 17 , 18 Furthermore, increased metabolic demands due to virally induced acute inflammation results in decreased myocardial oxygenation and ischemic damage and vascular dysfunction, with thrombotic complications. 17 Thus, it appears that traditional CVD risk factors may significantly contribute to the morbidity and mortality from SARS‐CoV‐2 infection.
No drug has proven to be effective for the routine prevention and or management of most causes of ARDS. Early administration of corticosteroids to septic patients usually does not prevent the development of ARDS. Numerous other pharmacologic therapies, including the use of inhaled synthetic surfactant, intravenous antibody to endotoxin, ketoconazole, simvastatin, and ibuprofen, have been tried and were largely found to be relatively ineffective and offered no survival benefit. 19 , 20 The current standard of care for the management of ARDS is largely supportive, with lung‐protective ventilation and a fluid conservative strategy. The search for effective medicines to treat COVID‐19 disease due to SARS‐CoV2 infection‐induced ARDS is of great interest. A variety of approaches to suppress the “cytokine storm” aiming to protect against lung damage, for example, by blocking individual inflammatory cytokines with anti‐IL‐1 or anti‐IL‐6 therapies are now being actively investigated. It is debatable, however, whether blockade of a single cytokine will be sufficient to suppress the “cytokine storm,” which involves multiple cytokines and redundant systems maintaining the inflammatory response.
Here, we hypothesize that targeting immune‐mediated inflammatory dyslipidemia in COVID‐19, alone or in combination with other therapeutics, could possibly improve the clinical course in these patients.
2. LIPOPROTEIN CHANGES IN COVID‐19
The measurement of plasma lipids and lipoproteins is a critical part of the current approach to CVD risk management. Low HDL‐cholesterol (HDL‐C) is a strong predictor of CVD progression 21 , 22 and serves as a biomarker for increased risk of all‐cause mortality and nonfatal myocardial infarction, even in statin‐treated patients. 23 The best understood function of HDL is to promote reverse cholesterol transport (RCT) from the periphery to the liver, which can be assessed in clinical settings by measuring the first step in RCT, cholesterol efflux capacity (CEC). 24 In addition to its function in RCT, HDL particles have several other properties that may be relevant to modulation of the immune system and the control of infectious diseases. Specifically, among the several lipoproteins, HDL particles have the greatest affinity for binding and neutralization of pathogen‐associated lipids (eg, lipopolysaccharide, lipoteichoic acid) that mediate the excessive immune activation in sepsis. HDL particles also have immunomodulatory, antithrombotic, and antioxidant effects. 25 For example, HDL‐C levels have been shown to be inversely related to the frequency of several autoimmune diseases. 26
High‐density lipoprotein is a heterogeneous collection of particles with different sizes and apolipoprotein compositions. ApoA‐I, the major protein constituent of HDL, is present in most HDL particles, whereas other apolipoproteins, such as ApoE, are associated with specific HDL particle subspecies. Cellular cholesterol efflux, the first step in RCT, is driven mainly by the interaction between relatively lipid‐poor ApoA‐I in small discoidal (pre‐beta) forms of HDL and cell‐bound transporters (ie, ABCA1, ABCG1, and SR‐BI) (Figure 1).
FIGURE 1.
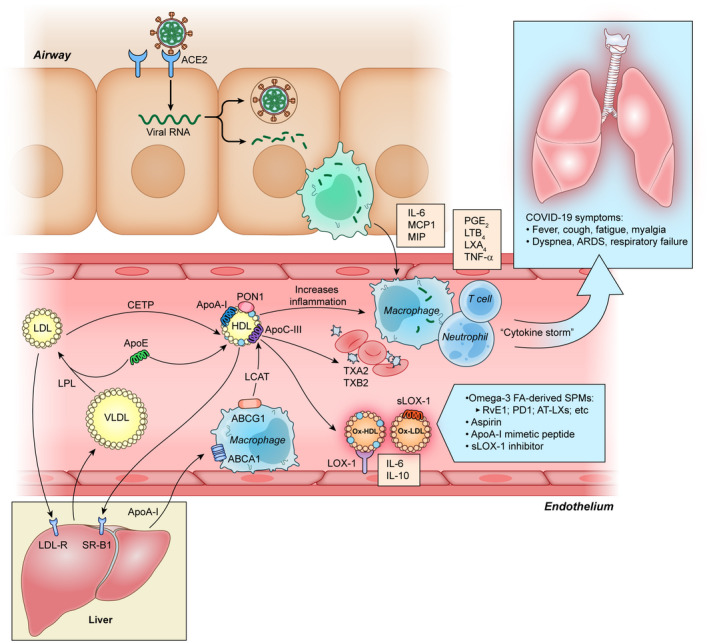
Proposed mechanism of COVID‐19—associated dyslipidemia and impaired resolution of infection. Severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) binds to angiotensin‐converting enzyme 2 (ACE2) via spike protein, which facilitates entry into the cell with subsequent damage by alveolar macrophages. Subsequently, tissue microenvironment releases pro‐inflammatory cytokines and chemokines (IL‐6, MCP1, and MIP) promoting attraction of macrophages, neutrophils, and T cells. This cell activation leads to uncontrolled inflammation and immune dysregulation with further accumulation of eicosanoids like PGE2, TXB2, LTB4, and LXA4. Persistent inflammation culminates in the modulation of HDL‐associated apolipoproteins, such as a decrease in apolipoprotein A‐I (ApoA‐I), ApoE, and an increase serum amyloid protein A, which adversely affects the anti‐inflammatory, antioxidant, and immunomodulatory function of HDL. The imbalance in the antioxidant system also causes oxHDL modification via intracellular lectin‐like oxLDL (LOX‐1) receptor. The extracellular portion of LOX‐1, serum‐soluble form (sLOX‐1), additionally stimulate interaction between oxidized lipids, and circulating macrophages resulting in pro‐inflammatory cytokines release, such as IL‐6, IL‐10, and tumor necrosis factor‐alpha (TNF‐α). Impaired paraoxonase 1 (PON1) enzyme function on HDL, and excessive inflammatory response leads to further lipid oxidation. Excessive development of oxLDL and oxHDL results in lipoprotein transport alteration and impairment of the reverse‐cholesterol transport (RCT) pathway (shown at left) characterized by insufficient ApoA‐I interaction with the adenosine triphosphate‐binding cassette transporter A1 (ABCA1) on macrophages and decreased cholesterol esterification by lecithin cholesterol acyltransferase (LCAT). This culminates in decreased return of cholesteryl esters to the liver either directly after interaction with hepatic scavenger receptor‐B1 (SR‐B1) receptors or indirectly after transfer to LDL by cholesteryl ester transfer protein (CETP) and uptake by hepatic LDL receptors (LDL‐R). Low levels of ApoE and ApoC‐III on HDL result in decreased lipoprotein lipase (LPL) activity, which in turn leads to VLDL and TGs accumulation
High‐density lipoprotein‐associated apolipoproteins, such as apolipoprotein A‐I (ApoA‐I) and apolipoprotein M (ApoM), interact with lipid rafts on cellular membranes that are enriched in immune cell receptors, such as Toll‐like receptors on macrophages 27 and T‐cell receptors 28 and modulate the immune responses. Interestingly, previous and more recent literature suggest a causal inference for an inverse relationship between HDL‐C, but not LDL‐C or triglycerides, and risk of an infectious disease hospitalization. 29 Furthermore, the anti‐inflammatory and antioxidant properties of HDL are significantly reduced during influenza 30 and HIV infection. 31 Indeed, lower levels of both HDL‐C and LDL‐C were detected in HIV patients, with restored lipoprotein levels after treatment. 27 In the context of COVID‐19, it has been recently reported that low levels of total cholesterol (TC), HDL‐C and LDL‐C are associated with disease severity and mortality. 32 , 33 Increased plasma triglyceride levels during infection and inflammation is also a well‐known phenomenon. 29 , 34
Illustrative of what has already been described in COVID‐19, 32 , 33 we observed a similar trend of lipid changes in our clinical case of COVID‐19 (Figure 2, Table 1). The patient was a 40‐year‐old male without a history of CVD and was not on any lipid‐lowering treatment. After acute onset of COVID‐19, the TC levels reduced by about half, accompanied by low HDL‐C (22 mg/dL on Day 3) and low LDL‐C (20 mg/dL at Day 3). The severity of the disease, including hypoxic pneumonia, reached its peak at Day 3. Changes in lipid levels appeared to parallel with increases in C‐reactive protein (from 0.1 to 21.0 mg/dL at Day 3). These changes were accompanied by lymphocytopenia (from 909/μL onset to 400/μL at Day 3) and basopenia (from 64/μL onset to 23/μL at Day 3). Following treatment with an artificial respirator and supportive therapy, the patient's condition markedly improved, and TC returned to preadmission state at discharge (Day 60).
FIGURE 2.
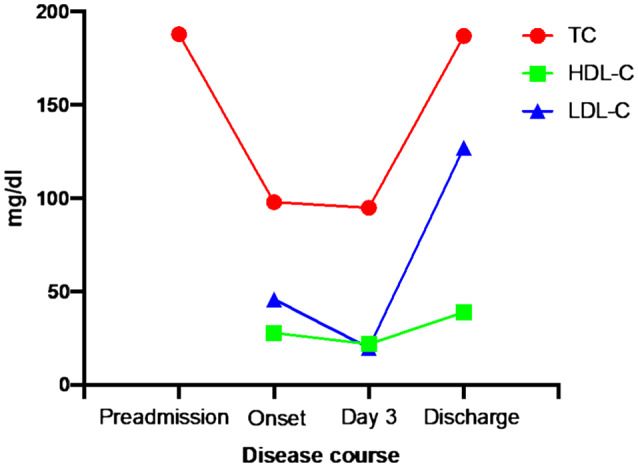
Lipid changes in COVID‐19 patient over the course of disease
TABLE 1.
The changes of laboratory data in a COVID‐19 case
| Parameters | Preadmission | Onset | Day 3 | Day 60 (discharge) |
|---|---|---|---|---|
| White blood cells (/μL) | 5600 | 6400 | 4600 | 7200 |
| Neutrophils (/μL) | – | 4845 | 3671 | – |
| Lymphocytes (/μL) | – | 909 | 400 | – |
| Eosinophils (U/L) | – | 6 | 55 | – |
| Basophils (U/L) | – | 64 | 23 | – |
| Monocytes (U/L) | – | 576 | 446 | – |
| Lactate dehydrogenase (U/L) | 232 | 354 | 370 | 174 |
| Total cholesterol (mg/dL) | 188 | 98 | 95 | 187 |
| HDL cholesterol (mg/dL) | – | 28 | 22 | 39 |
| LDL cholesterol (mg/dL) | – | 46 | 20 | 127 |
| Triglycerides (mg/dL) | – | 119 | 264 | 120 |
| C‐reactive protein (mg/dL) | 0.1 | 13.1 | 21.0 | 0.0 |
The mechanisms by which increased inflammation reduces HDL functionality are not clearly defined. It has been previously observed that inflammation alters HDL apolipoprotein composition. It has been described that inflammation alters hepatic apolipoprotein gene expression 35 and promotes binding of the pro‐inflammatory serum amyloid protein A (SAA) which, in turn, displaces and decreases ApoA‐I levels in HDL. 36 Moreover, in the setting of acute inflammation, decreased plasma levels of lecithin cholesterol acyltransferase (LCAT) may also alter HDL function and further deteriorate the inflammatory response. 37 Interestingly, it was recently reported that treatment of inflammation‐altered HDL with LCAT ex‐vivo reduces HDL‐bound SAA, while increasing HDL‐bound ApoA‐I, and HDL function. 38 It was recently shown that SAA plasma levels are dynamically elevated with COVID‐19 disease severity and SAA has been proposed as a biomarker for evaluating the severity and prognosis of COVID‐19. 39 These findings suggest that HDL protein composition and function are altered in COVID‐19 patients, raising the possibility that interventions, such as the LCAT treatment mentioned above, may improve HDL function and reduce disease burden.
Additional mechanisms leading to the dysfunction of HDL involve inflammation‐induced oxidative modification of ApoA‐I which reduces RCT. 40 An antioxidant enzyme present in HDL, paraoxonase 1 (PON1), is also inactivated under inflammation‐induced oxidative stress, 41 which further compromises HDL function. In fact, low PON1 activity is associated with worse prognosis of CVD and was found to be decreased in various inflammatory 42 and infectious 43 diseases. Notably, the treatment of inflammation‐altered HDL with LCAT ex‐vivo, mentioned above, also significantly decreased HDL‐bound PON1. 38 IL‐10, which under certain circumstances may promote an inflammatory phenotype, can also decrease plasma HDL‐C levels 44 by increasing micropinocytosis. 45
Impaired HDL antioxidant activity further results in lipid oxidation, specifically generating oxidized LDL (oxLDL). As discussed further below, oxLDL and oxidized HDL (oxHDL) are potent activators of the oxidized LDL scavenge receptor (LOX‐1), inducing further inflammation and aggravating tissue damage. Interventions to either improve HDL functionality, such as increasing LCAT activity mentioned above, or replenishment with functional HDL or relevant apolipoproteins, such as ApoA‐I mimetic peptides may be effective in improving HDL functionality for the management of SARS‐CoV‐2 related complications.
3. ApoE AND COVID‐19
Besides ApoA‐I, ApoE is also found on HDL, as well as ApoB‐containing lipoproteins. ApoE serves as a ligand for the clearance of triglyceride‐rich ApoB‐containing lipoproteins by binding to membrane lipoprotein receptors in the LDL receptor family. 46 , 47 , 48 , 49 ApoE has many genetic variants affecting health and disease. 50 , 51 The most common isoform, ApoE3 has a Cys at codon 112 and Arg at codon 158. The ApoE2 isoform (Arg158Cys) is associated with decreased LDL‐C but increased chylomicron and VLDL remnants, whereas the ApoE4 isoform (Cys112Arg) is associated with elevated plasma LDL‐C and Alzheimer's disease. 46 Complete ApoE deficiency in humans results in increased plasma levels of triglycerides and cholesterol in ApoB‐containing lipoproteins, decreased HDL‐cholesterol, palmar‐tuberoeruptive xanthoma, and premature cardiovascular disease. 52 , 53 It is well known that lack of ApoE in murine models also provokes atherogenesis accompanied by low HDL‐C and high TG‐rich lipoproteins. 54 Increases in both ApoE and phospholipid transfer protein (PLTP) activity have been shown to improve the delivery of energy substrates and phospholipids to tissues for sustaining cellular membrane homeostasis in intensive care patients. 55
ApoE has additional functions beyond ApoB‐lipoprotein uptake into cells. It has been shown to protect LDL from oxidation through its receptor‐binding domain 56 and may also be involved in RCT. 57 ApoE‐containing HDL particles promote cholesterol efflux from extrahepatic cells 58 by ABCA1‐ and ABCG1‐dependent processes and this process is antagonized by the presence of ApoC‐III. 59 ApoE is also expressed and secreted from monocytes and macrophages, with anti‐atherosclerotic effects, and in other tissues such as adipose tissue, brain, kidney and adrenal glands. 46 , 60 Interestingly, in the vascular endothelium, although endothelial cells do not produce ApoE, its local expression by macrophages has a paracrine effect on these cells, inhibiting VCAM‐1, stimulating NO synthesis, suppressing endothelial activation, and decreasing adhesion of monocytes to the endothelium, whereas ApoE4 was found to antagonize these anti‐inflammatory effects. 46
Several findings suggest that deficits in ApoE function in SARS‐CoV‐2 dyslipidemia may contribute to disease progression and complications. Notably, ApoE is expressed in lung macrophages and also alveolar epithelial cells (both type I and type II). 61 ApoE‐KO mice are highly susceptible to acute lung injury through an IL‐6–dependent mechanism that increases endothelial cell permeability to oxLDL; 62 IL‐6 is a major cytokine released during the COVID‐19 cytokine storm. 63 In humans, ApoE can potentially function as an endogenous signal that primes the NLRPS inflammasome in alveolar macrophages from asthmatic subjects. 64 Moreover, the APOE gene is associated with modified lung physiology in humans. 65 The ApoE4 variant was also reported to predict COVID‐19 severity. ApoE4/E4 homozygotes in the UK Biobank were more likely to test positive for COVID‐19 (OR 2.31, CI 1.65 to 3.24, P = 1.19 × 106). The association between ApoE4/E4 genotype and COVID19 was independent of pre‐existing dementia, CVD or Type 2 diabetes. 61
4. OXIDIZED LIPOPROTEINS AND SCAVENGER RECEPTORS IN COVID‐19
Low‐density lipoprotein is the main vehicle for transporting cholesterol and phospholipids in the human circulation. During acute inflammation, LDL and its major apolipoprotein, apolipoprotein B (apoB) are oxidized (oxLDL). Lipid hydroperoxides derived from the lipoxygenase pathway, 66 and hydroxy fatty acids derived from arachidonic acid (AA) and linoleic acid (LA) accumulate and some are esterified into cholesterol esters, triacylglycerol, and phospholipids in oxLDL. 67 Oxidized phospholipids (OxPLs) in oxLDL are recognized as danger‐associated molecular patterns (DAMPs) by cell scavenger receptors, inducing a cascade of intracellular signaling events, culminating in inflammasome activation and endothelial cell dysfunction, both of which contribute to atherosclerosis initiation and progression. 68 In addition, oxPL production is increased in the lungs of virus‐infected humans and animals and oxPL induces macrophage cytokine production and acute lung inflammation in mice. 69
The oxLDL scavenger receptor lectin‐like oxLDL receptor (LOX‐1), expressed in endothelial cells, macrophages, and smooth muscle cells, binds multiple ligands, including oxLDL, oxHDL, C‐reactive protein, and advanced glycated end products. 70 , 71 oxLDL binding to LOX‐1 results in oxLDL internalization and oxLDL cellular accumulation, which is thought to contribute to early atherosclerotic lesion development. In addition, ligand binding to LOX‐1 triggers intracellular signaling processes leading to pro‐apoptotic, pro‐oxidant, and pro‐inflammatory pathways, causing cell dysfunction associated with atherosclerosis and increased CVD risk. 70 , 72 Because of its binding and response to dysfunctional lipids, such as oxLDL and oxHDL, LOX‐1 may be a key mediator of CVD by inducing inflammation‐triggered atheroma growth and eventually plaque erosion and rupture. 73
The serum‐soluble form of LOX‐1 (sLOX‐1), the extracellular portion of LOX‐1 produced by proteolytic shedding of the receptor, reflects membrane‐bound intact receptor levels and is elevated in acute coronary syndrome (ACS), 74 stable coronary artery disease 75 and stroke. 76 The association of LOX‐1 activation and acute inflammatory conditions raises the possibility that LOX‐1 is also activated and may contribute to COVID‐19 complications. In fact, recent clinical data suggest that SARS‐CoV‐2 may cause a pediatric multisystem inflammatory syndrome reminiscent of Kawasaki Disease (KD) in children. 77 , 78 Previous work has provided evidence for LOX‐1 overactivation in KD patients. 79 These data, together with recent human pathology findings suggest that SARS‐CoV‐2 infection could trigger endothelial damage in multiple organs throughout the body. 80 Furthermore, LOX‐1 blockade has been shown to protect mice from LPS‐induced ALI, 81 suggesting that LOX‐1 may play an important role in COVID‐19 complications and is another potential target for therapy.
5. SPECIALIZED PRORESOLVING MEDIATORS IN COVID‐19
The continuous inflammatory response driven by the “cytokine storm,” which includes release of the pro‐inflammatory cytokines (TNF‐α, IL‐6, IL‐8, and IL‐10) 82 and lymphopenia, are considered to be one of the main cause of life‐threatening complications in SARS‐CoV‐2 patients. The immunological phenotype characterizing COVID‐19 highlights the importance of T lymphocytes, with decreased numbers of circulating CD4+/CD8+ T cells in particular. 83 However, the direct effect of the released cytokines and chemokines results in massive cell death that provokes a cascade of biological reactions, including production of macrophage‐derived eicosanoids that further potentiate inflammation. 84
Until recently, inflammation resolution, the process by which an inflamed system returns to homeostasis and reduces inflammation, was thought to be a passive process relying mainly in the biological “decay” of pro‐inflammatory factors. It is now known, however, that inflammation resolution is, in fact, an active process mediated by specific molecules collectively referred to as specialized proresolving mediators (SPMs). 85
Omega‐3 polyunsaturated fatty acids (PUFAs), which are transported in plasma on lipoproteins, serve as precursors to SPMs production by macrophages and neutrophils and treatment with PUFAs increase SPM levels in circulation. 86 The main omega‐3–derived SPMs include lipid mediators called E‐series resolvins from eicosapentaenoic acid (EPA) and D‐series resolvins, protectins and maresins from docosahexaenoic acid (DHA). A key mechanism of action of SPMs to suppress inflammation is the induction of efferocytosis, the process by which macrophages and other phagocytotic cells engulf and eliminate apoptotic cells. For example, it was shown recently that the EPA‐derived resolvin E1 (RvE1) promotes phagocytosis of apoptotic neutrophils and reduces acute lung inflammation in a murine model. 87 Moreover, the DHA‐derived protectin D1 (PD1), is an innate suppressor of influenza virus replication. 88 There are numerous studies now showing divergent properties for EPA and DHA in modulating inflammation. For example, EPA is considered to be a more potent inhibitor than DHA of the inflammatory response in human asthmatic alveolar macrophages 89 and is more efficient in reducing AA products. 90 In contrast, endogenous PD1 has been described as a pivotal counterregulatory signal in allergic airway inflammation 41 with overall DHA derivatives reported to be biologically more stable as compared to SPMs from EPA and AA. 91 In addition, EPA‐rich fish oil increased ApoM abundance in plasma and HDL versus DHA‐rich fish oil supplement. 92
Recently it has been found that ApoE‐KO mice consuming an omega‐3 fatty acid‐deficient diet have significant accumulation of pro‐inflammatory eicosanoids in lungs. 93 Supplementing the diet with omega‐3 fatty acids reduced accumulation of these pro‐inflammatory eicosanoids and, interestingly, supplementation with omega‐3 fatty acids and aspirin significantly reduced these mediators even further. Moreover, combining the omega‐3 fatty acid diet with aspirin led to greater abundance of the main EPA‐ and DHA‐derived pathway metabolites, anti‐inflammatory 18‐hydroxyeicosapentaenoic acid (18‐HEPE), and 17‐ hydroxydocosahexaenoic acid (17‐HDHA). 84 In addition to respiratory symptoms, abnormal coagulation with thromboembolic disease is considered to be a significant factor in COVID‐19 deterioration and clinical outcomes. 94 Indeed, ApoE‐deficient mice showed evident endothelial dysfunction, which along with high ACE2 receptor expression and TxB2 accumulation can lead to excessive vascular permeability and increased blood coagulation. It should also be mentioned that endothelial cells represent a significant proportion of lung cells. 95 Of interest, omega‐3 fatty acid supplementation and aspirin treatment significantly suppressed lung TxB2 levels in ApoE‐KO mice. 93
Taking together, these observations suggest that direct treatment with either EPA‐ or DHA‐derived SPMs in the course of COVID‐19 might have potential therapeutic effect. 96 , 97 In fact, omega‐3 fatty acids are currently being tested in several ongoing clinical trials using EPA monotherapy or as a supplement mixture containing EPA, gamma‐linolenic acid, and antioxidants in COVID‐19 patients (Table 2).
TABLE 2.
Adjunctive therapies currently under investigation for COVID‐19
| Therapy | Identifiers ClinicalTrials.gov/ChiCTR.org.cn/clinicaltrialsregister.eu (EudraCT) | Mechanism of action |
|---|---|---|
| NSAID | COX‐1 and COX‐2 inhibitor | |
| Aspirin (acetylsalicylic acid) | NCT04365309 | |
| + Losartan + Simvastatin | NCT04343001 | |
| + Vitamin D | NCT04363840 | |
| Naproxen | 2020‐001301‐23 | |
| Corticosteroids | NCT04273321 | Multifactorial |
| NCT04323592 | ||
| Statin | HMG‐CoA reductase inhibitor | |
| Ulinastatin | CHICTR2000030779 | |
| Ulinastatin | CHICTR2000032135 | |
| Atorvastatin | NCT04380402 | |
| Omega‐3 PUFAs | Multifactorial | |
| EPA | NCT04335032 | |
| EPA + gamma‐linolenic acid and antioxidants | NCT04323228 | |
| Sitagliptin | NCT04365517 | Dipeptidyl peptidase‐4 (DPP‐4) inhibitor |
| Colchicine | NCT04355143 | Inhibition of microtubule polymerization |
| NCT04322565 | ||
| NCT04326790 | ||
| NCT04363437 | ||
| NCT04375202 | ||
| NCT04350320 | ||
| NCT04367168 | ||
| NCT04328480 | ||
| NCT04360980 | ||
| NCT04322682 | ||
| 2020‐001603‐16 |
6. CONCLUSIONS
The “cytokine storm” underlying COVID‐19 produces immune‐mediated inflammatory dyslipoproteinemia, leading to low HDL‐C and LDL‐C levels, elevated triglycerides, increased lipoprotein oxidation, low ApoE levels, and impaired inflammation resolution due to decreased SPMs biosynthesis. These lipid abnormalities might be modified by pharmacological agents that increase ApoA‐I and HDL plasma levels. For example, ApoA‐I mimetic peptides are known to inhibit inflammation induced by influenza infection in a model of human pneumocytes 98 and modulate the severity of neutrophilic airway inflammation in a mouse asthma model. 99 Raising HDL may also restore lipid transport function, along with improving the antioxidant properties of HDL. 59 We also propose that low levels of ApoA‐I and ApoE might be in part responsible for distinct lung inflammation, as shown previously in ApoE‐deficient mice and available but limited human data.
Other HDL‐raising pharmacological compounds that could be considered as adjunctive therapy for COVID‐19, include fibrates, CETP‐inhibitors, recombinant LCAT, 38 and small molecules that upregulate ApoA‐I production. 100 Increasing HDL would have the added benefit of decreasing platelet hyperreactivity by limiting intraplatelet cholesterol overload, along with suppressing the coagulation cascade and inhibiting platelet activation. 101
The rise in eicosanoids and hypercoagulation that occurs in COVID‐19 may possibly controlled by combined therapy with omega‐3 fatty acids and aspirin, which by itself has anticoagulant properties. This drug combination has been clinically validated and presents with low risk of interactions with other COVID‐19 treatments. Omega‐3 fatty acids in combination with aspirin might represent a valuable alternative treatment by producing proresolving aspirin‐triggered lipoxins (AT‐LXs) and decreasing eicosanoids such as PGE2 and TxB2. 102 Treatment with SPMs themselves, such as E‐ and D‐series resolvins, may also be effective in patients with severe COVID‐19, due to their effective tissue availability and fast biological action. 96
Any pharmacological agent added for the treatment of COVID‐19 will require, however, close monitoring for possible drug interactions or any other unanticipated consequences. 103 This requires the need to perform randomized placebo control clinical trials for ultimately establishing the safety and efficacy of any new drug added to the treatment of COVID‐19.
CONFLICT OF INTEREST
The author declares no conflicts of interest to disclose.
AUTHOR CONTRIBUTIONS
A.V. Sorokin conceived and researched the hypothesis proposed in this manuscript and wrote the manuscript. A.T. Remaley, S.T. Karathanasis, Z.‐H. Yang, L. Freeman, and K. Kotani critically reviewed the manuscript and contributed to the hypothesis exploration. K. Kotani analyzed clinical case data.
ACKNOWLEDGMENT
We thank Dr Nagata for providing clinical case data.
Sorokin AV, Karathanasis SK, Yang Z‐H, Freeman L, Kotani K, Remaley AT. COVID‐19—Associated dyslipidemia: Implications for mechanism of impaired resolution and novel therapeutic approaches. The FASEB Journal. 2020;34:9843–9853. 10.1096/fj.202001451
This article was fast‐tracked under a recently instituted interim policy in which editors may, at their discretion, accept coronavirus‐related manuscripts submitted for the Review, Perspectives, and Hypotheses categories without additional review.
Kazuhiko Kotani and Alan T. Remaley have equal senior authorship.
Contributor Information
Alexander V. Sorokin, Email: sorokinav2@nhlbi.nih.gov.
Alan T. Remaley, Email: alan.remaley@nih.gov.
REFERENCES
- 1. Cheng VC, Lau SK, Woo PC, Yuen KY. Severe acute respiratory syndrome coronavirus as an agent of emerging and reemerging infection. Clin Microbiol Rev. 2007;20(4):660‐694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wu C, Chen X, Cai Y, et al. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan, China. JAMA Intern Med. 2020. 10.1001/jamainternmed.2020.0994. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bernard GR, Artigas A, Brigham KL, et al. The American‐European Consensus Conference on ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am J Respir Crit Care Med. 1994;149(3 Pt 1):818‐824. [DOI] [PubMed] [Google Scholar]
- 4. Tay MZ, Poh CM, Rénia L, MacAry PA, Ng LFP. The trinity of COVID‐19: immunity, inflammation and intervention. Nat Rev Immunol. 2020;20(6):363‐374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sun X, Wang T, Cai D, et al. Cytokine storm intervention in the early stages of COVID‐19 pneumonia. Cytokine Growth Factor Rev. 2020;53:38‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Leng Z, Zhu R, Hou W, et al. Transplantation of ACE2(‐) mesenchymal stem cells improves the outcome of patients with COVID‐19 pneumonia. Aging Dis. 2020;11(2):216‐228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, function, and antigenicity of the SARS‐CoV‐2 spike glycoprotein. Cell. 2020;181(2):281‐292.e286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Xu H, Zhong L, Deng J, et al. High expression of ACE2 receptor of 2019‐nCoV on the epithelial cells of oral mucosa. Int J Oral Sci. 2020;12(1):8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zou X, Chen K, Zou J, Han P, Hao J, Han Z. Single‐cell RNA‐seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019‐nCoV infection. Front Med. 2020;14(2):185‐192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sluimer JC, Gasc JM, Hamming I, et al. Angiotensin‐converting enzyme 2 (ACE2) expression and activity in human carotid atherosclerotic lesions. J Pathol. 2008;215(3):273‐279. [DOI] [PubMed] [Google Scholar]
- 11. Chen L, Li X, Chen M, Feng Y, Xiong C. The ACE2 expression in human heart indicates new potential mechanism of heart injury among patients infected with SARS‐CoV‐2. Cardiovasc Res. 2020;116(6):1097‐1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Iwai M, Horiuchi M. Devil and angel in the renin‐angiotensin system: ACE‐angiotensin II‐AT1 receptor axis vs. ACE2‐angiotensin‐(1–7)‐Mas receptor axis. Hypertens Res. 2009;32(7):533‐536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Diaz JH. Hypothesis: angiotensin‐converting enzyme inhibitors and angiotensin receptor blockers may increase the risk of severe COVID‐19. J Travel Med. 2020;27(3):taaa041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Danser AHJ, Epstein M, Batlle D. Renin‐angiotensin system blockers and the COVID‐19 pandemic: at present there is no evidence to abandon renin‐angiotensin system blockers. Hypertension. 2020;75(6):1382‐1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Huang C, Wang Y, Li X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395(10223):497‐506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Soares MP, Teixeira L, Moita LF. Disease tolerance and immunity in host protection against infection. Nat Rev Immunol. 2017;17(2):83‐96. [DOI] [PubMed] [Google Scholar]
- 17. Libby P, Loscalzo J, Ridker PM, et al. Inflammation, immunity, and infection in atherothrombosis: JACC review topic of the week. J Am Coll Cardiol. 2018;72(17):2071‐2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Goeijenbier M, van Sloten TT, Slobbe L, et al. Benefits of flu vaccination for persons with diabetes mellitus: a review. Vaccine. 2017;35(38):5095‐5101. [DOI] [PubMed] [Google Scholar]
- 19. Cepkova M, Matthay MA. Pharmacotherapy of acute lung injury and the acute respiratory distress syndrome. J Intensive Care Med. 2006;21(3):119‐143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Walkey AJ, Wiener RS. Utilization patterns and patient outcomes associated with use of rescue therapies in acute lung injury. Crit Care Med. 2011;39(6):1322‐1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Desforges JF, Gordon DJ, Rifkind BM. High‐density lipoprotein—the clinical implications of recent studies. N Engl J Med. 1989;321(19):1311‐1316. [DOI] [PubMed] [Google Scholar]
- 22. Pencina MJ, D'Agostino RB Sr, Larson MG, Massaro JM, Vasan RS. Predicting the 30‐year risk of cardiovascular disease: the framingham heart study. Circulation. 2009;119(24):3078‐3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Acharjee S, Boden WE, Hartigan PM, et al. Low levels of high‐density lipoprotein cholesterol and increased risk of cardiovascular events in stable ischemic heart disease patients: a post‐hoc analysis from the COURAGE trial (Clinical Outcomes Utilizing Revascularization and Aggressive Drug Evaluation). J Am Coll Cardiol. 2013;62(20):1826‐1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rader DJ, Alexander ET, Weibel GL, Billheimer J, Rothblat GH. The role of reverse cholesterol transport in animals and humans and relationship to atherosclerosis. J Lipid Res. 2009;50(suppl):S189‐S194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Karathanasis SK, Freeman LA, Gordon SM, Remaley AT. The changing face of HDL and the best way to measure it. Clin Chem. 2017;63(1):196‐210. [DOI] [PubMed] [Google Scholar]
- 26. Madsen CM, Varbo A, Nordestgaard BG. Low HDL cholesterol and high risk of autoimmune disease: two population‐based cohort studies including 117341 individuals. Clin Chem. 2019;65(5):644‐652. [DOI] [PubMed] [Google Scholar]
- 27. Suzuki M, Pritchard DK, Becker L, et al. High‐density lipoprotein suppresses the type I interferon response, a family of potent antiviral immunoregulators, in macrophages challenged with lipopolysaccharide. Circulation. 2010;122(19):1919‐1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kabouridis PS, Jury EC. Lipid rafts and T‐lymphocyte function: implications for autoimmunity. FEBS Lett. 2008;582(27):3711‐3718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Trinder M, Walley KR, Boyd JH, Brunham LR. Causal inference for genetically determined levels of high‐density lipoprotein cholesterol and risk of infectious disease. Arterioscler Thromb Vasc Biol. 2020;40(1):267‐278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Van Lenten BJ, Wagner AC, Nayak DP, Hama S, Navab M, Fogelman AM. High‐density lipoprotein loses its anti‐inflammatory properties during acute influenza a infection. Circulation. 2001;103(18):2283‐2288. [DOI] [PubMed] [Google Scholar]
- 31. Feingold KR, Krauss RM, Pang M, Doerrler W, Jensen P, Grunfeld C. The hypertriglyceridemia of acquired immunodeficiency syndrome is associated with an increased prevalence of low density lipoprotein subclass pattern B. J Clin Endocrinol Metab. 1993;76(6):1423‐1427. [DOI] [PubMed] [Google Scholar]
- 32. Xingzhong H, Dong C, Lianpeng W, Guiqing H, Wei Y. Low serum cholesterol level among patients with COVID‐19 infection in Wenzhou, China. Lancet. 2020. 10.2139/ssrn.3544826. [Epub ahead of print]. [DOI] [Google Scholar]
- 33. Fan J, Wang H, Ye G, et al. Letter to the Editor: Low‐density lipoprotein is a potential predictor of poor prognosis in patients with coronavirus disease 2019. Metabolism. 2020;107:154243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Khovidhunkit W, Kim MS, Memon RA, et al. Effects of infection and inflammation on lipid and lipoprotein metabolism: mechanisms and consequences to the host. J Lipid Res. 2004;45(7):1169‐1196. [DOI] [PubMed] [Google Scholar]
- 35. Han CY, Chiba T, Campbell JS, et al. Reciprocal and coordinate regulation of serum amyloid A versus apolipoprotein A‐I and paraoxonase‐1 by inflammation in murine hepatocytes. Arterioscler Thromb Vasc Biol. 2006;26(8):1806‐1813. [DOI] [PubMed] [Google Scholar]
- 36. Han CY, Tang C, Guevara ME, et al. Serum amyloid A impairs the antiinflammatory properties of HDL. J Clin Invest. 2016;126(1):266‐281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Khovidhunkit W, Shigenaga JK, Moser AH, Feingold KR, Grunfeld C. Cholesterol efflux by acute‐phase high density lipoprotein: role of lecithin: cholesterol acyltransferase. J Lipid Res. 2001;42(6):967‐975. [PubMed] [Google Scholar]
- 38. Ossoli A, Simonelli S, Varrenti M, et al. Recombinant LCAT (lecithin: cholesterol acyltransferase) rescues defective HDL (high‐density lipoprotein)‐mediated endothelial protection in acute coronary syndrome. Arterioscler Thromb Vasc Biol. 2019;39(5):915‐924. [DOI] [PubMed] [Google Scholar]
- 39. Li H, Xiang X, Ren H, et al. Serum amyloid A is a biomarker of severe coronavirus disease and poor prognosis. J Infect. 2020;80(6):646‐655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bergt C, Oram JF, Heinecke JW. Oxidized HDL: the paradox‐idation of lipoproteins. Arterioscler Thromb Vasc Biol. 2003;23(9):1488‐1490. [DOI] [PubMed] [Google Scholar]
- 41. Levy BD, Kohli P, Gotlinger K, et al. Protectin D1 is generated in asthma and dampens airway inflammation and hyperresponsiveness. J Immunol. 2007;178(1):496‐502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bacchetti T, Campanati A, Ferretti G, Simonetti O, Liberati G, Offidani AM. Oxidative stress and psoriasis: the effect of antitumour necrosis factor‐α inhibitor treatment. Br J Dermatol. 2013;168(5):984‐989. [DOI] [PubMed] [Google Scholar]
- 43. Farid AS, Horii Y. Modulation of paraoxonases during infectious diseases and its potential impact on atherosclerosis. Lipids Health Dis. 2012;11:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Moraitis AG, Freeman LA, Shamburek RD, et al. Elevated interleukin‐10: a new cause of dyslipidemia leading to severe HDL deficiency. J Clin Lipidol. 2015;9(1):81‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lucero D, Islam P, Freeman LA, et al. Interleukin 10 promotes macrophage uptake of HDL and LDL by stimulating fluid‐phase endocytosis. Biochim Biophys Acta Mol Cell Biol Lipids. 2020;1865(2):158537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Martínez‐Martínez AB, Torres‐Perez E, Devanney N, Del Moral R, Johnson LA, Arbones‐Mainar JM. Beyond the CNS: the many peripheral roles of APOE. Neurobiol Dis. 2020;138:104809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pamir N, Hutchins P, Ronsein G, et al. Proteomic analysis of HDL from inbred mouse strains implicates APOE associated with HDL in reduced cholesterol efflux capacity via the ABCA1 pathway. J Lipid Res. 2016;57(2):246‐257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Raffaï RL, Hasty AH, Wang Y, et al. Hepatocyte‐derived ApoE is more effective than non‐hepatocyte‐derived ApoE in remnant lipoprotein clearance. J Biol Chem. 2003;278(13):11670‐11675. [DOI] [PubMed] [Google Scholar]
- 49. Babin PJ, Thisse C, Durliat M, Andre M, Akimenko MA, Thisse B. Both apolipoprotein E and A‐I genes are present in a nonmammalian vertebrate and are highly expressed during embryonic development. Proc Natl Acad Sci U S A. 1997;94(16):8622‐8627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Marais AD. Apolipoprotein E in lipoprotein metabolism, health and cardiovascular disease. Pathology. 2019;51(2):165‐176. [DOI] [PubMed] [Google Scholar]
- 51. Low‐Kam C, Rhainds D, Lo KS, et al. Variants at the APOE/C1/C2/C4 locus modulate cholesterol efflux capacity independently of high‐density lipoprotein cholesterol. J Am Heart Assoc. 2018;7(16):e009545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Brewer HB Jr, Zech LA, Gregg RE, Schwartz D, Schaefer EJ. NIH conference. Type III hyperlipoproteinemia: diagnosis, molecular defects, pathology, and treatment. Ann Intern Med. 1983;98(5 Pt 1):623‐640. [DOI] [PubMed] [Google Scholar]
- 53. Koopal C, Marais AD, Visseren FL. Familial dysbetalipoproteinemia: an underdiagnosed lipid disorder. Curr Opin Endocrinol Diabetes Obes. 2017;24(2):133‐139. [DOI] [PubMed] [Google Scholar]
- 54. Meyrelles SS, Peotta VA, Pereira TMC, Vasquez EC. Endothelial dysfunction in the apolipoprotein E‐deficient mouse: insights into the influence of diet, gender and aging. Lipids Health Dis. 2011;10(1):211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Barlage S, Fröhlich D, Böttcher A, et al. ApoE‐containing high density lipoproteins and phospholipid transfer protein activity increase in patients with a systemic inflammatory response. J Lipid Res. 2001;42(2):281‐290. [PubMed] [Google Scholar]
- 56. Pham T, Kodvawala A, Hui DY. The receptor binding domain of apolipoprotein E is responsible for its antioxidant activity. Biochemistry. 2005;44(20):7577‐7582. [DOI] [PubMed] [Google Scholar]
- 57. Getz GS, Reardon CA. Apoprotein E and reverse cholesterol transport. Int J Mol Sci. 2018;19(11):3479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kypreos KE, Zannis VI. Pathway of biogenesis of apolipoprotein E‐containing HDL in vivo with the participation of ABCA1 and LCAT. Biochem J. 2007;403(2):359‐367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Morton AM, Koch M, Mendivil CO, et al. Apolipoproteins E and CIII interact to regulate HDL metabolism and coronary heart disease risk. JCI Insight. 2018;3(4):98045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kockx M, Traini M, Kritharides L. Cell‐specific production, secretion, and function of apolipoprotein E. J Mol Med (Berl). 2018;96(5):361‐371. [DOI] [PubMed] [Google Scholar]
- 61. Goldstein MR, Poland GA, Graeber CW. Are certain drugs associated with enhanced mortality in COVID‐19? QJM. 2020. 10.1093/qjmed/hcaa103. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yamashita CM, Fessler MB, Vasanthamohan L, et al. Apolipoprotein E‐deficient mice are susceptible to the development of acute lung injury. Respiration. 2014;87(5):416‐427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Moore JB, June CH. Cytokine release syndrome in severe COVID‐19. Science. 2020;368(6490):473‐474. [DOI] [PubMed] [Google Scholar]
- 64. Gordon EM, Yao X, Xu H, et al. Apolipoprotein E is a concentration‐dependent pulmonary danger signal that activates the NLRP3 inflammasome and IL‐1β secretion by bronchoalveolar fluid macrophages from asthmatic subjects. J Allergy Clin Immunol. 2019;144(2):426‐441.e423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kulminski AM, Barochia AV, Loika Y, et al. The APOE ε4 allele is associated with a reduction in FEV1/FVC in women: a cross‐sectional analysis of the Long Life Family study. PLoS One. 2018;13(11):e0206873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Navab M, Hama SY, Cooke CJ, et al. Normal high density lipoprotein inhibits three steps in the formation of mildly oxidized low density lipoprotein: step 1. J Lipid Res. 2000;41(9):1481‐1494. [PubMed] [Google Scholar]
- 67. Jira W, Spiteller G, Carson W, Schramm A. Strong increase in hydroxy fatty acids derived from linoleic acid in human low density lipoproteins of atherosclerotic patients. Chem Phys Lipids. 1998;91(1):1‐11. [DOI] [PubMed] [Google Scholar]
- 68. Jin Y, Fu J. Novel insights into the NLRP 3 inflammasome in atherosclerosis. J Am Heart Assoc. 2019;8(12):e012219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Imai Y, Kuba K, Neely GG, et al. Identification of oxidative stress and Toll‐like receptor 4 signaling as a key pathway of acute lung injury. Cell. 2008;133(2):235‐249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Pothineni NVK, Karathanasis SK, Ding Z, Arulandu A, Varughese KI, Mehta JL. LOX‐1 in atherosclerosis and myocardial ischemia: biology, genetics, and modulation. J Am Coll Cardiol. 2017;69(22):2759‐2768. [DOI] [PubMed] [Google Scholar]
- 71. Ryoo S, Bhunia A, Chang F, Shoukas A, Berkowitz DE, Romer LH. OxLDL‐dependent activation of arginase II is dependent on the LOX‐1 receptor and downstream RhoA signaling. Atherosclerosis. 2011;214(2):279‐287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Balzan S, Lubrano V. LOX‐1 receptor: a potential link in atherosclerosis and cancer. Life Sci. 2018;198:79‐86. [DOI] [PubMed] [Google Scholar]
- 73. Stancel N, Chen C‐C, Ke L‐Y, et al. Interplay between CRP, atherogenic LDL, and LOX‐1 and its potential role in the pathogenesis of atherosclerosis. Clin Chem. 2016;62(2):320‐327. [DOI] [PubMed] [Google Scholar]
- 74. Hayashida K, Kume N, Murase T, et al. Serum soluble lectin‐like oxidized low‐density lipoprotein receptor‐1 levels are elevated in acute coronary syndrome: a novel marker for early diagnosis. Circulation. 2005;112(6):812‐818. [DOI] [PubMed] [Google Scholar]
- 75. Lubrano V, Del Turco S, Nicolini G, Di Cecco P, Basta G. Circulating levels of lectin‐like oxidized low‐density lipoprotein receptor‐1 are associated with inflammatory markers. Lipids. 2008;43(10):945‐950. [DOI] [PubMed] [Google Scholar]
- 76. Skarpengland T, Skjelland M, Kong XY, et al. Increased levels of lectin‐like oxidized low‐density lipoprotein receptor‐1 in ischemic stroke and transient ischemic attack. J Am Heart Assoc. 2018;7(2):e006479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Riphagen S, Gomez X, Gonzalez‐Martinez C, Wilkinson N, Theocharis P. Hyperinflammatory shock in children during COVID‐19 pandemic. Lancet. 2020;395(10237):1607‐1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Verdoni L, Mazza A, Gervasoni A, et al. An outbreak of severe Kawasaki‐like disease at the Italian epicentre of the SARS‐CoV‐2 epidemic: an observational cohort study. Lancet. 2020;395(10239):1771‐1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. He Y‐E, Qiu H‐X, Wu R‐Z, et al. Oxidised low‐density lipoprotein and its receptor‐mediated endothelial dysfunction are associated with coronary artery lesions in kawasaki disease. J Cardiovasc Transl Res. 2020;13(2):204‐214. [DOI] [PubMed] [Google Scholar]
- 80. Varga Z, Flammer AJ, Steiger P, et al. Endothelial cell infection and endotheliitis in COVID‐19. Lancet. 2020;395(10234):1417‐1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Zhang P, Liu MC, Cheng L, Liang M, Ji HL, Fu J. Blockade of LOX‐1 prevents endotoxin‐induced acute lung inflammation and injury in mice. J Innate Immun. 2009;1(4):358‐365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ. COVID‐19: consider cytokine storm syndromes and immunosuppression. Lancet. 2020;395(10229):1033‐1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Chen G, Wu DI, Guo W, et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J Clin Invest. 2020;130(5):2620‐2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Serhan CN. Pro‐resolving lipid mediators are leads for resolution physiology. Nature. 2014;510(7503):92‐101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti‐inflammatory and pro‐resolution lipid mediators. Nat Rev Immunol. 2008;8(5):349‐361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Souza PR, Marques RM, Gomez EA, et al. Enriched marine oil supplements increase peripheral blood specialized pro‐resolving mediators concentrations and reprogram host immune responses: a randomized double‐blind placebo‐controlled study. Circ Res. 2020;126(1):75‐90. [DOI] [PubMed] [Google Scholar]
- 87. El Kebir D, Gjorstrup P, Filep JG. Resolvin E1 promotes phagocytosis‐induced neutrophil apoptosis and accelerates resolution of pulmonary inflammation. Proc Natl Acad Sci U S A. 2012;109(37):14983‐14988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Morita M, Kuba K, Ichikawa A, et al. The lipid mediator protectin D1 inhibits influenza virus replication and improves severe influenza. Cell. 2013;153(1):112‐125. [DOI] [PubMed] [Google Scholar]
- 89. Mickleborough TD, Tecklenburg SL, Montgomery GS, Lindley MR. Eicosapentaenoic acid is more effective than docosahexaenoic acid in inhibiting proinflammatory mediator production and transcription from LPS‐induced human asthmatic alveolar macrophage cells. Clin Nutr. 2009;28(1):71‐77. [DOI] [PubMed] [Google Scholar]
- 90. Kendall AC, Pilkington SM, Murphy SA, et al. Dynamics of the human skin mediator lipidome in response to dietary ω‐3 fatty acid supplementation. FASEB J. 2019;33(11):13014‐13027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Homann J, Suo J, Schmidt M, et al. In vivo availability of pro‐resolving lipid mediators in oxazolone induced dermal inflammation in the mouse. PLoS One. 2015;10(11):e0143141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Yang Z‐H, Amar M, Sampson M, et al. Comparison of omega‐3 eicosapentaenoic acid versus docosahexaenoic acid‐rich fish oil supplementation on plasma lipids and lipoproteins in normolipidemic adults. Nutrients. 2020;12(3):749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Sorokin AV, Yang Z‐H, Vaisman BL, et al. Addition of aspirin to a fish oil‐rich diet decreases inflammation and atherosclerosis in ApoE‐null mice. J Nutr Biochem. 2016;35:58‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Tang N, Li D, Wang X, Sun Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J Thromb Haemost. 2020;18(4):844‐847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Zeng H, Pappas C, Belser JA, et al. Human pulmonary microvascular endothelial cells support productive replication of highly pathogenic avian influenza viruses: possible involvement in the pathogenesis of human H5N1 virus infection. J Virol. 2012;86(2):667‐678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Panigrahy D, Gilligan MM, Huang S, et al. Inflammation resolution: a dual‐pronged approach to averting cytokine storms in COVID‐19? Cancer Metastasis Rev. 2020:1‐4. 10.1007/s10555-020-09889-4. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Das UN. Can bioactive lipids inactivate coronavirus (COVID‐19)? Arch Med Res. 2020;51(3):282‐286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Van Lenten BJ, Wagner AC, Navab M, et al. D‐4F, an apolipoprotein A‐I mimetic peptide, inhibits the inflammatory response induced by influenza A infection of human type II pneumocytes. Circulation. 2004;110(20):3252‐3258. [DOI] [PubMed] [Google Scholar]
- 99. Dai C, Yao X, Keeran KJ, et al. Apolipoprotein A‐I attenuates ovalbumin‐induced neutrophilic airway inflammation via a granulocyte colony‐stimulating factor‐dependent mechanism. Am J Respir Cell Mol Biol. 2012;47(2):186‐195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Kingwell BA, Chapman MJ, Kontush A, Miller NE. HDL‐targeted therapies: progress, failures and future. Nat Rev Drug Discov. 2014;13(6):445‐464. [DOI] [PubMed] [Google Scholar]
- 101. van der Stoep M, Korporaal SJ, Van Eck M. High‐density lipoprotein as a modulator of platelet and coagulation responses. Cardiovasc Res. 2014;103(3):362‐371. [DOI] [PubMed] [Google Scholar]
- 102. Ortiz‐Muñoz G, Mallavia B, Bins A, Headley M, Krummel MF, Looney MR. Aspirin‐triggered 15‐epi‐lipoxin A4 regulates neutrophil‐platelet aggregation and attenuates acute lung injury in mice. Blood. 2014;124(17):2625‐2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Driggin E, Madhavan MV, Bikdeli B, et al. Cardiovascular considerations for patients, health care workers, and health systems during the COVID‐19 pandemic. J Am Coll Cardiol. 2020;75(18):2352‐2371. [DOI] [PMC free article] [PubMed] [Google Scholar]