

Abstract
Microbial pathogens often target the host mitogen-activated protein kinase (MAPK) network to suppress host immune responses. We previously identified a bacterial type III secretion system effector, termed NleD, a metalloprotease that inactivates MAPKs by specifically cleaving their activation loop. Here, we show that NleDs form a growing family of virulence factors harbored by human and plant pathogens as well as insect symbionts. These NleDs disable specifically Jun N-terminal kinases (JNKs) and p38s that are required for host immune response, whereas extracellular signal-regulated kinase (ERK), which is essential for host cell viability, remains intact. We investigated the mechanism that makes ERK resistant to NleD cleavage. Biochemical and structural analyses revealed that NleD exclusively targets activation loops with high conformational flexibility. Accordingly, NleD cleaved the flexible loops of JNK and p38 but not the rigid loop of ERK. Our findings elucidate a compelling mechanism of native substrate proteolysis that is promoted by entropy-driven specificity. We propose that such entropy-based selectivity is a general attribute of proteolytic enzymes.
Keywords: mitogen-activated protein kinase (MAPK), extracellular signal-regulated kinase (ERK), p38, Jun N-terminal kinase (JNK), NleD, metalloprotease, effector protein, type III secretion system (T3SS), bacterial pathogen, protein structure, crystallography, bacterial pathogenesis, structural biology, proteolysis
Mitogen-activated protein kinases (MAPKs) are serine/threonine kinases conserved in all eukaryotes (1–4). MAPKs are involved in fundamental cell processes, such as growth, survival, and response to stressors (2–4). The MAPK family comprises three main subfamilies, each including several isoforms. These subfamilies include the extracellular signal-regulated kinases (ERK1 and ERK2), the c-Jun N-terminal kinases (JNK1, JNK2, and JNK3), and the p38 kinases (p38α, p38β, p38γ, and p38δ). MAPKs are activated in response to a variety of stimuli. ERK1 and ERK2 are preferentially activated in response to growth factors and mitogenic stimuli (5), whereas JNK and p38 kinases are more responsive to stress-associated stimuli. The latter include response to proinflammatory cytokines, such as tumor necrosis factor α and interleukin-1β, whose production is typically associated with bacterial infection (6). All MAPKs share a similar structure, consisting of the N and C lobes, with the active site and the activation loop in a crevice between them. The activation loop contains a TXY motif that includes two adjacent phosphor-acceptors, threonine and tyrosine residues, separated by a third variable residue. The X in the TXY motif is used as a hallmark for the specific MAPK, i.e. proline for JNKs, glutamate for ERKs, and glycine for p38s (7–9). Phosphorylation of both threonine and tyrosine residues is essential for the maximal activity of the MAPKs. The activated MAPKs phosphorylate a substantial set of cellular and nuclear proteins, such as other kinases, tumor suppressors, and transcription and translation factors (10). The activation loop in nonphosphorylated p38 and JNK often exhibits high flexibility, whereas that of ERK typically displays a defined conformation (11–13).
Pathogenic bacteria often produce toxins that target the MAPK network to manipulate the host immune response (14–16). Among these pathogens is enteropathogenic Escherichia coli (EPEC), which employs its type III secretion system (T3SS) (1) to inject into infected host cells effector proteins that manipulate MAPKs. These effectors include NleE, which inhibits the activation of MAP3K7 (also called TAK1, for transforming growth factor beta-activated kinase 1) (17, 18) and NleD, which targets the JNK and p38 MAPKs. NleD is a Zn-dependent protease that rapidly inactivates JNK and p38 by cleaving them in their TXY motif between the X and Y residues (19). Notably, in contrast to JNK and p38, ERK remains unaffected by NleD (19). Thus, by employing NleD, EPEC inactivates p38 and JNK, which are required for the host immune response, without affecting the ERK-dependent host cell viability.
Here, we show that NleD belongs to a large family of proteins harbored by various bacterial species, including human and plant pathogens and insect symbionts, all of which discriminate between JNK/p38 and ERK. We revealed that NleD distinguishes between the different MAPKs, disabling only p38s and JNKs by a mechanism involving entropy-driven selectivity. Accordingly, it targets only flexible activation loops containing the TXY motif. Our results provide the first example of a proteolytic enzyme bearing entropy-driven selectivity toward its native substrate. We estimate, however, that a similar mechanism is a widespread strategy for enhancing substrate selectivity.
Results
NleDEPEC cleaves all p38s and JNKs but not ERKs
We have previously shown that NleD of EPEC (NleDEPEC) cleaves the activation loops of JNK2 and p38α at the TXY motif (TPY and TGY, respectively) between Pro/Gly and Tyr (19). On the other hand, NleD does not proteolyze ERK2 (19). To further examine NleD specificity, we assayed its activity toward the different isoforms of ERK, JNK, and p38 (i.e. JNK1, JNK2, p38α, p38β, p38γ, p38δ, ERK1, and ERK2) (20). For this purpose, we expressed an epitope-tagged form of each of the above MAPK isoforms in HEK293T cells and then infected them with WT EPEC or an nleD-deficient mutant. Western blot analysis of proteins extracted from the infected cells shows that NleDEPEC cleaved all JNKs and p38s but none of the ERKs (Fig. 1, A–C). To further verify these findings, we assayed the in vitro activity and specificity of purified NleDEPEC against purified MAPK substrates. Similarly, we found that JNK2 and p38α, but not ERK2, were cleaved by NleD (Fig. 1D), strengthening the premise that NleDEPEC is incapable of cleaving ERKs.
Figure 1.

Cleavage of MAPK isoforms by NleDEPEC. A–C, HEK293T cells, transiently expressing different epitope-tagged isoforms of JNK (A), p38 (B), and ERK (C), were infected with WT EPEC (WT) or the EPEC ΔnleD mutant, as indicated above the lanes. Proteins were extracted from the cells and subjected to Western blot analysis using anti-HA (HA-JNKs and HA-ERK2) or anti-His antibody (His-p38s and His-ERK1). D, Recombinant purified NleDEPEC was incubated with recombinant p38a, JNK2, or ERK2. Proteins were then resolved using SDS-PAGE and visualized by Coomassie staining. The cleavage of p38a results in a single product band, as the two fragments exhibit almost identical molecular masses of 20.8 and 20.4 kDa. JNK2, however, has a C-terminal extension region that is 49 residues larger than that of p38a; thus, its cleavage results in two distinct bands. Intact and fragmented MAPKs are indicated by blue and red arrowheads, respectively, and a green arrowhead indicates NleD. The incomplete cleavage of JNK and p38 proteins is likely due to the presence of misfolded subpopulations of the MAPKs.
Inability to cleave ERK is a conserved property of NleDs
A database search using BLAST (21) identified numerous NleD orthologues from different species that exhibit high sequence identity. These NleD proteins were aligned and analyzed for their phylogenetic relationships (22) (Fig. S1 and Fig. 2A). We then selected four NleDs for further analysis, including those of Salmonella enterica serovar Arizonae (NleDSEA), the insect symbiont Hamiltonella defensa (NleDHD), and the two NleD copies of Citrobacter rodentium (NleDCR1 and NleDCR2). These NleD orthologues were cloned in an expression vector, and their proteolytic activity of cleaving selected MAP kinases (JNK2, p38β, and ERK1) was assayed using coexpression in E. coli BL21. We found that similarly to NleDEPEC, all the assayed orthologues cleaved JNK2 and p38β, but none of them cleaved ERK1 (Fig. 2B). These results suggest that the inability to cleave ERK proteins is conserved throughout the NleD family. The question that arises is how NleD distinguishes ERK from the highly similar p38 and JNK.
Figure 2.
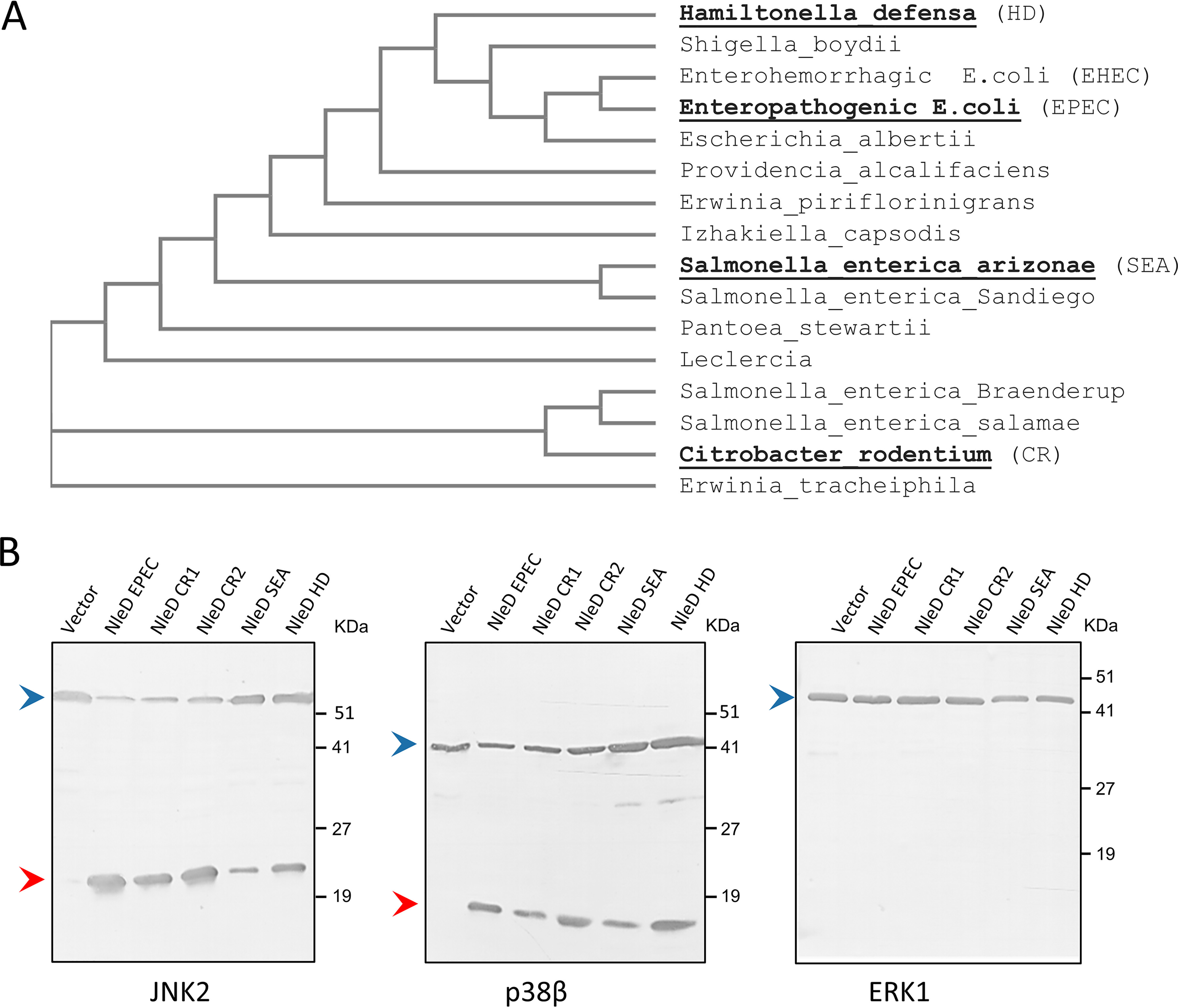
NleD orthologues cleave JNK and p38 but not ERK. A, An NleD orthologue phylogenetic tree was created with Clustal Omega. Proteins selected for further analysis are highlighted in boldface. B, NleD orthologues of C. rodentium (NleDCR1 and NleDCR2), S. enterica serovar Arizonae (NleDSEA), and H. defensa (NleDHD) were coexpressed in E. coli BL21 together with hexahistidine-tagged MAP kinases (i.e. p38β, JNK2, or ERK1). Proteins were then extracted from the bacteria, and MAPK cleavage was probed by Western blot analysis using the anti-6His antibody. Intact and fragmented MAPKs are indicated by blue and red arrowheads, respectively.
Cleavage of the ERK activation loop is restricted specifically in its intrinsic context
The MAPKs differ at the X residue of their TXY motif. In ERK, X is the negatively charged Glu, whereas in p38 and JNK, X is the neutral Gly or Pro residue, respectively (Fig. S2). To examine if the X residue plays a role in NleD selectivity, we generated a JNK2 mutant whose TPY motif was converted to TEY (P184E), as in ERK. The results showed that upon coexpression with NleD, the JNK2 P184E mutant was cleaved with efficiency similar to that of the WT JNK2 (Fig. 3A), indicating that the Glu in the ERK TEY motif is not the factor that impairs the NleD ability to cleave ERK. These results also suggest that the recognition of JNK2 by NleD is mediated by other determinants, possibly related to the activation loop.
Figure 3.

The impact of the X residue within the TXY motif and the activation loop context on NleD substrate specificity. A, JNK2 WT or JNK2-P184E mutant was coexpressed in E. coli BL21 with NleD (+) or vector control (−). Proteins were extracted from the expressing bacteria and subjected to Western blot analysis using an anti-JNK antibody. Intact and cleaved JNKs are indicated by blue and red arrowheads, respectively. B, Schematic view of native p38a, ERK2, and JNK2 and the activation loop swapped variants. C, Purified proteins variants were incubated with purified NleDEPEC, and, as negative controls, the MAPK variants and NleD were incubated separately. For cleavage analysis, proteins were subjected to SDS-PAGE and visualized by Coomassie staining. Intact and cleaved MAPKs are indicated by blue and red arrowheads, respectively. A green arrowhead indicates NleD protein.
Since the size and composition of the activation loops of the MAP kinases differ (Fig. S2), we also examined their role in imposing the inhibitory effect of ERK on NleD. To this end, we swapped the complete activation loops between the MAPKs and assayed NleD activity. We generated three variants, including ERK2, replacing the activation loops with that of JNK2 (ERKA-JNK) or that of p38α (ERKA-p38). Similarly, we constructed a p38α containing the activation loop of ERK2 (p38A-ERK) (Fig. 3B). We next tested the capability of NleD to cleave the purified MPAK variants (i.e. ERKA-JNK, ERKA-p38, and p38A-ERK). NleD cleaved all swapped variants, including p38A-ERK, containing the ERK activation loop in the context of p38 (Fig. 3C). These results indicate that the size and composition of the activation loops per se are not the prevailing factors that restrict ERK cleavage by NleD.
The search for MAPK regions accountable for NleD specificity
Since the ERK activation loop resists cleavage by NleD only within its native context, we assumed that other regions of ERK play a role in restricting NleD activity. We further predicted that these ERK regions differ from the corresponding JNK and p38 regions, which might even promote p38 and JNK cleavage. The first candidate region that we assayed was the common docking motif (CD), localized at the N-lobe distal from the active site (Fig. S2) (23–26). The CD is the primary recognition site of MAPKs for interactions with upstream activators, substrates, and phosphatases. To test whether the CD is required for targeting MAPKs by NleD, we mutated the CD of JNK2 and p38α and examined NleD activity. To this end, we constructed a mutant in the p38 CD (D316N) (27) and four mutants of the JNK CD, including three single-residue replacements (D326A, E329A, and E331A) and a triple mutant (28). The results show that all assayed JNK2 and p38α variants were efficiently cleaved by NleDEPEC (Fig. 4A), emphasizing that the CD region does not play a role in NleD specificity.
Figure 4.
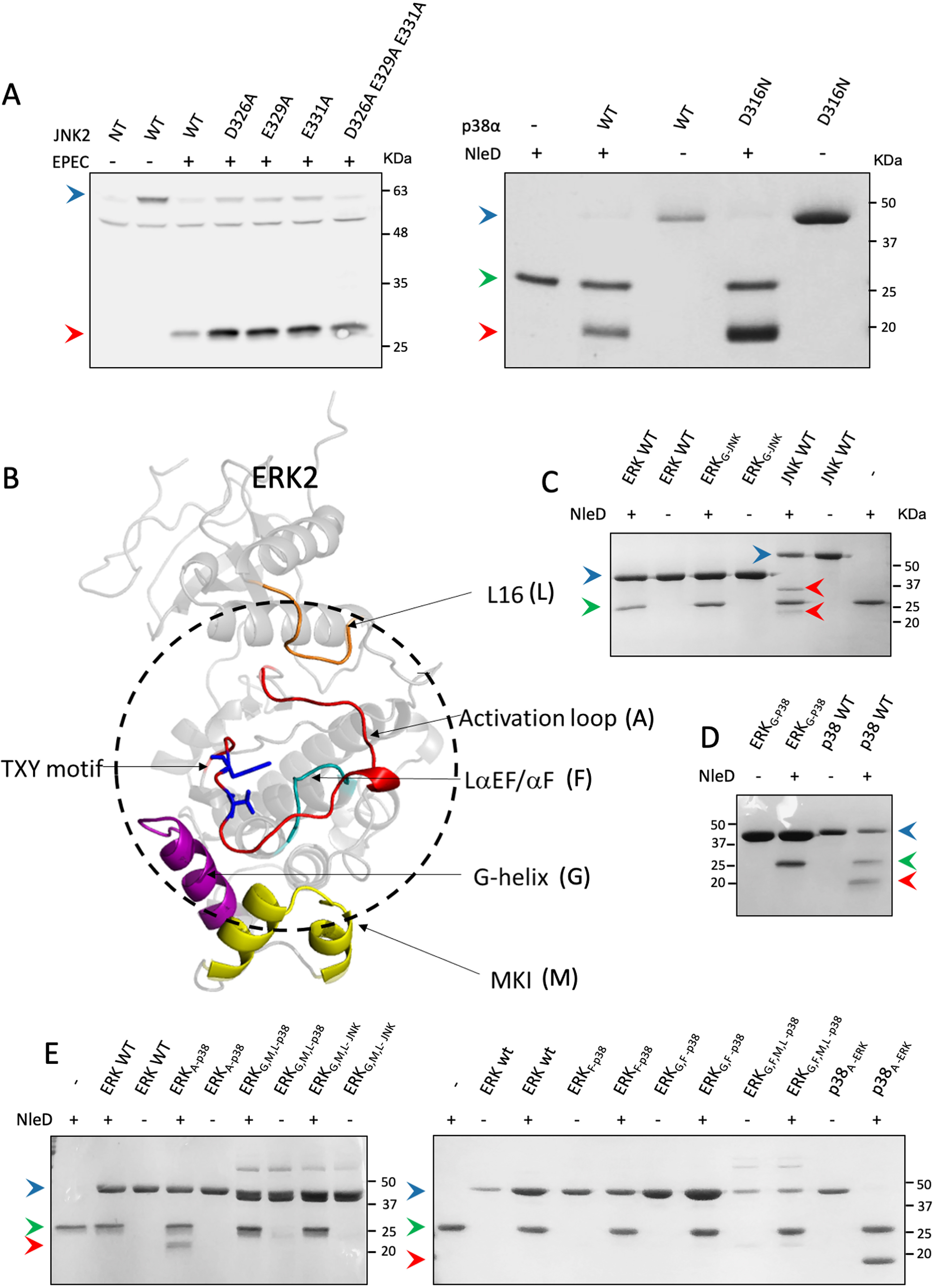
Region replacements between ERK and p38/JNK did not alter cleavage preferences of NleD. A, HEK293T cells were transfected, or not (NT), with plasmids expressing WT JNK2 carrying an HA tag (WT) or JNK2 mutated in critical CD domain residues, as indicated above the lanes (left). The cells were then infected with WT EPEC and subjected to Western blot analysis using an anti-HA antibody. (Right) In addition, WT p38a (WT) or p38 mutated in a critical CD domain residue (D316N) was purified and incubated in vitro with NleDEPEC. Samples were subjected to SDS-PAGE and visualized by Coomassie staining. B, Structure model of ERK2 (PDB entry 4S31). The activation loop is colored red, and threonine and tyrosine of the TXY motif are highlighted in blue. The dashed circle highlights the regions that are in close proximity to the activation loop. Regions that were replaced in the variants include MKI (M), colored yellow, G-helix (G), colored purple, L16 (L), colored orange, and LaEF/aF (F), colored cyan. C–E, Purified MAPK variants were incubated with or without purified NleDEPEC and then resolved by SDS-PAGE, followed by Coomassie staining. The variants used are indicated above the respective lanes and include WT ERK2 (ERKwt), WT JNK2 (JNKwt), WT p38α (p38wt), ERK2 with G-helix of JNK2 or p38α (ERKG-JNK and ERKG-p38, respectively), ERK2 with the p38α activation loop (ERKA-p38), p38α with the ERK2 activation loop (p38A-ERK), ERK2 with the LαEF/αF region of p38α (ERKF-p38), double mutant ERK2 with G-helix and LαEF/αF regions of p38α (ERKG,F-p38), triple mutant ERK2 with G-helix, MKI, and L16 regions of p38α (ERKG,M,l-p38), and quadruple mutant ERK2 with G-helix, LαEF/αF, MKI, and L16 regions of p38α (ERKG,F,M,l-p38). The presence (+) or absence (−) of NleD is indicated. Intact and fragmented MAPKs are indicated by blue and red arrowheads, respectively. A green arrowhead indicates NleD protein.
We then conducted a structure-based analysis to identify ERK sequences positioned in spatial proximity to the activation loop cleavage site and identified four such regions. These included short segments of the G-helix (G), the MAP kinase insert MKI (M), and the L16 (L) and αEF/αF (F) loops (Fig. 4B, Fig. S2). We subsequently generated several ERK2 replacement variants, or combinations of them, producing altogether nine variants, described in detail in the supporting information (Table S2). We next expressed and purified each of the variants and assayed for their proteolysis by NleD. The results clearly showed that none of these variants were cleaved by NleD (Fig. 4, C–E), indicating that the four regions neighboring the ERK activation loop (L16, LαEF/αF, G-helix, and MKI) are not accountable for NleD selectivity.
MAPK cleavage by NleD correlates with the flexibility of the activation loop
Analysis of the PDB database revealed that the activation loops of p38 and JNK are mostly unstructured (flexible), whereas those of ERK exhibit generally a defined structure (11–13). Thus, we investigated whether only flexible loops are recognized and proteolyzed by NleD. To this end, we determined the crystal structure of ERKA-p38 and p38A-ERK (Table 1, Fig. 5), both shown to be cleavable NleD substrates (Fig. 3C). We also solved the structure of the ERK variant containing the G helix of p38 (ERKG-p38), which was shown to be resistant to NleD proteolysis (Fig. 4E). In the latter variant, the ERK2 G-helix residues 229KHY231 were replaced by the respective 226TDH228 residues from p38α. Structural analysis showed that the three chimeric MAPKs consist of the canonical two-lobe topology, highly similar to the WT MAPKs (Fig. 5A). The structure of p38A-ERK showed that the activation loop originating from ERK did not have indicative electron density, exhibiting high flexibility (Fig. 5B). Similarly, the structure of ERKA-p38 showed that its activation loop, derived from p38, also exhibits high flexibility, similar to that of the p38A-ERK loop (Fig. 5B). In contrast, the loop of ERKG-p38 showed a defined and rigid conformation (Fig. 5). Taken together, our results showed that ERKA-p38 and p38A-ERK, which exhibit a disordered (flexible) activation loop, were cleaved by NleD, whereas ERKG-p38, which has a loop with defined conformation, remained intact (Fig. 3C, Fig. 5, and Table 2). The observed conformation and the rigidity of the activation loop in ERK2 and ERKG-p38 is not a result of crystal packing, as the majority of its interactions are intramolecular. The introduction of the loop of p38α into ERK2 (ERKA-p38) also results in the same crystal packing, yet the loop displays flexibility. Thus, we postulated that local conformational flexibility of the activation loop is a primary factor promoting JNK/p38 cleavage by NleD and that the rigidity of the ERK loop hinders its cleavage.
Table 1.
Data collection and refinement statistics
| Parameter | p38A-ERK | ERKA-p38 | ERΚG-p38 |
|---|---|---|---|
| PDB entry | 6QYX | 6RFO | 6RFP |
| ESRF beamline | ID23-2 | ID23-1 | ID23-1 |
| Wavelength (Å) | 0.873 | 0.972 | 0.972 |
| Space group | P212121 | P21 | P21 |
| Unit cell parameters (Å) | a = 68.86, b = 74.50, c = 75.28 | a = 48.00, b = 68.69, c = 60.23, β = 109.13° | a = 48.74, b = 68.81, c = 60.81, β = 110.14° |
| Resolution range (Å) (outer shell) | 52.95–1.66 (1.72–1.66) | 45.35–1.70 (1.73–1.70) | 45.76–1.742 (1.79–1.74) |
| No. of unique reflections | 46,102 (4,589) | 40,460 (2,160) | 38,438 (2,840) |
| Redundancy | 4.2 | 4.0 | 3.7 |
| Rsym(I)a | 0.068 (0.862) | 0.062 (0.716) | 0.047 (0.661) |
| Completeness | 99.1 (99.06) | 99.4 (98.8) | 99.4 (99.8) |
| I/σ | 9.8 (1.0) | 10.0 (1.0) | 16.10 (1.92) |
| CC (1/2) | 99.8 (39.6) | 99.8 (48.9) | 99.9 (77.8) |
| No. of protein atoms | 2,731 | 2,590 | 2,762 |
| No. of ligand atoms | 20, 15 | ||
| No. of solvent atoms | 204 | 173 | 227 |
| R factor | 18.5 | 15.6 | 14.6 |
| Rfreeb | 24.2 | 21.9 | 19.7 |
| Average B factor | |||
| Protein | 38.2 | 38.7 | 37.2 |
| Ligands (βOG, HEPES) | 39.6, 46,6 | ||
| Solvent | 44.2 | 44.2 | 46.8 |
| RMSD from ideality | |||
| Bond length | 0.017 | 0.013 | 0.016 |
| Bond angle | 1.80 | 1.89 | 1.97 |
| Ramachandran plot (Rampage) (%) | |||
| Favored | 96.4 | 96.4 | 97.8 |
| Generously allowed | 3.6 | 3.0 | 2.2 |
| Disallowed | 0.0 | 0.7 | 0.0 |
a Rsym(I)=Σ|I − |/ΣI.
b Test set consists of 5% for all data.
Figure 5.

Structure comparison between WT MAPKs and activation loop-swapped variants. Structural superposition of the ERK molecules resolved in this study, including overall structures (A) and view of the activation loop vicinity (B). The overall structures of ERKG-p38 (cyan and blue) and ERKA-p38 (magenta) remain similar without significant conformation changes other than in the activation loops. The activation loop (blue) in ERKG-p38 and the αEF/αF loop (yellow) maintain the conformation of the WT structure (not shown). The activation loop of ERKG-p38 is disordered, and its edges are indicated in panel B by red arrows. The αEF/αF loop of ERKG-p38, known to be conjugated with the contour of the activation loop, is also disordered (shown by a dotted red line in panel B).
Table 2.
Summary of NleD proteolysis results
| MAP kinase substrate | Result for NleDEPEC |
|
|---|---|---|
| In vitro | In vivo | |
| Wild type | ||
| ERK | No | No |
| p38 | Yes | Yes |
| JNK | Yes | Yes |
| Activation loop | ||
| JNK P184E | Yes | |
| ERKA-p38 | Yes | |
| ERKA-JNK | Yes | |
| p38A-ERK | Yes | |
| GST-JNK2169-192-GFP | Yes | |
| GST-JNK2176-192-GFP | Yes | |
| GST-JNK2180-192-GFP | Yes | |
| GST-JNK2176-188-GFP | Yes | |
| GST-JNK2180-186-GFP | Yes | |
| GST-JNK2182-188-GFP | Yes | |
| GST-ERKA-GFP | Yes | |
| G-helix | ||
| ERΚG-p38 | No | |
| ERKG-JNK | No | |
| MKI | ||
| ERKm-p38 | No | |
| ERKm-JNK | No | |
| G-helix + MKI + L16 | ||
| ERKG,M,l-p38 | No | No |
| ERKG,M,l-JNK | No | No |
| αEF/αF loop | ||
| ERΚF-p38 | No | |
| G-helix + αEF/αF loop | ||
| ERKG,F-p38 | No | |
| G-helix + MKI + L16 + αEF/αF loop | ||
| ERKG,F,M,l-p38 | No | |
NleD recognizes a specific sequence within the flexible activation loops
Given the above-described results, we predicted that inducing flexibility in the ERK activation loop should be sufficient to convert it into an NleD-sensitive state, leading to its proteolysis. To test this assumption, we cloned the ERK1 activation loop (residues C183 to W209) and the JNK2 loop (residues D169 to R192) between two nonrelated proteins, GST and GFP, that should not impose constraints on the loop conformation (Fig. 6). We then probed for cleavability by NleD and found that in this artificial context, NleD readily cleaved both the ERK1 and JNK2 activation loops (Fig. 6). These results strengthen the premise that the conformational flexibility of the ERK activation loop is sufficient for efficient cleavage by NleD.
Figure 6.
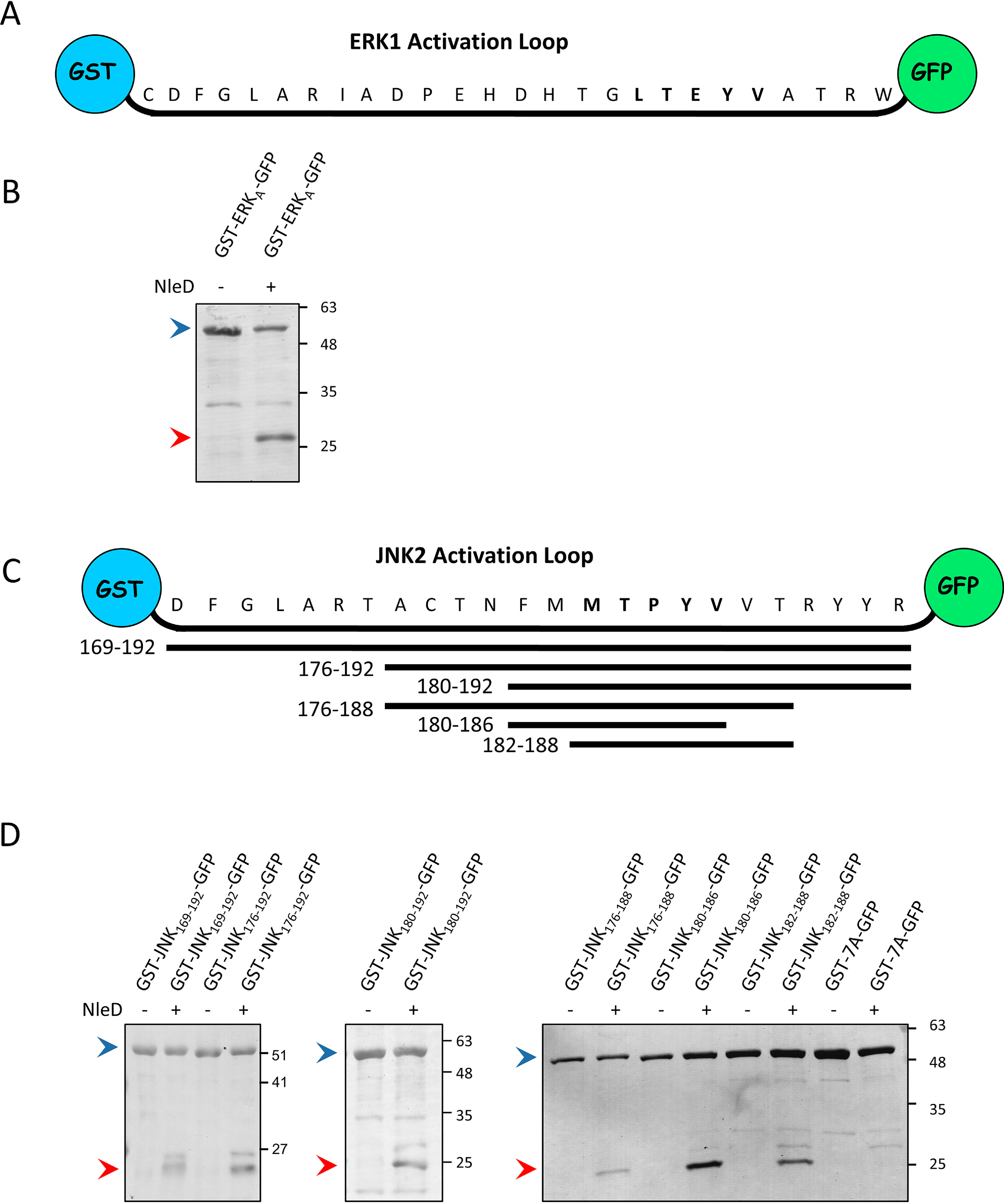
Activation loops of JNK2 and ERK1 are recognized and cleaved by NleD when inserted between GST and GFP. A, Schematic presentation of the ERK1 activation loop (ERKA) cloned between the GST and GFP proteins. B, The ERK1 activation loop flanked by GST and GFP (GST-ERKA-GFP) was coexpressed in E. coli BL21 with or without NleDEPEC. The extracted proteins were pulled down using GSH beads and subjected to SDS-PAGE, followed by Coomassie staining. Intact and fragmented GST-ALERK-GFP proteins are indicated by blue and red arrowheads, respectively. C, Schematic view of the JNK2 activation loop (residues 169 to 192) cloned between GST and GFP proteins. Below are shown the different fragments of the JNK2 activation loop and the corresponding coordinates, which were cloned between the GST and GFP proteins. D, GST-GFP bridged by different segments of the JNK2 activation loop shown in panel C or by seven alanine residues (7A) were coexpressed in E. coli BL21 with or without NleDEPEC. The extracted proteins were pulled down using GSH beads and subjected to SDS-PAGE, followed by Coomassie staining. The variants used and the presence or absence of NleD are indicated above the lanes. Intact and fragmented proteins are indicated by blue and red arrowheads, respectively.
The demonstration that the ERK1 and JNK2 activation loops are cleaved by NleD provided an approach to define further the sequence within the loop that is recognized by NleD. To address this issue, we constructed a set of plasmids expressing successively shorter segments of the JNK2 loop, all flanked by GST and GFP (Fig. 6C). The coexpression of these hybrid proteins and NleD in E. coli, followed by cleavage analysis, identified two short overlapping sequences, MTPYVVT and FMMTPYV, that were sufficient to promote proteolysis by NleD (Fig. 6D). Notably, a replacement of this sequence with seven sequential alanine residues abrogated the cleavage of the hybrid protein by NleD (Fig. 6D). Taken together, these results suggest that NleD recognizes and cleaves specific sequences that overlap the TXY motif and are located within flexible MAPK activation loops.
Discussion
A remarkable common feature of the NleD family of proteins is their ability to specifically target JNK and p38 MAPKs while leaving ERK intact. We examined the reason for ERK remaining entirely resistant to cleavage by NleD, even upon prolonged incubation at high concentrations of NleD. In contrast, the closely related JNK and p38 are specifically and rapidly cleaved. Our data, based on biochemical and structural analyses, suggest that NleD targets flexible MAPK activation loops. We show here that the activation loop of ERK exhibits a defined structure that restricts the accessibility of NleD to the TEY motif, leading to resistance to NleD. Most of the ERK2 structures deposited in the PDB (more than 80%) exhibit a defined conformation of the activation loop. In contrast, the loops of p38 and JNK alternate between several defined structures; some of them are selected to be cleaved by NleD. In agreement with our data, the examination of deposited (PDB) p38 and JNK structures showed that these MAPKs regularly display high loop flexibility (11–13).
Additionally, solution NMR studies of p38α further indicate that the activation loop exhibit flexibility in the nonphosphorylated form (29). Interestingly, we obtained preliminary results indicating that NelD cannot proteolyze the dually phosphory-lated forms of p38 and JNK. These findings further support our hypothesis, since conserved interactions of the phosphorylated Thr and Tyr impose activation loop rigidity, as observed in the available phospho-p38 X-ray structure (9). Thus, our data show that NleD evolved to take advantage of the differences in the activation loop flexibility–rigidity interplay to specifically target JNK and p38 and evade the cleavage of ERK. Further analysis revealed that NleD recognizes the short overlapping sequence MTPYVVT or FMMTPYV within the loop, which is highly similar in all MAPKs. As a result, NleD recognizes these minimal sequences embedded within flexible MAPK activation loops, cleaving them between the X and Tyr residues, resulting in irreversible MAPK inactivation.
The MAPK network is targeted by a large number of effectors and toxins produced by pathogenic bacteria. The anthrax lethal factor toxin is a metalloprotease that targets MAP2K (30). YopJ, AvrA, and VopA represent a family of T3SS effector proteins that acetylate MAP2K and MAP3K proteins (31–35). NleE and OspZ are T3SS effectors that prevent the activation of the MAP3K TAK1 (17, 18). OspF, SpvC, and HopA1 form a family of effector proteins that, like NleD, inactivate MAPKs (1, 36). These effectors target the phosphorylated TXY motif (pT-X-pY) and irreversibly remove the phosphate from the Thr residue by phosphothreonine lyase activity (25). The physiological substrate of OspF and SpvC proteins is mainly p38 and, to a lesser extent, ERK, while JNK is the least preferred substrate (25). NleD and OspF employ different strategies to recognize the TXY or pT-X-pY motif of MAPKs. OspF and SpvC contain an N-terminal D motif, which interacts with the CD of p38 to position the catalytic site correctly (25). In contrast, our data show that NleD lacks a D motif and that the MAPK CD is not required for cleavage. Instead, NleD recognizes a short sequence containing the TXY motif in the context of a flexible loop.
The combination of virulence factors carried by a given pathogen is shaped through evolution to fit its specific needs and relationships with the eukaryotic host. Here, we show that NleD represents a growing family of metalloproteases consisting either of T3SS effector proteins or a domain within a larger exotoxin. While the biochemical function of NleD in JNK and p38 inactivation is well defined, elucidating its biological function is more complex. The biological function is most likely dependent on the specific pathogen, the associated repertoire of T3SS effectors carried by this pathogen, and the infected organism and tissue. For example, the consequences of the activity of identical NleDs within human or plant cells are expected to vary. In mammalian pathogens, NleD is likely to downregulate stress and inflammatory responses associated with p38 and JNK while maintaining host cell viability, which requires functional ERK. An additional key factor that impacts the biological role of NleD is the composition of other effectors and exotoxins that are codelivered with it. For example, most clinical isolates of EPEC and enterohemorrhagic E. coli inject NleD into the host cell together with additional MAPK-modulating effectors, such as NleE, which targets the MAP3K TAK1 (18). Some isolates also carry a gene encoding an AvrA-like protein (37), which likely acetylates MAP2Ks. Harboring distinct combinations of anti-MAPK effectors could provide the pathogen with enhanced flexibility to accurately manipulate the MAPK signaling. It is conceivable that, by horizontal gene transfer combined with fine-tuning through mutational evolution, a given pathogen collects and shapes a set of T3SS effectors to manipulate the MAPK pathway as well as other host processes in a way that is compatible with its virulence strategy.
In conclusion, here we demonstrate that NleD represents a growing family of T3SS metalloprotease effectors that target JNK and p38 but not ERK. We show that NleD specifically evolved to take advantage of differences in entropy between activation loops of MAPKs, avoiding ERK cleavage while efficiently disabling JNK and p38. This concept of protease activity driven by peptidic substrate entropy, shown in this study for the first time in a native system, can explain how NleD discriminates between very similar substrates. Notably, recent studies utilizing synthetic systems, including single-molecule force-dependent kinetics and molecular dynamics of conformational transitions (38, 39), support the notion that substrate flexibility has a profound impact on the rate of proteolysis. Thus, we anticipate that substrate flexibility-driven proteolysis is not unique to NleD and possibly represents a widespread mechanism utilized by proteases to enable and promote their corresponding selectivity.
Experimental procedures
Bacterial strains, plasmids, and primers
Bacterial strains, plasmids, and primers used in this study are listed in Tables S1–S3 in the supporting information, respectively. Cloning was performed using either restriction enzymes or the Gibson assembly method. Inserts integrated into plasmids were generated using either a Pfu-Ultra II fusion Hot Start DNA polymerase (Agilent) or KOD Hot Start DNA polymerase kit (Novagen). PCR was performed on the human p38αwt cDNA, NleDEPEC subcloned into pET-28a, and rat ERK2wt cDNA subcloned into pET-15b. All PCR reactions were conducted downstream and in-frame with the hexahistidine coding sequence. 5′-Phosphorylated primers were used for producing the mutants, and DNA sequencing was used to verify the constructs. To search for NleD orthologues, the NleDEPEC protein sequence was compared using PSI-BLAST at NCBI. The resulting NleD orthologues were aligned and clustered into a phylogenetic tree using Clustal Omega. The DNA of the NleD orthologues was obtained by PCR using suitable primers and template genomic DNA extracted from the corresponding bacterial strains or intact aphids colonized by symbiotic H. defensa.
Transfection, infection, and bacterial cell assays
For transfection, HEK293T cells were plated onto 35-mm dishes (0.5 × 106 cells per well) and cultured in DMEM with 10% FCS and penicillin-streptomycin antibiotics for 18 h. Cells were transfected with 6 μg DNA per well, using Turbofact (Thermo Scientific, R0531) according to the manufacturer's protocol. Cells were analyzed 24 h posttransfection.
For infection, the epithelial cells grown in 35-mm dishes to ∼80% confluence were washed twice with antibiotic-free DMEM and infected at 37 °C for 3 h with 1 ml DMEM supplemented with 10 μl EPEC taken from an overnight standing culture in LB medium. After infection, cells were washed and lysed using RIPA buffer (Sigma, R0278) and centrifuged, and the supernatant was collected.
For the bacterial cell assay, E. coli BL21 was transformed with the chosen plasmids. Bacteria from an overnight culture were diluted 1:100 and incubated at 30 °C to an optical density at 600 nm of 0.4–0.6, and then 0.5 mm isopropyl-β-d-thiogalactopyranoside was added. The proteins were expressed at 20 °C overnight and then extracted.
Protein extracts were separated by SDS-PAGE. Membranes were either stained with Coomassie brilliant blue staining (Bio-Rad 6160436) or probed by Western blot analysis using α-JNK (BD Pharmingen, 554285), α-HA (Sigma, A2095), or α-hexahistidine (GE Healthcare, 27471001).
Protein expression and purification for in vitro assays and crystallization
All proteins were expressed in E. coli strains as N-terminal hexahistidine-tagged proteins. JNK2, p38α WT, and the D316N variant were produced as previously described (40, 41). For the other proteins, cells were grown at 37 °C until they reached an optical density at 600 nm of 0.4–0.6, and protein expression was induced by the addition of 0.2 mm isopropyl-β-d-thiogalactopyranoside. After induction, NleDEPEC and p38A-ERK were transferred to 21 °C and ERK2 variants to 17 °C overnight. Cells were collected by centrifugation, and the pellets were stored at −80 °C. All frozen pellets were gently thawed on ice and suspended in appropriate lysis buffer (described below), supplemented with protease inhibitor mixture (Sigma, p8849), 0.2 mg/ml lysozyme, 10 μg DNase I (Sigma), and 10 mm MgCl2. After mechanical disruption of the cells using a Microfluidizer device at 20,000 psi (LV1; Microfluidics Corporation, Newton, MA), the lysates were centrifuged at 16,000 × g for 30 min at 4 °C. The supernatant was filtered (0.45 μm; Millex®-HV) and loaded onto a Ni Sepharose FF 4-ml column (Amersham Biosciences) using the ÄKTATM explorer system (GE Healthcare Life Sciences). The eluted proteins were dialyzed overnight as described below and then loaded on a Resource 15Q 23-ml (Amersham Biosciences) anion exchange column. The eluted proteins were concentrated using Vivaspin (5000 MWCO PES; Sartorius), divided into aliquots, and stored at −80 °C.
The NleDEPEC protein was expressed in a one-liter culture of E. coli BL21(DE3) in 2× YT medium supplemented with NPS buffer contains 25 mm (NH4)2SO4, 50 mm KH2PO4, 50 mm Na2HPO4 (pH 6.75), 1% glucose, and antibiotics. Cells were lysed before purification with buffer containing 50 mm Tris-HCl, pH 8, 0.3 m NaCl, 10 mm imidazole, 10% glycerol, and 10 mm β-mercaptoethanol. The same buffer was used for equilibration of the Ni Sepharose FF 4-ml column (Amersham Biosciences), and the proteins were eluted in 300 mm imidazole and then dialyzed overnight against 50 mm glycine-NaOH, pH 9.5, 0.05 m NaCl, 10% glycerol, and 10 mm β-mercaptoethanol to reduce concentrations of imidazole and NaCl. A Resource 15Q 23-ml (Amersham Biosciences) anion exchange column was equilibrated in dialysis buffer. NleDEPEC was eluted in two peaks at ∼140 mm and 170 mm NaCl.
ERK2 variants (Table S2) were transformed into BL21(DE3) containing λ-ppase in 2 L of LB supplemented with NPS buffer contains 25 mm (NH4)2SO4, 50 mm KH2PO4, 50 mm Na2HPO4 (pH 6.75), 1% glucose, and antibiotics. Cells were lysed before purification with buffer containing 50 mm Tris-HCl, pH 8, 0.3 m NaCl, and 10 mm imidazole. The same buffer was used for equilibration of the Ni Sepharose FF 4-ml column (Amersham Biosciences) and the proteins eluted in 250 mm imidazole. The eluted proteins were then dialyzed twice for 1 h with 50 mm Tris-HCl, pH 8, 10 mm NaCl, 5% glycerol, and 1 mm EDTA and then overnight against 50 mm Tris-HCl, pH 8, 10 mm NaCl, 5% glycerol, and 1 mm DTT. A Resource 15Q 23-ml (Amersham Biosciences) anion exchange column was equilibrated in dialysis buffer, and proteins were eluted in two peaks at ∼60–70 mm and 100 mm NaCl. In this regard, the presence of these two peaks is a regular occurrence in ERK2 purification protocols.p38A-ERK was transformed into E. coli (strain Rosetta) in 0.5 L LB supplemented with antibiotics. Cells were lysed before purification with buffer containing 50 mm Tris-HCl, pH 7.4, 0.5 m NaCl, and 10 mm imidazole. The same buffer was used for equilibration of the Ni Sepharose FF 4-ml column (Amersham Biosciences), and the proteins were eluted in 231 mm imidazole. The eluted protein was then diluted 1:5.5 in 50 mm Tris-HCl, pH 7.4, 5% glycerol, 10 mm MgCl2, and 1 mm DTT and loaded on a Resource 15Q 23-ml (Amersham Biosciences) anion exchange column that was equilibrated in dilution buffer containing 0.1 m NaCl. The protein was eluted in one peak at ∼120 mm NaCl.
In vitro NleD proteolysis assays of MAP kinases
In vitro assays were conducted to determine NleD activity. Freshly thawed, purified proteins were mixed with NleDEPEC at a ratio of 1:2.76 NleD:MAPK. The reaction buffer contained 50 mm NaCl, 50 mm Tris-HCl, pH 7.4, and 2 mm CaCl2 (19). Reactions were carried out for 1.5 h at 37 °C and stopped by the addition of the chelating agent EDTA. Digestion products were visualized using SDS-PAGE and Coomassie staining.
Crystallization, data collection, structure solution, and refinement
The MAPK variants ERKA-p38 and ERαGp38 were crystallized using the vapor diffusion sitting-drop method at 20 °C with 0.6 ml reservoir solution containing 1.8 m ammonium sulfate, 0.1 m Bis-Tris, pH 6.8 or 6.5, 2% (w/v) PEG 550 at a 1:1 ratio. The 3-μl drops contained equal amounts of protein solution, at a concentration of 3.75–4.5 mg/ml, and reservoir solution. After approximately four hours, streak seeding was performed (42), using WT ERK2wt as the source crystals. Crystals of the mutants appeared within 3 days and reached their final size within 7–10 days. The p38A-ERK mutant was crystallized by sitting-drop vapor diffusion at 4 °C with 0.6 ml reservoir solution containing 0.1 m HEPES, pH 7.5, 0.2 m KF, 13–17% (w/v) PEG 3350, and 25 mm β-d-octyl glucoside (βOG). The 3-μl drops contained equal amounts of protein solution, at a concentration of 11 mg/ml, and reservoir solution. After approximately one hour, streak seeding (42) from p38wt crystals was performed, and crystals appeared within three hours. Before data collection, the crystals were suspended in a cryopreserving solution containing the respective reservoir solution described above and 20% ethylene glycol for ERK2 mutants, or 20% glycerol for p38α mutants, and immediately flash cooled in liquid N2.
Crystallographic data for the p38α and ERK2 variants were collected at the European Synchrotron Radiation Facility (ESRF), Grenoble, France, at 100K, using the Oxford Cryosystem Cryostream cooling device. For p38A-ERK, data were collected in beamline ID23-2 on a Pilatus 2M pixel detector to the highest resolution of 1.66 Ǻ (Table 1). Crystals belonged to the P212121 orthorhombic space group with one p38A-ERK molecule in the asymmetric unit, similar to WT p38 (43, 44). The data for the ERKA-p38 and ERαGp38 variants were obtained in beamline ID23-1 at 100K on a 6M Pilatus pixel detector to resolutions of 1.7 Å and 1.74 Å, respectively (Table 1). Crystals belonged to the monoclinic P21 space group with one kinase molecule in the asymmetric unit. All data were processed and scaled using XDS via EDNA (45, 46).
The structure of p38A-ERK was solved by molecular replacement using Molrep (47, 48), implemented in the CCP4 suite (49, 50), using a search model of p38wt (PDB entry 1p38) after removing all solvent molecules. It was initially refined by a restrained refinement protocol using Refmac (48, 50). After several iterative cycles of refinement and model building with Coot (51), which was also used to identify solvent molecules, the model converged to reasonable parameters. The refined structure contained residues 4–172 and 189–358 with 1 βOG and 1 HEPES and 204 water molecules having an R value of 18.5% and Rfree of 24.2% (Table 1). The structure of ERK variants was solved by molecular replacement using Molrep (48), implemented in the CCP4 suite (49, 50), using a search model of ERK2wt (PDB entry 4S31) after removing all solvent molecules. It was initially refined, and the model building was achieved via a protocol similar to that for p38A-ERK, converging to the final values shown in Table 1.
Data availability
The crystallographic data and coordinates were deposited in the RCSB-PDB with the accession codes 6QYX, 6RFO, and 6RFP for p38A-ERK, ERKA-p38, and ERKG-p38, respectively.
Supplementary Material
Acknowledgments
We thank D. Engleberg, E. Shaulian, Y. Gottlieb (The Hebrew University of Jerusalem), and G. Frankel (Imperial College, London), for providing DNA, plasmids, and bacterial strains and O. Furman and S. Ben Yehuda for critically reading the manuscript. We thank the staff of ESRF for their help in maintaining such a superb facility. I. R. is an Etta Rosensohn Professor in Bacteriology. O. L. is a Mathilda Marks-Kennedy Chair in Biochemistry.
This article contains supporting information.
Author contributions—L. G.-A., M. E.-W., N. W., and O. L. data curation; L. G.-A., N. W., I. R., and O. L. formal analysis; L. G.-A., M. E.-W., and N. W. investigation; L. G.-A., M. E.-W., I. R., and O. L. methodology; M. E.-W., I. R., and O. L. writing-original draft; I. R. and O. L. conceptualization; I. R. and O. L. resources; I. R. and O. L. supervision; I. R. and O. L. funding acquisition; I. R. and O. L. validation; O. L. project administration; O. L. writing-review and editing.
Funding and additional information—Work in the I. R. and O. L. laboratories was supported by grants from the Israel Science Foundation, funded by the Israel Academy of Science and Humanities, grants 617/15 and 1422/16, respectively, and in the I. R. laboratory also by a grant from the European Research Council (810186).
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- MAPK
- mitogen-activated protein kinase
- JNK
- Jun N-terminal kinase
- ERK
- extracellular signal-regulated kinase
- T3SS
- type III secretion system
- βOG
- β-d-octyl glucoside.
References
- 1. Arbibe L., Kim D. W., Batsche E., Pedron T., Mateescu B., Muchardt C., Parsot C., and Sansonetti P. J. (2007) An injected bacterial effector targets chromatin access for transcription factor NF-kappaB to alter transcription of host genes involved in immune responses. Nat. Immunol. 8, 47–56 10.1038/ni1423 [DOI] [PubMed] [Google Scholar]
- 2. Arthur J. S., and Ley S. C. (2013) Mitogen-activated protein kinases in innate immunity. Nat. Rev. Immunol. 13, 679–692 10.1038/nri3495 [DOI] [PubMed] [Google Scholar]
- 3. Peti W., and Page R. (2013) Molecular basis of MAP kinase regulation. Protein Sci. 22, 1698–1710 10.1002/pro.2374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zarubin T., and Han J. (2005) Activation and signaling of the p38 MAP kinase pathway. Cell Res. 15, 11–18 10.1038/sj.cr.7290257 [DOI] [PubMed] [Google Scholar]
- 5. Stofega M. R., Yu C. L., Wu J., and Jove R. (1997) Activation of extracellular signal-regulated kinase (ERK) by mitogenic stimuli is repressed in v-Src-transformed cells. Cell Growth Differ. 8, 113–119 [PubMed] [Google Scholar]
- 6. Pearson G., Robinson F., Beers Gibson T., Xu B. E., Karandikar M., Berman K., and Cobb M. H. (2001) Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr. Rev. 22, 153–183 10.1210/edrv.22.2.0428 [DOI] [PubMed] [Google Scholar]
- 7. Canagarajah B. J., Khokhlatchev A., Cobb M. H., and Goldsmith E. J. (1997) Activation mechanism of the MAP kinase ERK2 by dual phosphorylation. Cell 90, 859–869 10.1016/S0092-8674(00)80351-7 [DOI] [PubMed] [Google Scholar]
- 8. Lee J. C., Kumar S., Griswold D. E., Underwood D. C., Votta B. J., and Adams J. L. (2000) Inhibition of p38 MAP kinase as a therapeutic strategy. Immunopharmacology 47, 185–201 10.1016/S0162-3109(00)00206-X [DOI] [PubMed] [Google Scholar]
- 9. Zhang Y. Y., Wu J. W., and Wang Z. X. (2011) Mitogen-activated protein kinase (MAPK) phosphatase 3-mediated cross-talk between MAPKs ERK2 and p38alpha. J. Biol. Chem. 286, 16150–16162 10.1074/jbc.M110.203786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Plotnikov A., Zehorai E., Procaccia S., and Seger R. (2011) The MAPK cascades: signaling components, nuclear roles and mechanisms of nuclear translocation. Biochim. Biophys. Acta 1813, 1619–1633 10.1016/j.bbamcr.2010.12.012 [DOI] [PubMed] [Google Scholar]
- 11. Diskin R., Lebendiker M., Engelberg D., and Livnah O. (2007) Structures of p38alpha active mutants reveal conformational changes in L16 loop that induce autophosphorylation and activation. J. Mol. Biol. 365, 66–76 10.1016/j.jmb.2006.08.043 [DOI] [PubMed] [Google Scholar]
- 12. Zhang F., Strand A., Robbins D., Cobb M. H., and Goldsmith E. J. (1994) Atomic structure of the MAP kinase ERK2 at 2.3 A resolution. Nature 367, 704–711 10.1038/367704a0 [DOI] [PubMed] [Google Scholar]
- 13. Shaw D., Wang S. M., Villasenor A. G., Tsing S., Walter D., Browner M. F., Barnett J., and Kuglstatter A. (2008) The crystal structure of JNK2 reveals conformational flexibility in the MAP kinase insert and indicates its involvement in the regulation of catalytic activity. J. Mol. Biol. 383, 885–893 10.1016/j.jmb.2008.08.086 [DOI] [PubMed] [Google Scholar]
- 14. Pearson J. S., and Hartland E. L. (2014) The inflammatory response during enterohemorrhagic Escherichia coli infection. Microbiol. Spectr. 2, EHEC-0012-2013. 10.1128/microbiolspec.EHEC-0012-2013 [DOI] [PubMed] [Google Scholar]
- 15. Reddick L. E., and Alto N. M. (2014) Bacteria fighting back: how pathogens target and subvert the host innate immune system. Mol. Cell 54, 321–328 10.1016/j.molcel.2014.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Salomon D., and Orth K. (2013) Lost after translation: post-translational modifications by bacterial type III effectors. Curr. Opin. Microbiol. 16, 213–220 10.1016/j.mib.2013.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nadler C., Baruch K., Kobi S., Mills E., Haviv G., Farago M., Alkalay I., Bartfeld S., Meyer T. F., Ben-Neriah Y., and Rosenshine I. (2010) The type III secretion effector NleE inhibits NF-kappaB activation. PLoS Pathog. 6, e1000743 10.1371/journal.ppat.1000743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang L., Ding X., Cui J., Xu H., Chen J., Gong Y. N., Hu L., Zhou Y., Ge J., Lu Q., Liu L., Chen S., and Shao F. (2012) Cysteine methylation disrupts ubiquitin-chain sensing in NF-kappaB activation. Nature 481, 204–208 10.1038/nature10690 [DOI] [PubMed] [Google Scholar]
- 19. Baruch K., Gur-Arie L., Nadler C., Koby S., Yerushalmi G., Ben-Neriah Y., Yogev O., Shaulian E., Guttman C., Zarivach R., and Rosenshine I. (2011) Metalloprotease type III effectors that specifically cleave JNK and NF-kappaB. EMBO J. 30, 221–231 10.1038/emboj.2010.297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cargnello M., and Roux P. P. (2011) Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 75, 50–83 10.1128/MMBR.00031-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Altschul S. F., Madden T. L., Schaffer A. A., Zhang J., Zhang Z., Miller W., and Lipman D. J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 10.1093/nar/25.17.3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sievers F., Wilm A., Dineen D., Gibson T. J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Soding J., Thompson J. D., and Higgins D. G. (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539 10.1038/msb.2011.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tanoue T., Adachi M., Moriguchi T., and Nishida E. (2000) A conserved docking motif in MAP kinases common to substrates, activators and regulators. Nat. Cell Biol. 2, 110–116 10.1038/35000065 [DOI] [PubMed] [Google Scholar]
- 24. Chang C. I., Xu B. E., Akella R., Cobb M. H., and Goldsmith E. J. (2002) Crystal structures of MAP kinase p38 complexed to the docking sites on its nuclear substrate MEF2A and activator MKK3b. Mol. Cell 9, 1241–1249 10.1016/S1097-2765(02)00525-7 [DOI] [PubMed] [Google Scholar]
- 25. Zhu Y., Li H., Long C., Hu L., Xu H., Liu L., Chen S., Wang D. C., and Shao F. (2007) Structural insights into the enzymatic mechanism of the pathogenic MAPK phosphothreonine lyase. Mol. Cell 28, 899–913 10.1016/j.molcel.2007.11.011 [DOI] [PubMed] [Google Scholar]
- 26. English J., Pearson G., Wilsbacher J., Swantek J., Karandikar M., Xu S., and Cobb M. H. (1999) New insights into the control of MAP kinase pathways. Exp. Cell. Res. 253, 255–270 10.1006/excr.1999.4687 [DOI] [PubMed] [Google Scholar]
- 27. Hutter D., Chen P., Barnes J., and Liu Y. (2000) Catalytic activation of mitogen-activated protein (MAP) kinase phosphatase-1 by binding to p38 MAP kinase: critical role of the p38 C-terminal domain in its negative regulation. Biochem. J. 352, 155–163 10.1042/0264-6021:3520155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mooney L. M., and Whitmarsh A. J. (2004) Docking interactions in the c-Jun N-terminal kinase pathway. J. Biol. Chem. 279, 11843–11852 10.1074/jbc.M311841200 [DOI] [PubMed] [Google Scholar]
- 29. Kumar G. S., Clarkson M. W., Kunze M. B. A., Granata D., Wand A. J., Lindorff-Larsen K., Page R., and Peti W. (2018) Dynamic activation and regulation of the mitogen-activated protein kinase p38. Proc. Natl. Acad. Sci. U S A 115, 4655–4660 10.1073/pnas.1721441115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Duesbery N. S., Webb C. P., Leppla S. H., Gordon V. M., Klimpel K. R., Copeland T. D., Ahn N. G., Oskarsson M. K., Fukasawa K., Paull K. D., and Vande Woude G. F. (1998) Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science 280, 734–737 10.1126/science.280.5364.734 [DOI] [PubMed] [Google Scholar]
- 31. Mukherjee S., Keitany G., Li Y., Wang Y., Ball H. L., Goldsmith E. J., and Orth K. (2006) Yersinia YopJ acetylates and inhibits kinase activation by blocking phosphorylation. Science 312, 1211–1214 10.1126/science.1126867 [DOI] [PubMed] [Google Scholar]
- 32. Mittal R., Peak-Chew S. Y., and McMahon H. T. (2006) Acetylation of MEK2 and I kappa B kinase (IKK) activation loop residues by YopJ inhibits signaling. Proc. Natl. Acad. Sci. U S A 103, 18574–18579 10.1073/pnas.0608995103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jones R. M., Wu H., Wentworth C., Luo L., Collier-Hyams L., and Neish A. S. (2008) Salmonella AvrA coordinates suppression of host immune and apoptotic defenses via JNK pathway blockade. Cell Host Microbe 3, 233–244 10.1016/j.chom.2008.02.016 [DOI] [PubMed] [Google Scholar]
- 34. Trosky J. E., Li Y., Mukherjee S., Keitany G., Ball H., and Orth K. (2007) VopA inhibits ATP binding by acetylating the catalytic loop of MAPK kinases. J. Biol. Chem. 282, 34299–34305 10.1074/jbc.M706970200 [DOI] [PubMed] [Google Scholar]
- 35. Paquette N., Conlon J., Sweet C., Rus F., Wilson L., Pereira A., Rosadini C. V., Goutagny N., Weber A. N., Lane W. S., Shaffer S. A., Maniatis S., Fitzgerald K. A., Stuart L., and Silverman N. (2012) Serine/threonine acetylation of TGFbeta-activated kinase (TAK1) by Yersinia pestis YopJ inhibits innate immune signaling. Proc. Natl. Acad. Sci. U S A 109, 12710–12715 10.1073/pnas.1008203109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kramer R. W., Slagowski N. L., Eze N. A., Giddings K. S., Morrison M. F., Siggers K. A., Starnbach M. N., and Lesser C. F. (2007) Yeast functional genomic screens lead to identification of a role for a bacterial effector in innate immunity regulation. PLoS Pathog. 3, e21 10.1371/journal.ppat.0030021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Creuzburg K., Heeren S., Lis C. M., Kranz M., Hensel M., and Schmidt H. (2011) Genetic background and mobility of variants of the gene nleA in attaching and effacing Escherichia coli. Appl. Environ. Microbiol. 77, 8705–8713 10.1128/AEM.06492-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Guerin M. E., Stirnemann G., and Giganti D. (2018) Conformational entropy of a single peptide controlled under force governs protease recognition and catalysis. Proc. Natl. Acad. Sci. U S A 115, 11525–11530 10.1073/pnas.1803872115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kayode O., Wang R., Pendlebury D. F., Cohen I., Henin R. D., Hockla A., Soares A. S., Papo N., Caulfield T. R., and Radisky E. S. (2016) An acrobatic substrate metamorphosis reveals a requirement for substrate conformational dynamics in trypsin proteolysis. J. Biol. Chem. 291, 26304–26319 10.1074/jbc.M116.758417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tzarum N., Komornik N., Ben Chetrit D., Engelberg D., and Livnah O. (2013) DEF pocket in p38alpha facilitates substrate selectivity and mediates autophosphorylation. J. Biol. Chem. 288, 19537–19547 10.1074/jbc.M113.464511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tesker M., Selamat S. E., Beenstock J., Hayouka R., Livnah O., and Engelberg D. (2016) Tighter alphaC-helix-alphaL16-helix interactions seem to make p38alpha less prone to activation by autophosphorylation than Hog1. Biosci. Rep. 36, e00324. 10.1042/BSR20160020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wilson I. A., Rini J. M., Fremont D. H., Fieser G. G., and Stura E. A. (1991) X-ray crystallographic analysis of free and antigen-complexed Fab fragments to investigate structural basis of immune recognition. Methods Enzymol. 203, 153–176 10.1016/0076-6879(91)03009-6 [DOI] [PubMed] [Google Scholar]
- 43. Diskin R., Engelberg D., and Livnah O. (2008) A novel lipid binding site formed by the MAP kinase insert in p38 alpha. J. Mol. Biol. 375, 70–79 10.1016/j.jmb.2007.09.002 [DOI] [PubMed] [Google Scholar]
- 44. Tzarum N., Eisenberg-Domovich Y., Gills J. J., Dennis P. A., and Livnah O. (2012) Lipid molecules induce p38alpha activation via a novel molecular switch. J. Mol. Biol. 424, 339–353 10.1016/j.jmb.2012.10.007 [DOI] [PubMed] [Google Scholar]
- 45. Kabsch W. (2010) Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. D Biol. Crystallogr. 66, 133–144 10.1107/S0108767310004551, 10.1107/S0907444909047374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Incardona M. F., Bourenkov G. P., Levik K., Pieritz R. A., Popov A. N., and Svensson O. (2009) EDNA: a framework for plugin-based applications applied to X-ray experiment online data analysis. J Synchrotron Radiat. 16, 872–879 10.1107/S0909049509036681 [DOI] [PubMed] [Google Scholar]
- 47. Murshudov G. N., Vagin A. A., and Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 10.1107/S0907444996012255 [DOI] [PubMed] [Google Scholar]
- 48. Vagin A. A., Steiner R. A., Lebedev A. A., Potterton L., McNicholas S., Long F., and Murshudov G. N. (2004) REFMAC5 dictionary: organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr. D Biol. Crystallogr. 60, 2184–2195 10.1107/S0907444904023510 [DOI] [PubMed] [Google Scholar]
- 49. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 10.1107/S0907444910045749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The crystallographic data and coordinates were deposited in the RCSB-PDB with the accession codes 6QYX, 6RFO, and 6RFP for p38A-ERK, ERKA-p38, and ERKG-p38, respectively.