

Abstract
The pandemic caused by novel severe acute respiratory syndrome coronavirus (SARS‐CoV‐2) has resulted in over 452 822 deaths in the first 20 days of June 2020 due to the coronavirus virus disease 2019 (COVID‐19). The SARS‐CoV‐2 uses the host angiotensin‐converting enzyme 2 (ACE2) receptor to gain entry inside the human cells where it replicates by using the cell protein synthesis mechanisms. The knowledge of the tissue distribution of ACE2 in human organs is therefore important to predict the clinical course of the COVID‐19. Also important is the understanding of the viral receptor‐binding domain (RBD), a region within the spike (S) proteins, that enables the entry of the virus into the host cells to synthesize vaccine and monoclonal antibodies (mAbs). We performed an exhaustive search of human protein databases to establish the tissues that express ACE2 and performed an in‐depth analysis like sequence alignments and homology modeling of the spike protein (S) of the SARS‐CoV‐2 to identify antigenic regions in the RBD that can be exploited to synthesize vaccine and mAbs. Our results show that ACE2 is widely expressed in human organs that may explain the pulmonary, systemic, and neurological deficits seen in COVID‐19 patients. We show that though the S protein of the SARS‐CoV‐2 is a homolog of S protein of SARS‐CoV‐1, it has regions of dissimilarities in the RBD and transmembrane segments. We show peptide sequences in the RBD of SARS‐CoV‐2 that can bind to the major histocompatibility complex alleles and serve as effective epitopes for vaccine and mAbs synthesis.
Keywords: 2019‐nCoV, bat virus, biological agents, BSL‐4, COVID‐19, MERS virus, SARS virus, SARS‐CoV‐2, vaccine and antibody against SARS‐CoV‐2, viral pandemics, Wuhan coronavirus outbreak, zoonotic infections
Highlights
On the basis of our results in the present study, the distribution and density of ACE2 receptor can be computed to understand the multisystem and specific organ attacks as seen clinically in COVID‐19 caused by SARS‐CoV‐2. The expression of ACE2 in the brain provides and explanation to the neurological deficits seen in COVID‐19 as has been widely reported now. We show the emergence of mutation (99.97% sequence identities) between the SARS‐CoV‐2 (Wuhan‐Hu‐1 virus) compared with the recently deposited genomes of diverse clinical isolates (May 2020). By the homology modeling of receptor‐binding domain (RBD) of SARS‐CoV‐1 and SARS‐CoV‐2, we show the structural similarities between both the proteins. For vaccine and monoclonal antibody synthesis against SARS‐CoV‐2 we identified peptides sequences in the receptor‐binding domain (RBD) of SARS‐CoV‐2 with strong binding predictions to the MHC class I and class II allele with log‐transformed binding affinity, nM affinity, and ranks. As the amino acids in the RBD sequences predicted in this study are known to interact with human ACE2 receptors, synthesis of monoclonal antibodies against them can prove to be of translational value. Our results could be useful for designing effective prevention and treatment strategies in COVID‐19.
1. INTRODUCTION
The recent outbreak of coronavirus virus disease 2019 (COVID‐19) caused by severe acute respiratory syndrome coronavirus (SARS‐CoV‐2) has resulted in widespread mortalities worldwide and has now been declared as the COVID‐19 pandemic. 1 , 2 , 3 The healthcare leadership, community, and scientists worldwide have come forward to fight SARS‐CoV‐2 that has started behaving almost like the once‐in‐a‐century pathogen we have been worried about. 3 Management of the outbreaks and attempts to contain the current COVID‐19 crisis has proven to be difficult with the rapid spread of SARS‐CoV‐2 worldwide. 2 , 4 With no vaccine and specific drugs available that target the SARS‐CoV‐2, the current COVID‐19 pandemic is annoying and threatening to the human population worldwide. The hard work of the scientists, 5 , 6 , 7 , 8 devoted scientific institutes, and organizations 8 , 9 , 10 , 11 has resulted in the identification of the genome of SARS‐CoV‐2, providing the structural details of the virus and determining the ligand‐binding attributes on the receptor‐binding domain (RBD) of the virus as quickly as possible, which has allowed exploration and elucidation the pathogenic proteins encoded by the SARS‐CoV‐2. Being taxonomically related to the betacoronaviridae group of the viral pathogens, 8 , 9 the SARS‐CoV‐2 shares a significant similarity with other members of the coronaviridae group, which have caused similar if not identical cross‐species viral diseases in humans in the past. 5 , 6 , 7 Examples of the latter include SARS‐CoV‐1 (2002 to 2004) and the Middle East respiratory syndrome (MERS) that affected the human population in 2012. 12 , 13 Additionally, very little is known about the diversity of the expression of angiotensin‐converting enzyme 2 (ACE2) in human organs and tissues. An investigation into the quantitative and qualitative distribution of ACE2 in different tissues is needed that can predict the possible organs involved and hint toward the expected outcome of localized and systemic forms of COVID‐19 presenting in the clinics. The COVID‐19 genome (NCBI Reference Sequence: NC_004718.3) is known to encode several proteins, of which the surface protein S (NCBI Reference Sequence: NP_828851.1) is known to be essential to dock the virus to ACE2 receptor on host cells. 6 , 7 , 14 It was inferred that as both, SARS‐nCoV‐1 and SARS‐CoV‐2, taxonomically belong to the betacoronavirus 5 , 6 they would have a similar if not identical composition of the S protein and the RBD that helps to dock the SARS‐CoV‐2 on to the ACE2 receptors. 6 , 14 It is important to mention here that although we know the target molecular receptor that is needed for the virus to gain entry into human cells, the RBD of SARS‐CoV‐2 is very similar but not identical to the RBD of SARS‐CoV‐1. 6 The functional significance of the regions of disparity between the S protein of SARS‐CoV‐2 and SARS‐CoV‐1 remains to be determined and exploited for therapeutic gains. The structural model of SARS‐CoV‐1 is already studied in detail for the binding of neutralizing antibodies and therefore identification of the differences between the S proteins of both the SARS‐CoV viruses can be of help in designing more specific neutralizing monoclonal antibodies (mAbs) against SARS‐CoV‐2.
We at first focused on drawing an organ‐based distribution of ACE2 receptor followed by an in‐depth analysis of the S protein of SARS‐CoV‐2 and its RBD for identification of short sequences that can be used for vaccine development and synthesis of mAbs against SARS‐CoV‐2. By performing sequence alignments and homology modeling the understating of the RBD of SARS‐CoV‐2, it is expected that we will gain an in‐depth understanding of the pathogenesis of the COVID‐19 and assemble effective ways to counter the mode of infection of SARS‐CoV‐2.
Finally, we performed in silico methods by using automated epitope‐detecting databases that extract sequences of variable length with the potential of binding to class I and class II major histocompatibility complex (MHC) alleles on antigen‐presenting cells and therefore capable of evoking a B/T‐killer cell immune response. For the epitopes generation, the S protein and the RBD of SARS‐CoV‐2 were selected and the epitopes that had shown a strong binding (SB) prediction with MHC alleles were proposed for vaccination and mAbs synthesis.
2. METHODOLOGY
2.1. Messenger RNA encoding and protein expression of ACE2 in normal human tissues
Online databases that are an integrative web resource on human and other mammalian tissue expressions 12 , 15 were used to identify the ACE2 encoding messenger RNA (mRNA). The tissue associations in these databases for ACE2 were retrieved from manually curated knowledge in UniProtKB (https://www.uniprot.org/uniprot/?query=ACE2&sort=score) 13 and via automatic text mining of the biomedical works of literature. The confidence of each association is signified by signs, where +++++ is the highest confidence and +++, ++, + as the lowest. Human tissues and Protein Atlas (HPA) 12 (www.proteinatlas.org/ENSG00000130234-ACE2/tissue) were used to retrieve the mRNA expression levels encoding ACE2 in forms of histograms. Both the servers have a user‐friendly interface wherein protein (ACE2) and other protein molecules can be queried, which results in a list of organs expressing the protein in question.
2.2. Databases of human protein expression of ACE2 at the cellular level by immunohistochemistry
Online databases that are a huge resource of data of expression of human proteins in a healthy and diseased state like HPA was used to retrieve and study the expression levels of ACE2 in normal body tissues as compared with diseased states. Immunohistochemistry of ACE2‐stained tissue was investigated for mild, moderate, and strong staining.
2.3. Genomics, phylogenetics, and transcriptomics of COVID‐19 coronavirus
The genome of COVID‐19 was obtained from the NCBI database 9 , 16 and the proteins encoded by this virus were retrieved.
NCBI automated server (https://www.ncbi.nlm.nih.gov/tools/msaviewer/) was used for multiple sequence alignment (MSA) and building an evolutionary tree of the genome of viruses and the proteins that they encode. Uniprot database 13 was also used for MSA and UniprotKB and NCBI were used to develop the coronavirus phylogenetic tree of SARS‐CoV‐2. For comparison, the S‐protein sequence of both the viruses was retrieved from the NCBI database. Pfam and NCBI database 16 were used to obtain circular and rectangular cladograms of evolutionary proteins of the SARS‐CoV‐2 family.
2.4. Sequence analysis of nucleotide, S protein, and RBD encoded by SARS‐CoV and COVID‐19
Bioinformatics computational tools were used to search for protein homologs of the RBD of S protein of SARS‐CoV‐1 and SARS‐CoV‐2. BLASTn and BLASTp tools were used to uncover nucleotide and protein homologs in between SARS‐CoV and SARS‐CoV‐2, respectively. BLASTn was selected for nucleotide similarity searches and BLASTp was selected for the determination of S protein and RBD homologs. The align tool available in the Uniprot server 13 was used for MSA of different protein sequences of SARS‐CoV‐1 and SARS‐CoV‐2 in general and S protein and its RBD in particular. In sequence alignments, particular attention was directed toward identifying the mutations in amino acid residues of the RBD of SARS‐CoV‐2 that are known to engage with ACE2 receptor during the process of host‐virus interaction and initiating viral entry into the cells. During comparisons of S protein and RBD, we specifically focused on spotting the occurrence of mutations in the transmembrane regions and motif segments of the S protein of the SARS‐CoV‐2 as compared with SARS‐CoV‐1.
2.5. Homology modeling of proteins
Sequences of the SARS‐CoV‐2 S protein and RBD were submitted to the automated SWISS‐MODEL database 17 to develop template‐based models. Similarly, sequences of the SARS‐CoV‐1 S protein and RBD were submitted to the automated SWISS‐MODEL database for the development of template‐based models so that the differences and similarities between the proteins of the viruses can be identified. Previous models of SARS‐CoV‐1 S protein and RBD were downloaded and compared with a similar region in the SARS‐CoV‐2 virus.
2.6. Identification of epitopes predicted to bind the MHC class I and II allele
Though the SARS‐CoV‐2 is declared to be a novel variant of betacoronavirus, its S protein has homologs (evolutionarily related) to the taxon of betacoronaviruses. The Immune Epitope Database and Analysis Resource (IEDB) 18 and DTU bioinformatic servers 19 , 20 were used for epitope mapping in the S protein RBD sequence in SARS‐CoV‐2. Filters were applied to search within betacoronavirus, SARS, and MERS members of the taxa only. Other filters that were selected included epitope type, host, assays type, MHC restrictions, and infectious disease. 19 Also, the amino acid sequence of S protein and RBD of SARS‐CoV‐1 and SARS‐CoV‐2 was used to generate epitopes of sequence lengths between 5mer and 9mer where a peptide‐MHC class I binding could occur for provoking antibody production in vivo.
2.7. Epitope prediction method summary
2.7.1. NetMHC‐4.0 method for the prediction of peptide
The NetMHC‐4.0 method for the prediction of peptide‐MHC class I binding affinity is publicly available (http://www.cbs.dtu.dk/services/NetMHC-4.0). The computer‐learning algorithm used by NetMHC‐4.0 is based on artificial neural networks, namely the NNAlign method. By bringing together the training instances onto a shared window of fixed length, the NNAlign algorithm efficiently creates an MSA representing the minimal binding core of each peptide submitted as a query. The novelty of the algorithm is the introduction of insertions and deletions into the NNAlign learning framework, fundamentally permitting the creation of gapped sequence alignments. In the context of the MHC class I scheme, where the length of ligands is usually not constant, insertions and deletions allow reconciling peptides of different lengths to a binding core of a common size. The prediction of the core location can provide insight on the binding mode of linear peptides to their receptors, such as in the case of binders bulging out from the middle of the MHC groove or noncanonical binders protruding at the terminal ends. The NetMHC‐4.0 automated server uses a proteome scan in search of potential T‐cell epitopes; therefore, peptides of optimal length for the alleles of interest are inherently prioritized. The user inserts a sequence, as in our case the S‐protein FASTA sequence, followed by the selection of peptide length of (8mer‐14mer) range, species, and HLA class type, and subtype selection. There are options to sort by predicted affinity and threshold for strong binders: %Rank is set to 0.5 by default. Output can be retrieved in XLS format or simple text. NetMHC‐4.0 ranks the predicted affinity by comparing it to a set of 400.000 random natural peptides. This measure is not affected by the inherent bias of certain molecules toward higher or lower mean predicted affinities. Strong binders are defined as having %Rank less than 0.5 and weak binders with %Rank less than 2. The selection of the candidate binders was based on %Rank rather than nM affinity, which is predicted by NetMHC‐4.0. A parameter of BindLevel (SB: strong binder, WB: weak binder) is also attributed to the sequence predicted as an epitope. The peptide will be identified as a strong binder if the %Rank is below the specified threshold for the strong binders, by default 0.5%. The details of the NetMHC‐4.0 usage are available at the abovementioned link, which is user friendly.
2.7.2. The Immune Epitope Database
IEDB is funded by the National Institute of Allergy and Infectious Diseases (NIAID) and is a freely available resource for users worldwide. It catalogs experimental data on antibody and T‐cell epitopes studied in the context of infectious disease, allergy, autoimmunity, and transplantation. The IEDB also hosts tools to assist in the prediction and analysis of epitopes. We used the IEDB for the prediction of epitopes that could interact with the MHC class II allele on antigen‐presenting cells and T lymphocytes. The database is interactive and user friendly as the user has to choose one of the radio buttons to select protein sequence (RBD of SARS‐CoV‐2) that contains the MHC class II epitopes for which predictions get generated. The sequence will be transferred to the MHC class II binding predictions on clicking the “Submit” button. These test data sets are meant to demonstrate the functionality of the tools and are by no means considered equivalent to a formal performance evaluation. For the prediction of MHC class II epitopes, it has a consensus approach that combines NN‐align, SMM‐align, and combinatorial library methods. An automated server from Immune Epitope Database Analysis Resource 18 was selected and predictions were generated for RBD of SARS‐CoV‐1 and SARS‐CoV‐2 and compared. The automated IEDB interface shows options like species/locus selection, prediction of linear B‐cell or T‐cell epitopes, choice of prediction methods, selection of MHC allele(s), select option for length(s), and output formats as XML or text. Details of the use and options can be found at the automated webserver site at https://www.iedb.org/.
3. RESULTS
3.1. ACE2 is expressed in diverse organs with a predominance in the lungs, endothelium, heart muscle, kidney, gastrointestinal tract, testicular tissue, brain, and adrenal glands
The mRNA encoding ACE2 dominates in the endothelial cell, lungs, heart muscle, kidney, gastrointestinal tract, testicular tissue, adrenal glands, and nervous system when compared with the rest of the organs and tissues in the human body. The ACE2 (Figure 1A) protein is expressed in different organs and tissues (Figure 1B,C) with its structure (Figure 1D) deposited in a protein database that can be a target of the SARS‐CoV‐2. The data shown (Figure 1B) were derived from the data retrieved from HPA, tissue expression database, and UniProtKB as mentioned in Section 2.
Figure 1.
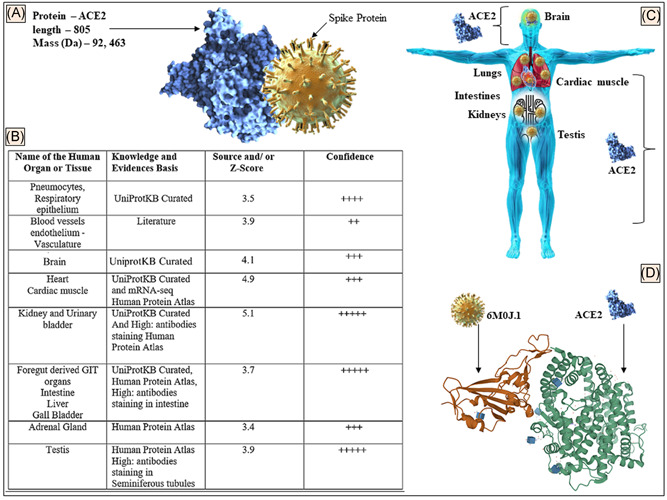
A, Details of the ACE2 protein showing schematic interaction with S protein. B, The distribution of ACE2 protein in different tissues based on the evidence of microarray, the protein expression data, and published literature. A scoring and confidence level of tissue distribution is also shown. C, Animated image to show the ACE2 distribution in organs of the human body. D, SARS‐CoV‐2 spike protein (6m0j.1) bound to the human ACE2 complex (https://doi.org/10.2210/pdb6M0J/PDB) is shown (data retrieved from References 15, 12, and 11, respectively in accord with the policy of the database resource, which enables the third parties to have access to the data shown). ACE2, angiotensin‐converting enzyme 2; GIT, gastrointestinal tract; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus‐2
3.2. Immunostaining comparison of ACE2 receptor in different organs showed predominant expression in the intestines, kidney, adrenals, gastrointestinal tract, and testis
The representative immunohistochemistry staining of ACE2 in different tissues (Figure 2) was retrieved from https://www.proteinatlas.org/ENSG00000130234-ACE2/tissue. The retrieved images show the differential staining of ACE2 in foregut derivatives like the intestine (duodenum, ileum, colon, and rectum) and gall bladder, which showed stain with maximum intensity (Figure 2). Kidney and testicular cells and tubules also showed a high expression of ACE2 receptors. Cardiac muscle and adrenal glands stained with moderate intensity in the retrieved images (Figure 2 histogram and image).
Figure 2.
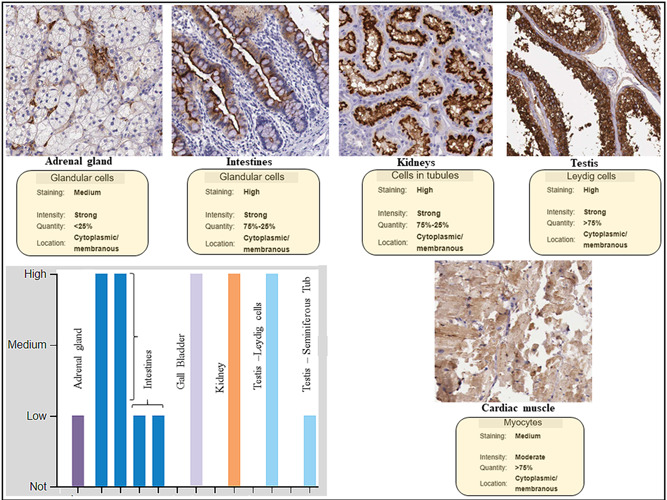
Immunocytochemistry data of quantitative expression, location, and staining intensity of ACE2 in human tissues. Protein expression overview of ACE2 showed it to be expressed highly in the intestines, gall bladder, kidney, and testicular tissue. Human Protein Atlas (HPA) did not have neuronal tissue staining in its archive (data retrieved from the HPA database in accord with the policy of the database resource retrieval which enables the third parties to have access to the data shown, http://www.proteinatlas.org/ENSG00000130234-ACE2/tissue)
3.3. SARS‐CoV‐2 and SARS‐CoV‐1 share a common ancestor and belong to the same taxon
The BLASTn results showed that the genome of SARS‐CoV‐2 (GenBank ID: QHD34416.1) had 100% sequence identities with related SARS‐CoV‐2 clinical isolates reported from other countries (with few exceptions, see below) and shared 82% to 99% sequence identities with SARS‐CoV‐1 genome. The NBCI automated server 9 , 16 was used to perform an MSA and build an evolutionary tree for the top matches with SARS‐CoV‐1 and the bat coronaviruses (Figure 3A). The phylogenetic cladogram shows the COVID‐19 Wuhan CoV‐Hu‐1 A and SARS‐CoV‐1 share a common ancestor (Figure 3B, black arrow). The BLASTn search with coronaviruses selected in the NCBI server showed (Figure 3B) the genomes of a diverse SARS‐CoV‐1 and bat coronavirus (Figure 3A, black star) as homologs, which justifies it to be named as SARS‐CoV‐2 based on its taxonomy. With the recently deposited SARS‐CoV‐2 genomes from different clinical isolates from around the world, the occurrence of mutations in the genome of SARS‐CoV‐2 has been noticed, some of which have sequences identities of 99.97% (Figure 3C, top node and blue stars) and the 00.03% difference could be of pathogenic significance.
Figure 3.
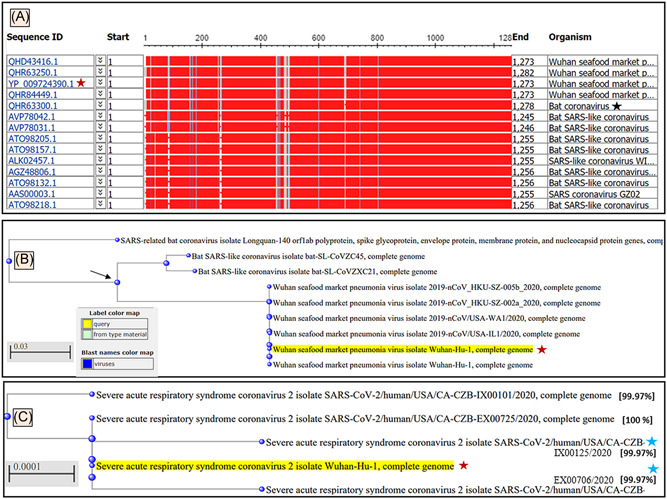
A, Graphic summary of sequences producing significant alignments of severe acute respiratory syndrome coronavirus‐2 (SARS‐CoV‐2) isolate Wuhan‐Hu‐1, complete genome. B, The phylogenetic tree developed for SARS‐CoV‐2 Wuhan‐Hu‐1 virus in the form of a rectangular cladogram showed its root of origins in the taxon of coronaviruses. B, The bat coronavirus, SARS‐CoV‐1 and SARS‐CoV‐2 Wuhan‐Hu‐1 virus showed a common ancestor. The BLASTn results of SARS‐CoV‐2 Wuhan‐Hu‐1 virus (Wuhan seafood coronavirus, GenBank ID: QHD34416.1) nucleotides showed bat coronavirus (black star) and bat SARS‐like CoV as the top homologs of this virus. C, The phylogenetic tree of the SARS‐CoV‐2 Wuhan‐Hu‐1 virus (yellow highlighted with red stars) compared with the recently deposited genomes of diverse clinical isolate (May 2020) show the occurrence of mutation (99.97% sequence identities) at early (top node) proximal node and recently after the outbreak (blue stars) coronavirus (wild type, black star)
3.4. MSA of the RBD encoded by SARS‐CoV‐1 and SARS‐CoV‐2
The sequence of the S protein of the SARS‐CoV‐1 and SARS‐CoV‐2 is well‐established. The sequence alignment of the RBD of the S protein SARS‐CoV‐1 (NP_828851.1) with the SARS‐CoV‐2 (YP_009724390.1) showed a remarkable homology with a sequence identity of 73.09%. The pairwise alignment also showed the differences between the S protein sequences. The RBD of SARS‐CoV‐1 that stretches between the 306th and 527th amino acids (aa) is located within the S protein sequence of the protein length of 1255 aa. While the RBD (319th‐541st aa) of S protein in SARS‐CoV‐2, which is composed of 1273 aa has now been elucidated. 21 , 22 We aligned the RBD (319th‐541st aa) of SARS‐CoV‐2 (YP_009724390.1) with the RBD (306th‐527th aa) of SARS‐CoV‐1 (NP_82885.1.1) (Figure 4A) and pairwise alignments showed 73.09% sequence identities. Interestingly, we found that the RBD of SARS‐CoV‐2 had mutations at five (Figure 4A, red stars) of the six amino acids that are needed to engage with the human ACE2 receptors, as has also been reported by others. 1
Figure 4.
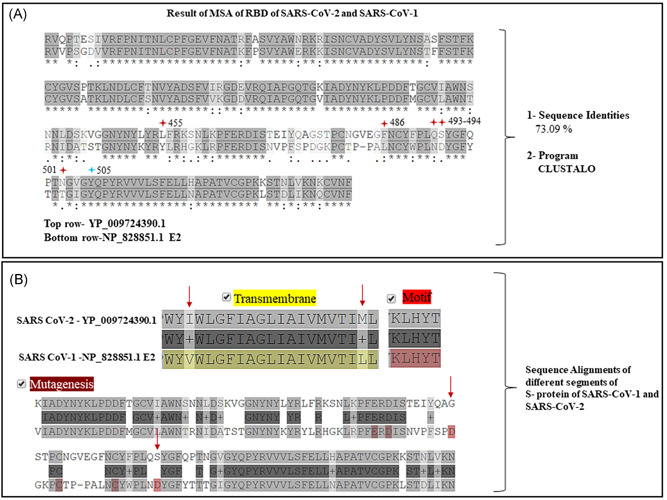
A, Multiple sequence alignment (MSA) results of the receptor‐binding domain (RBD) (319th‐541st aa) (top row) of severe acute respiratory syndrome coronavirus‐2 (SARS‐CoV‐2) (accession# YP_009724390) and the RBD of S protein (306th‐527th aa) of SARS‐CoV‐1 (accession# NP_828851) showed 73.09% of sequence identities. Note five (red stars) of the six amino acid residues mutated in (top row) in RBD of SARS‐CoV‐2. B, Except for the motif, segments of transmembrane regions and points of mutagenesis were noted in between the S protein of and SARS‐CoV‐2 (top row) and of SARS‐CoV‐1 (bottom row)
3.5. Homology modeling RBD of S protein in SARS‐CoV‐2 and SARS‐CoV‐1
The SARS‐CoV‐2 RBD (319th‐541st aa) was subjected to template‐based model developments. The FASTA sequence of the above amino acids of RBD of SARS‐CoV‐2 was submitted to the SWISS‐MODEL automated server 17 for homology modeling, which developed a template (5x5b.1)‐based model of SARS‐CoV‐1 S protein (Figure 5A) RBD (Figure 5A, circle) with 73.42% sequence identities. The RBD of SARS‐CoV‐1 (306th‐527th aa) was also subjected to homology modeling (Figure 5B) to determine its homology with SARS‐CoV‐2 RBD. The automated server developed a template (6vw.1)‐based model of SARS‐CoV‐2 RBD in complex with human ACE2 (Figure 5B, green ribbons) with 87.50% sequence identities (Figure 5B, brown ribbons).
Figure 5.
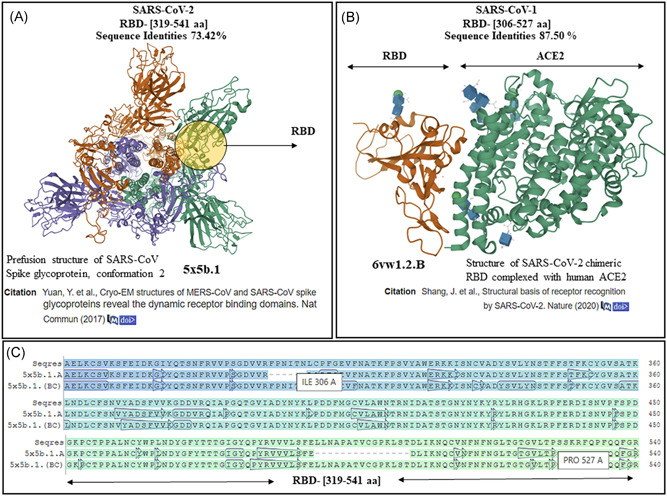
Homology modeling results of receptor‐binding domain (RBD) of severe acute respiratory syndrome coronavirus‐1 (SARS‐CoV‐1) and SARS‐CoV‐2. A, The results for SARS‐CoV‐2 RBD (319th‐541st aa) showed a template‐based model of perfusion structure of RBD of S protein of SARS‐CoV‐1. B, On homology modeling, the RBD of SARS‐CoV‐1 (306th‐527th aa) developed a template‐based model of SARS‐CoV‐2 RBD (brown ribbons) in complex with ACE2 (green ribbons) with PDB ID (6vw.1). C, The sequence of RBD of SARS‐CoV‐2 aligned with the template (5b5x chain A, B, C) sequences show the near‐identical amino acid similarities between the B and C chains of the template and the Seqres S protein (top row) (template‐based models were developed by SWISS‐MODEL automated and accessible at the database: https://swissmodel.expasy.org/templates/6vw1.1 and PDB ID at https://www.rcsb.org/3d-view/6VW1/1)
3.6. Epitopes identified in SARS‐CoV‐2 RBD for vaccine and monoclonal antibody development
The entire sequence of S protein of SARS‐CoV‐2 (1273 aa) (YP_009724390.1) was submitted to the healthtech dtu dk server, 18 , 19 and the IEDB 20 for the generation of epitopes. The results from the healthtech dtu dk server fetched four epitopes within the RBD (319‐541 aa) with SB prediction to the HLA‐A and HLA‐B class I allele (Figure 6A, stars). The IEDB automated server was used to fetch sequences in the RBD of SARS‐CoV‐2 for predicted binding to the MHC class II allele. Shown are the sequences that fall between 319 and 541 aa of the RBD of SARS‐CoV‐2 as a result (Figure 7A,B). The RBD regions of the SARS‐CoV‐2 that contain these sequences are shown on the model of SARS‐CoV‐2 spike protein (Figure 1, 2, 7).
Figure 6.
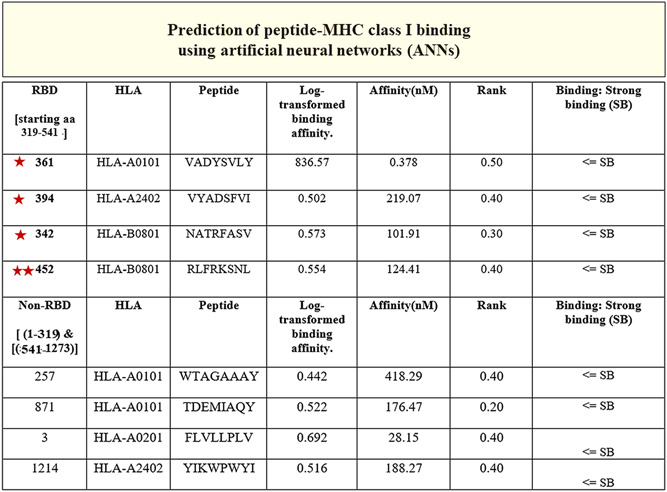
The peptides (third column) identified in the RBD (319‐541 aa) region of the S of severe acute respiratory syndrome coronavirus‐2 (SARS‐CoV‐2) (stars) had SB predictions (seventh column). The binding prediction to the MHC class I allele (second column) with log‐transformed binding affinity, nM affinity, and rank (fourth, fifth, and sixth column, respectively) are shown. The non‐RBD resident sequences with SB and nM affinity are shown in the last four columns that were predicted to bind the MHC class I allele. Note the double stars (452‐459 aa) of which 455 is a known residue in the RBD of S protein that engages with the ACE2 receptor. 21 HLA, human leukocyte antigen; MHC, major histocompatibility complex; RBD, receptor‐binding domain; SB, strong binding
Figure 7.
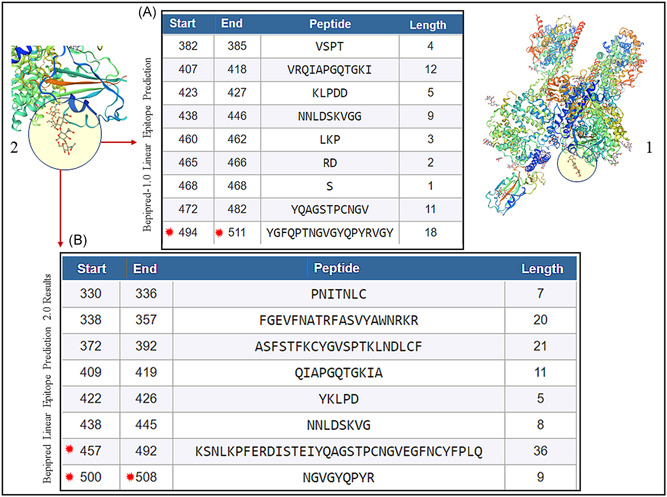
A, BepiPred 1.0 prediction software in the automated Immune Epitope Database and Analysis Resource (IEDB) server was used that predicted the location of linear B‐cell epitopes using a combination of a hidden Markov model and a propensity scale method. Results are based on a large benchmark calculation containing close to 85 B‐cell epitopes. Note stars (494‐511 aa), which contain 501 and 505 amino acids that are known to engage with ACE2 receptor. 23 B, BepiPred‐2.0: Sequential B‐Cell Epitope Predictor automated serve was used to predict B‐cell epitopes from receptor‐binding domain (RBD) of the S of protein of SARS‐CoV‐2, using a random forest algorithm trained on epitopes and non‐epitope amino acids that are determined from crystal structures. Note stars (457‐492 aa) which contain 486, 501, and 505 amino acids that are known to engage with ACE2 receptor. 23 The structure of SARS‐CoV‐2 RBD (PDB ID: 6vsb.1) is shown (1) with the peptides predicted (A, B) to be located within the RBD (two zoomed circles) of the template‐based model of SARS‐CoV‐2
4. DISCUSSION
The knowledge of the pathogenesis of the SARS‐CoV‐2 and the understanding of the diversity of its clinical presentation is still sketchy and much remains to be elucidated. 22 , 23 We know very little about the translational significance of the genomic, transcriptomic, and proteomic discoveries on SARS‐CoV‐2 that have emerged recently. Though we know the tissue distribution of the ACE2, a molecular target of RBD of SARS‐CoV‐2, 7 the cascade of host and virus protein interaction in human organs and tissue remains obscure; therefore, an understanding of the pathogenesis of SARS‐CoV‐2 is urgently needed. 23 It is noteworthy that though the SARS‐CoV‐2 uses the same ACE2 receptor for human cell infection in various organs and tissues that express this receptor subtype (Figures 1 and 2), the RBD of SARS‐CoV‐2 is different when compared with SARS‐CoV‐1 7 , 21 , 24 , 25 and besides different ACE2 alleles may show perceptible variations in the molecular interactions with the RBD of SARS‐CoV‐2. 32 Phylogenetic analysis clearly showed the origin of SARS‐CoV‐2 from the linage of betacoronaviruses (Figure 3A,B), 9 , 21 and its strong association with different clinical isolates that have been identified during the recent pandemic (Figure 3C). Regarding the mutations occurring in proteins encoded by SARS‐CoV‐2, we show that the in clinical isolates from the US mutations have emerged (Figure 3C, blue stars) as compared with the original isolate SARS‐CoV‐2 (GenBank ID: YP_009724390.1) identified and archived in NCBI 9 (Figure 3C, red star). Others have also reported mutations involving RNA‐dependent RNA polymerase. 26
The significance of the mutations in RBD of the S protein of SARS‐CoV‐2 as compared with SARS‐CoV‐1 has just begun to be elucidated with the insight that the mutations in the RBD of S protein of SARS‐CoV‐2 has increased its affinity for the ACE2 by several folds. 7 In contrast, different ACE2 alleles could reduce the affinity of RBD of SARS‐CoV‐2 to infect human cells as mentioned above. We, like the other, 21 report mutations in five out of six key amino acids of the RBD of SARS‐CoV‐2 compared with SARS‐CoV‐1, which are cardinal for engaging the ACE2 receptors (Figure 4A, red stars). Sequence alignments showed other regions of mutagenesis in SARS‐CoV‐2 in regions like transmembrane, while the motif has shown to be remained conserved (Figure 4B, arrows and brown alphabets). The next step in our study was to explore the structural details of the S protein and its RBD in SARS‐CoV‐2 to see how it differs from SARS‐CoV‐1, which could help uncover the rate of transmission and infectivity of SARS‐CoV‐2 in human cells. As the structural details of the S protein and RBD of SARS‐CoV‐2 got resolved and deposited early after the outbreak of COVID‐19, 7 , 24 , 25 we performed homology modeling of the structures of the RBD of SARS‐CoV‐1 and SARS‐CoV‐2 to compare them for homology and compute their interaction with ACE2. The RBD sequence of SARS‐CoV‐2 and SARS‐CoV‐1 was subjected to homology modeling (Figure 5). The RBD of SARS‐CoV‐2 developed a template‐based model of SARS‐CoV‐1 (Figure 5A) and the RBD of SARS‐CoV‐1 developed a template‐based model of SARS‐CoV‐2 RBD in complex with human ACE2 (Figure 5B), which showed them to be homologs (evolutionarily related) reinforcing the taxonomical relation between the two viruses. We show that in contrast to the A chain, the B and C chains of the template (5x5b.1) developed for SARS‐CoV‐2 RBD (319th‐541st aa) have identical amino acids when compared with the model developed (Figure 5C, top row‐Seqres). After the identification of the RBD sequence of SARS‐CoV‐2 and their differences with SARS‐CoV‐1, we were curious to identify sequences in the RBD of SARS‐CoV‐2 that are capable of serving as epitopes to bind the HLA class I and class II allele in macrophages to mount an immune response against RBD of SARS‐CoV‐2 neutralizing its capability to infect human cells. As expected, any short or long sequence extracted from the S protein would not match any human protein as we checked (data not are shown). There is no certainty that sequences picked in this way would interact with the HLA class I and class II allele in macrophages to mount an immune response. The sequences that were identified to have the potential to bind the HLA class I (Figure 6) and class II allele (Figure 7) in macrophages for antigen presentation are shown. The DTU bioinformatics portal, 19 recognizes a minimal five to nine amino acid length peptides with a binding core directly in contact with the MHC allele, predicted binding affinity is in nanomolar units, a rank of the predicted affinity (Figure 6) with strong binders (SB, defined as having %Rank <0.5). 19 The automated IEDB 20 server Bepipred‐1.0 Linear Epitope Predictor and BepiPred‐2.0: Sequential B‐Cell Epitope Predictor identified the peptide sequence with the predictions of binding to the MHC class II allele (Figure 7). An important observation that was needed to be taken into account was to spot an epitope(s) that would attack the ACE2 binding amino acid residues 21 in the RBD of the S protein of SARS‐CoV‐2. We show (Figure 7A,B, stars) the epitopes we present in this study can perhaps mount a T‐cell response in particular by binding HLA class II allele(s), which was previously reported by using the IEDB server by us for SARS‐CoV‐2 in a preprint 27 and others studies on the Middle East Respiratory Syndrome Coronavirus (MERS‐CoV). 28 The mechanism of T‐cell–mediated cytotoxicity unlike IgG‐type humoral response is expected to cause a limited cytokine formation and therefore lesser tissue damage. We infer that the peptides we are reporting in this study have stronger MHC class I and II allele binding predictions with their affinity in nanomolar units that make them superior over other peptides that are proposed for vaccine development reported previously by us. 27 Also, reverse vaccinology and machine learnings have been applied to generate epitopes for a vaccine against SARS‐CoV‐2, SARS‐CoV‐1, and MERS‐CoV. 29 It is obvious now that this is the right time to consider investing in vaccines against emerging viruses, which if delayed could lead to loss of human lives 31 as evident in the ongoing pandemic. Though vaccines against other proteins and targets including the S protein have been detailed, 30 , 31 epitopes reported by us are niched around the RBD of the S‐protein with more importance positioned around prophylaxis and prevention of reinfection by SARS‐CoV‐2 in COVID‐19.
5. CONCLUSIONS AND FUTURE DIRECTIONS
The areas that need attention in our fight against COVID‐19 are the differences in tissue distribution of ACE2 in different organs, the differential binding affinities of RBD of S‐protein to ACE2 in diverse ethnic populations, 32 the understanding of the RBD basis of the SARS‐CoV‐2 infection caused by mutations in this segment 7 , 24 , 25 and an urgent attempt to prepare vaccine 30 /mAbs to combat COVID‐19. 27 It would be ideal if the mAbs generated against the epitopes could be produced in mass in vitro to be given by infusions, as this could lessen the need for the convalescent plasma of patients recovered from COVID‐19 infections. Unraveling these pivotal components is likely to make us capable to fight the COVID‐19. We implore to speed up the process of vaccine development and synthesis of mAbs against SARS‐CoV‐2 to fight the pandemic caused by COVID‐19, by testing the sequences submitted in this study and other similar reports. 29 , 30 Also, much needed is a safe and efficacious antiviral agent that could eradicate COVID‐19 with minimal adverse effects.
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
Baig AM, Khaleeq A, Syeda H. Elucidation of cellular targets and exploitation of the receptor‐binding domain of SARS‐CoV‐2 for vaccine and monoclonal antibody synthesis. J Med Virol. 2020;92:2792–2803. 10.1002/jmv.26212
REFERENCES
- 1.US Centers for Disease Control and Prevention. Coronavirus disease 2019 (Covid‐19): situation summary. https://www.cdc.gov/coronavirus/2019-nCoV/summary.html. Accessed June 1, 2020.
- 2. Watkins J. Preventing a COVID‐19 pandemic. BMJ. 2020;368:m810. 10.1136/bmj.m810 [DOI] [PubMed] [Google Scholar]
- 3. Gates B. Responding to Covid‐19—a once‐in‐a‐century pandemic? N Engl J Med. 2020;382:1677‐1679. 10.1056/NEJMp2003762 [DOI] [PubMed] [Google Scholar]
- 4. Day M. Covid‐19: surge in cases in Italy and South Korea makes pandemic look more likely. BMJ. 2020;368:m751. 10.1136/bmj.m751 [DOI] [PubMed] [Google Scholar]
- 5. Wahba L, Jain N, Fire AZ, et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;5. 10.1038/s41586-020-2008-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yan R, Zhang Y, Li Y, Xia L, Guo Y, Zhou Q. Structural basis for the recognition of the SARS‐CoV‐2 by full‐length human ACE2. Science. 2020;367:1444‐1448. 10.1126/science.abb2762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wrapp D, Wang N, Corbett KS, et al. Cryo‐EM structure of the 2019‐nCoV spike in the prefusion conformation. Science. 2020;367:1260‐1263. 10.1126/science.abb2507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Roy A, Kucukural A, Zhang Y. I‐TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 2010;5:725‐738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. National Center for Biotechnology Information (NCBI) . Bethesda (MD): National Library of Medicine (US), National Center for Biotechnology Information; 1988. https://www.ncbi.nlm.nih.gov/nuccore/1798174254. Accessed June 1, 2020.
- 10. Bertoni M, Kiefer F, Biasini M, Bordoli L, Schwede T. Modeling protein quaternary structure of homo‐ and hetero‐oligomers beyond binary interactions by homology. Sci Rep. 2017;7:10480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Berman HM, Westbrook J, Feng Z, et al. The Protein Data Bank. Nucleic Acids Res. 2000;28:235‐242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Uhlén M, Fagerberg L, Hallström BM, et al. Tissue‐based map of the human proteome. Science. 2015;347. 10.1126/science.1260419 [DOI] [PubMed] [Google Scholar]
- 13. The UniProt Consortium . UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019;47:D506‐D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen Y, Guo Y, Pan Y, Zhao ZJ. Structure analysis of the receptor binding of 2019‐nCoV. Biochem Biophys Res Commun. 2020;525(1):135‐140. 10.1016/j.bbrc.2020.02.071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Palasca O, Santos A, Stolte C, Gorodkin J, Jensen LJ. TISSUES 2.0: an integrative web resource on mammalian tissue expression. Database. 2018;2018:bay003. 10.1093/database/bay003 [DOI] [PMC free article] [PubMed]
- 16. National Center for Biotechnology Information . U.S. National Library of Medicine 8600 Rockville Pike, Bethesda, MD. https://guides.nnlm.gov/tutorial/ncbi-gene-sequence-. Accessed February 16, 2020.
- 17. Waterhouse A, Bertoni M, Bienert S, et al. SWISS‐MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46(W1):W296‐W303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kim Y, Ponomarenko J, Zhu Z, et al. Immune epitope database analysis resource. Nucleic Acids Res. 2012;40(Web Server issue):W525‐W530. 10.1093/nar/gks438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Andreatta M, Nielsen M. Gapped sequence alignment using artificial neural networks: application to the MHC class I system. Bioinformatics. 2016;32(4):511‐517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nielsen M, Lundegaard C, Worning P, et al. Reliable prediction of T‐cell epitopes using neural networks with novel sequence representations. Protein Sci. 2003;12:1007‐1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Andersen KG, Rambaut A, Lipkin WI, Holmes EC, Garry RF. The proximal origin of SARS‐CoV‐2. Nat Med. 2020;26(4):450‐452. 10.1038/s41591-020-0820-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tadini E, Papamidimitriou‐Olivgeris M, Opota O, et al. SARS‐CoV‐2, a point in the storm. Rev Med Suisse. 2020;16(692):917‐923. [PubMed] [Google Scholar]
- 23. Zhao N, Zhou ZL, Wu L, et al. An update on the status of COVID‐19: a comprehensive review. Eur Rev Med Pharmacol Sci. 2020;24(8):4597‐4606. 10.26355/eurrev_202004_21046 [DOI] [PubMed] [Google Scholar]
- 24. Lan J, Ge J, Yu J, et al. Structure of the SARS‐CoV‐2 spike receptor‐binding domain bound to the ACE2 receptor. Nature. 2020;581:215‐220. 10.1038/s41586-020-2180-5 [DOI] [PubMed] [Google Scholar]
- 25. Shang J, Ye G, Shi K, et al. Structural basis of receptor recognition by SARS‐CoV‐2. Nature. 2020;581:221‐224. 10.1038/s41586-020-2179-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pachetti M, Marini B, Benedetti F, et al. Emerging SARS‐CoV‐2 mutation hot spots include a novel RNA‐dependent‐RNA polymerase variant. J Transl Med. 2020;18(1):179. 10.1186/s12967-020-02344-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Baig AM. The devil in its details: unravelling the epitopes in COVID‐19 virus surface glycoprotein with the potential for vaccination and antibody synthesis. Available from Research square: Preprint Nature Research. 10.21203/rs.3.rs-19497/v1
- 28. Tahir ul Qamar M, Saleem S, Ashfaq UA, Bari A, Anwar F, Alqahtani S. Epitope‐based peptide vaccine design and target site depiction against Middle East Respiratory Syndrome Coronavirus: an immune‐informatics study. J Transl Med. 2019;17:362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ong E, Wong MU, Huffman A, He Y. COVID‐19 coronavirus vaccine design using reverse vaccinology and machine learning. bioRxiv. 2020. 10.1101/2020.03.20.000141. Accessed June 1, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Amanat F, Krammer F. SARS‐CoV‐2 vaccines: status report. Immunity. 2020;52(4):583‐589. 10.1016/j.immuni.2020.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ou J, Zhou Z, Dai R, Zhang J, Lan W, Zhao S, Wu J, Seto D, Cui L, Zhang G, Zhang Q. Emergence of RBD mutations in circulating SARS‐CoV‐2 strains enhancing the structural stability and human ACE2 receptor affinity of the spike protein. bioRxiv. 2020. 10.1101/2020.03.15.991844. Accessed June 1, 2020. [DOI] [Google Scholar]
- 32. Hussain M, Jabeen N, Raza F, et al. Structural variations in human ACE2 may influence its binding with SARS‐CoV‐2 spike protein. J Med Virol. 2020. 10.1002/jmv.25832 [DOI] [PMC free article] [PubMed] [Google Scholar]