

Abstract
G-protein-coupled receptors (GPCRs)—the largest family of cell-surface membrane proteins—mediate the intracellular signal transduction of many external ligands. Thus, GPCRs have become important drug targets. X-ray crystal structures of GPCRs are very useful for structure-based drug design (SBDD). Herein, we produced a new antibody (SRP2070) targeting the thermostabilised apocytochrome b562 from Escherichia coli M7W/H102I/R106L (BRIL). We found that a fragment of this antibody (SRP2070Fab) facilitated the crystallisation of the BRIL-tagged, ligand bound GPCRs, 5HT1B and AT2R. Furthermore, the electron densities of the ligands were resolved, suggesting that SPR2070Fab is versatile and adaptable for GPCR SBDD. We anticipate that this new tool will significantly accelerate structure determination of other GPCRs and the design of small molecular drugs targeting them.
Subject terms: Pharmaceutics, X-ray crystallography
Introduction
GPCRs are the largest transmembrane receptor family in humans, consisting of approximately 800 genes. They are involved in several physiological functions, such as immune response, blood pressure regulation, nerve system, vision system, and olfactory system1. Hence, drugs that target GPCRs have been developed to treat multiple human diseases, such as central nervous system disorders, inflammatory diseases, metabolic imbalances, cardiac diseases, and cancer2,3.
All GPCRs have conserved seven-pass transmembrane helices (TM 1–7) that are connected by three extracellular loops (ECL 1–3) and three intracellular loops (ICL 1–3). The third intracellular loop (ICL3) is very flexible and interacts with G proteins, which are necessary for intracellular signal transduction. Depending on the conditions within the cells, GPCRs adopt multiple conformational states, with the two major states being the active and inactive states4.
During small molecule drug development, the three-dimensional structure of the target protein with the candidate compound is very useful for refining the compound to improve its binding capacity. This method—structure-based drug design (SBDD)—has been widely used since the 1990s4–6. Using SBDD, rational drug design is possible, which greatly improves the success rates of drug development. However, structure determination of GPCRs is very difficult due to poor protein expression in native tissues or heterologous expression systems, low protein stability, and the presence of several receptor conformational states4, 7. In the last ten years, several technologies on protein expression, purification, crystallisation, and X-ray diffraction data collection have been developed8–13. For example, fusion proteins, such as T4 lysozyme14 and BRIL15, have enabled researchers to purify target GPCR proteins and determine their structures11. Our knowledge of GPCR structures has gradually increased, as evidenced by the more than 40 unique GPCR structures that have been determined to date13.
To improve the stability of GPCRs for crystallisation, ICLs are often replaced with soluble proteins, such as T4 lysozyme, BRIL, or rubredoxin11,15. In cases where the target GPCR cannot be successfully crystallised even after its fusion to soluble proteins, additional approaches are needed to enhance crystallisability, and antibodies that specifically bind to target GPCRs are often used for this purpose. Although this approach is useful to expand the soluble regions of the target for crystal packing11, antibodies must be screened for each target GPCR to identify those with high binding affinity, which is very difficult and laborious. Another common approach to increase the thermal stabilities of GPCRs is to mutate the transmembrane helices3,11. In this method, the mutation sites are originally specified based on our extensive knowledge of the active and inactive forms of the adenosine A2a receptor. However, screening mutation sites within the target GPCR is still necessary to determine those that stabilise the active or inactive state before the construct can be used for crystallisation experiments. As this approach is also very strenuous and time consuming, there is an urgent need for optimisation methods that are more efficient and can be widely applied to various GPCR targets for crystallography.
Here, we demonstrate that antibodies against fusion proteins can enhance the crystallisation of GPCRs with ligands in active or inactive conformational states. Based on the results presented herein, this approach may be widely applicable for various GPCRs, which would vastly streamline drug development efforts.
Results
Sample preparation and crystallisation
The constructs, 5HT1B-BRIL/ergotamine (ERG) and AT2R-BRIL/[Sar1, Ile8]-Angiotensin II (s-Ang II), were complexed with SRP2070Fab (the Fab fragment of SRP2070) for crystallography experiments. All crystallisation trials were performed by the lipidic cubic phase (LCP) method16. For 5HT1B-BRIL/ERG/SRP2070Fab, diffracting crystals grew within 1 week in 400 mM KSCN, 100 mM NaOAc pH 5.5, and 30% PEG 400. For AT2R-BRIL/s-Ang II/SRP2070Fab, diffracting crystals grew within 1 week in 50 mM KOAc, 100 mM MES-NaOH pH 6.5, 26–36% PEG 300, and 100 μM s-Ang II.
Structure determination of 5HT1B-BRIL/ERG/SRP2070Fab and AT2R-BRIL/s-Ang II/SRP2070Fab
The structures of both complexes were solved by molecular replacement and then refined. Following refinement, the Rwork/Rfree was 22.2%/28.0% for 5HT1B-BRIL/ERG/SRP2070 and 22.6%/27.8% for AT2R-BRIL/s-Ang II/SRP2070Fab (Table 1). Figure 1 shows the overall structures of both complexes. SRP2070Fab perpendicularly bound to the BRIL helices. We also observed that helices III and IV of BRIL served as epitopes for SRP2070Fab. In particular, helix III was recognised by both CDR-L3 and CDR-H3, suggesting that this region is a dominant epitope (Fig. 2). These findings indicate that SRP2070 recognises the tertiary structure of BRIL, which may be an important feature of SRP2070Fab as a crystallisation enhancer.
Table 1.
Data collection and refinement statistics.
| AT2R-SRP2070Fab complex (PDB:7C6A) | 5HT1B-SRP2070Fab complex (PDB:7C61) | |
|---|---|---|
| Data collection | ||
| Space group | P21 | C2 |
| Cell dimensions | ||
| a, b, c (Å) | 40.2, 131.0, 113.8 | 248.8, 65.7, 79.7 |
| α, β, γ (°) | 90, 97.1, 90 | 90, 98.8, 90 |
| Resolution (Å) | 42.8–3.4 (3.7–3.4)a | 48.5–3.0 (3.2–3.00) |
| Rpim (%) | 14.0 (72.0) | 9.0 (61.7) |
| I/σ (I) | 5.1 (1.1) | 9.2 (1.6) |
| CC1/2 | 0.99 (0.49) | 0.99 (0.62) |
| Completeness (%) | 100.0 (100.0) | 100.0 (100.0) |
| Redundancy | 18.8 (14.5) | 29.5 (28.2) |
| Refinement | ||
| Resolution (Å) | 42.8–3.4 (3.49–3.4) | 48.5–3.0 (3.1–3.0) |
| No. reflections | 15,370 (1,120) | 24,568 (1,796) |
| Rwork/Rfree (%) | 22.2/28.0 (37.7/33.2) | 21.3/26.2 (32.8/39.7) |
| No. atoms | ||
| Receptor | 2,252 | 2,105 |
| BRIL | 712 | 719 |
| SRP2070Fab fragment | 3,307 | 3,304 |
| Ligand | 69 | 43 |
| B factors | ||
| Receptor | 104.4 | 158.8 |
| BRIL | 110.6 | 76.9 |
| SRP2070Fab fragment | 103.5 | 68.3 |
| Ligand | 104.8 | 161.5 |
| R.M.S. deviations | ||
| Bond lengths (Å) | 0.008 | 0.011 |
| Bond angles (°) | 1.23 | 1.50 |
| Ramachandran plot | ||
| Favoured (%) | 94.5 | 95.1 |
| Allowed (%) | 5.4 | 4.8 |
| Disallowed (%) | 0.1 | 0.1 |
Data were collected from 144 and 118 crystals of AT2R-SRP2070Fab and 5HT1B-SRP2070Fab complexes, respectively.
aValues in parentheses are for the highest-resolution shell.
Figure 1.

Overall structure of 5HT1B-BRIL/ERG/SRP2070Fab and AT2R-BRIL/s-Ang II/SRP2070Fab. (A) Overall structure of 5HT1B-BRIL/ERG/SRP2070Fab. ERG is shown as sticks. Each molecule is coloured as follows: 5HT1B (gray), BRIL (light brown), SRP2070Fab (yellow). (B) Overall structure of AT2R-BRIL/s-Ang II/SRP2070Fab. s-Ang II is shown as sticks. Each molecule is coloured as follows: AT2R (purple), BRIL (light brown), SRP2070Fab (yellow).
Figure 2.
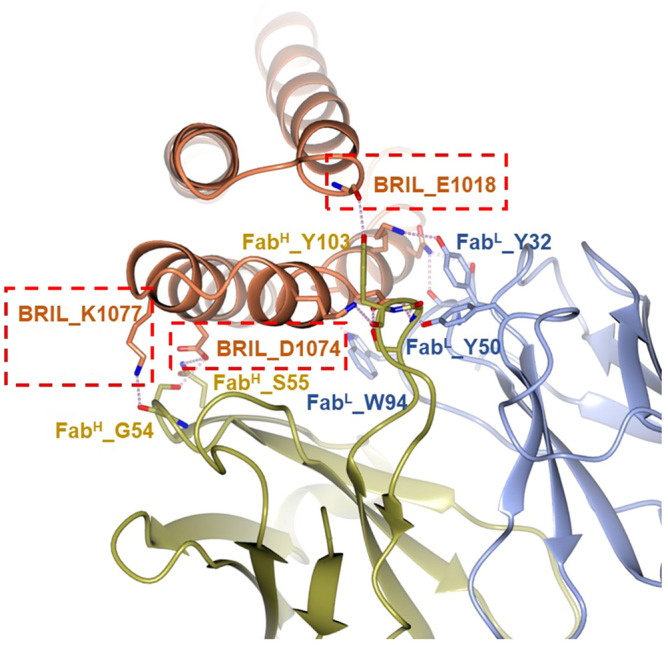
The binding interface between BRIL and SRP2070Fab. Interface-forming residues are coloured as follows: BRIL (brown), SRP2070Fab light chain (dark yellow), and SRP2070Fab heavy chain (light blue). Boxes drawn with red dashed lines indicate residues that are critical for BRIL binding.
To assess the effect of SRP2070Fab on crystallisation, the crystal packing interactions were investigated. As shown in Fig. 3, crystals of 5HT1B-BRIL/ERG/SRP2070Fab and AT2R-BRIL/s-Ang II/SRP2070Fab were formed by alternative stacking of the aqueous layers composed of SRP2070Fab and BRIL and the lipophilic layers composed of the receptor region. Crystal packing contacts were observed in the aqueous layers between SRP2070Fab molecules and also within the receptor region in the lipophilic layers. The crystal packing calculations for 5HT1B-BRIL/ERG/SRP2070Fab and AT2R-BRIL/s-Ang II/SRP2070Fab crystals are shown in Supplementary Table 1S and Supplementary Fig. 4S. In 5HT1B-BRIL/ERG/SRP2070Fab crystal, the accessible surface areas (ASA) buried due to crystal packing were 2,206.9 Å2 (5HT1B), 713.4 Å2 (BRIL), and 2,225.2 Å2 (SRP2070Fab). The 5HT1B-BRIL structure formed a head-to-tail dimer, which is often observed in GPCR crystals similar to the previously reported SRP2070Fab-free 5HT1B-BRIL/ERG structures (PDB ID: 4IAR) (Supplementary Fig. 5S). When the crystal packing contribution resulting from the head-to-tail dimerisation was excluded, it was clear that SRP2070Fab was the most dominant crystallisation promoter. These were also observed for AT2R-BRIL/s-Ang II/SRP2070Fab. The buried ASAs were 1,329.7 Å2 (AT2R), 510.1 Å2 (BRIL), and 2,293.8 Å2 (SRP2070Fab), suggesting that the crystal contacts among SRP2070Fab are the most dominant factor for the crystal formation.
Figure 3.

Crystal packing of 5HT1B-BRIL/ERG/SRP2070Fab and AT2R-BRIL/s-Ang II/SRP2070Fab. (A) The crystal packing of 5HT1B-BRIL/ERG/SRP2070Fab is shown from two directions. Each molecule is coloured as follows: 5HT1B (light purple), BRIL (light brown), SRP2070Fab (green). (B) The crystal packing of AT2R-BRIL/s-Ang II/SRP2070Fab is shown from two directions. Each molecule is coloured as follows: AT2R (purple), BRIL (light brown), SRP2070Fab (green).
Unambiguous electron densities corresponding to the ligands, ERG for 5HT1B and s-Ang II peptide for AT2R, were also observed within the orthosteric binding pockets of the receptors (Fig. 4A,B).
Figure 4.

Electron density maps of the ligand and structural comparison of the ligand binding site (A) The ligand omitted mFo–DFc polder electron density (light blue mesh) of ergotamine (ERG) contoured at 3.0 σ. (B) The ligand omitted mFo–DFc polder electron density (light blue mesh) of s-Ang II contoured at 3.0 σ. (C) Comparison of ligand binding cavity of 5HT1B/ERG/SRP2070Fab (left) and 5HT1B/ERG (PDB ID: 4IAR) (right). ERG is shown as sticks. (D) Comparison of ligand bindin cavity of AT2R/s-Ang II/SRP2070Fab (left) and AT2R/s-Ang II (PDB ID: 5XJM) (right). s-Ang II is shown as sticks.
Structure comparison
To confirm whether the structures of GPCR portion are affected by SRP2070 binding to BRIL, the structures of 5HT1B-BRIL/ERG/SRP2070Fab and AT2R-BRIL/s-Ang II/SRP2070Fab were compared to the previously reported structures of 5HT1B-BRIL/ERG (PDB ID: 4IAR)17 and AT2R-BRIL/s-Ang II in complex with an anti-AT2R antibody (PDB ID: 5XJM)18. Superimpositions of these structures showed that SRP2070Fab bound to BRIL had very little influence on the GPCR structures; i.e., the root mean square deviations (RMSDs) within the transmembrane regions of the GPCRs were 0.55 Å for 5HT1B-BRIL/ERG and 0.67 Å for AT2R-BRIL/s-Ang II between equivalent Cα atoms. Furthermore, no significant differences were found around the ligand binding sites in both AT2R-BRIL/s-Ang II and 5HT1B-BRIL/ERG as a result of SRP2070Fab binding (Fig. 4C,D). The three activation motifs—called microswitches—PIF19, NPxxY20, and DRY20, were also compared. It was confirmed that the three motifs showed very similar conformations in both comparisons (Supplementary Fig. 1S). Thus, the binding of SRP2070Fab to ICL BRIL fused GPCR has no significant effect on microswitch conformations. And also, it was clearly demonstrated that the activation motifs of AT2R-BRIL/s-Ang II in complex with an anti-AT2R antibody show an active state in the previous report18. Therefore, it is thought that AT2R-BRIL/s-Ang II/SRP2070Fab is also in an active form even though BRIL is fused to ICL3. The example of the structure that an agonist binds to the ICL3 BRIL fused GPCR has been already reported. The crystal structure of an active-state human angiotensin II (AngII) type I receptor (AT1R) bound to an AngII analog with partial agonist activity was determined even if BRIL was fused to ICL321. Hence, it is possible that the structure of AT2R-BRIL/s-AngII/SRP2070Fab shows an active form. It means that the SRP2070Fab is applicable to active state GPCR structure determination. On the other hand, it is very difficult to state clearly whether the 5HT1B-BRIL/ERG/SRP2070Fab and 5HT1B-BRIL/ERG show an active or an inactive form. Although the orientation of PIF motif was coincide with that of 5HT2B-BRIL structure in complex with ERG (PDB ID: 5TUD)22 whose structure was clearly demonstrated as an active form by comparing with the structure of β2 adrenergic receptor (β2AR)-Gs complex (PDB ID: 3SN6)22, the NPxxY and DRY motifs were different from 5HT2B-BRIL/ERG (Supplementary Fig. 2SA), which means 5HT1B-BRIL/ERG/SRP2070Fab structure does not show an active form. Furthermore, the three motifs of 5HT1B-BRIL/ERG/SRP2070Fab structure were not coincide with those of β2AR bound to the partial inverse agonist carazolol (PDB ID: 2RH1)23 that was known as an inactive form (Supplementary Fig. 2SB), which suggests that 5HT1B-BRIL/ERG/SRP2070Fab structure is not a complete inactive form. However, the orientation of TM6 helixes of 5HT1B-BRIL/ERG/SRP2070Fab showed more similar to that of carazolol bound β2AR than that of 5HT2B-BRIL/ERG (Supplementary Fig. 3S).
Based on these comparisons, the 5HT1B-BRIL/ERG/SRP2070Fab structure adopts an intermediate active state similar to that of the reported SRP2070Fab-free 5HT1B-BRIL/ERG structure. This is especially apparent based on the orientation of this intracellular region of TM6, which adopts a conformation observed in the inactive structure rather than active 5HT2B structure. Outward displacement of intracellular region of TM6 is known to be a structural hallmark of the inactive to active state transition, which results in formation of the cavity that recruits G proteins13. Thus, the orientation of fused-BRIL between TM5 and TM6 corresponding to the position of ICL3 is directly affected by the active or inactive state of the GPCR.
Discussion
In this study, we demonstrate that specific binders, such as antibodies, against soluble fusion partners of GPCRs can facilitate GPCR crystallisation. We discovered a new anti-BRIL antibody (SRP2070Fab), which promoted crystallisation of ICL BRIL-fused GPCRs. Three ICL BRIL-fused GPCR structures were solved in complex with SRP2070Fab. Two of them, 5HT1B-BRIL with ERG and AT2R-BRIL with s-Ang II, are described in this study; the other, ChemR23-BRIL apo form whose structure has not been reported yet, will be reported in a future manuscript. These results suggest that SRP2070Fab is a versatile tool for promoting the crystallisation and structure determination of various ICL BRIL-fused GPCRs. Importantly, this method of using ICL BRIL-fused proteins in complex with SRP2070Fab could also be used to solve the structures of previously unsolved GPCRs.
Based on our results, SRP2070Fab appears to promote crystallisation by affecting crystal packing. For 5HT1B-BRIL/ERG/SRP2070Fab, the overlapping layers of SRP2070Fab formed strong crystal packing by interacting at multiple sites (Fig. 3A). This was also observed in the crystal structure of AT2R-BRIL/s-Ang II SRP2070Fab (Fig. 3B). The crystal contacts between SRP2070Fab molecules were the most dominant contacts in both structures, suggesting that the range of the crystallisation conditions for ICL BRIL-fused GPCRs with SRP2070Fab is likely narrow. Thus, we predict that this system will increase the success rate of crystallisation and reduce the sample volume for crystallisation trials. Although we think the SRP2070Fab could be used for N- or C-terminal BRIL fused GPCR, further studies will be needed to demonstrate that.
Co-crystallisation with SRP2070Fab not only improves the success rate of crystallisation, but it is also advantageous for the structure determination. The phasing power of molecular replacement generally depends on the molecular mass of known search models (BRIL/SRP2070Fab in this study) relative to that of the protein of interest. Thus, the phasing power increases as a result of forming a complex with SRP2070Fab (~ 50 kDa) compared to performing phasing with BRIL (~ 12 kDa) alone. For both 5HT1B-BRIL/ERG/SRP2070Fab and AT2R-BRIL/s-Ang II/SRP2070Fab, phasing by molecular replacement with BRIL/SRP2070Fab resulted in electron densities that were interpretable even in the GPCR portion.
To use the SRP2070Fab for structure-based drug design (SBDD), it is very important to confirm that the SRP2070Fab does not adversely affect the electron density of the ligand. As demonstrated in a previous study, antibodies recognising ICLs are not suitable for SBDD, because the electron density of the ligand tends to be unclear24. BRIL is often inserted in place of ICL3 to stabilise GPCRs11, and thus SRP2070Fab also binds to the intracellular portion. To determine whether SRP2070Fab is available for SBDD, the B-factor and the ligand density map of structures in this study were compared with the previously reported structures, 5HT1B-BRIL/ERG (4IAR) and AT2R-BRIL/s-Ang II in complex with anti AT2R antibody (5XJM) (Fig. 4). For 5HT1B-BRIL/ERG/SRP2070Fab, although the B-factor was higher than that observed in 5HT1B-BRIL/ERG (4IAR) (Fig. 5A), the ligand electron density in the OMIT map25,26 was clearly recognised and was similar to that of 5HT1B-BRIL/ERG (4IAR) (Fig. 4A). Regarding AT2R-BRIL/s-Ang II/SRP2070Fab, the B-factors showed a little bit higher in the GPCR part (Fig. 5B), and the ligand electron density in the OMIT map was as clear as that of AT2R-BRIL/s-Ang II in complex with anti AT2R antibody (5XJM) (Fig. 4B). Thus, the ligand electron density maps between the structures with SRP2070Fab (intra-cellular recognition) and the antibody recognising extra-cellular domain (5XJM) were in good agreement (Fig. 5C). The B-factor of the GPCR parts of our structures are higher than the previously reported structures (Figs. 5A,B). It is because the interaction between SRP2070Fabs has the important role in the crystal packing and the interaction between GPCRs is relatively weak as shown in Fig. 3. However, we think this property is necessary to the versatility of SRP2070Fab. In short, although the SRP2070Fab do not give effects on GPCR structures, it can improve the crystal packing.
Figure 5.

B-factor analysis and comparison of the ligand electron density maps. (A) B-factor analysis for 5HT1B/ERG/SRP2070Fab (left) and 5HT1B/ERG (PDB ID: 4IAR) (right). Both the overall structure and the area around ligand are shown. (B) B-factor analysis for AT2R-BRIL/s-Ang II/SRP2070Fab (left) and AT2R/s-Ang II (PDB ID: 5XJM) (right). Both the overall structure and the area around ligand are shown. (C) The ligand electron density map of AT2R-BRIL/s-Ang II/SRP2070Fab (left) and AT2R/s-AngII (PDB ID: 5XJM) (right). The omitted mFo–DFc electron density map contoured at 3.0 σ (Polder map).
Based on these results, SRP2070Fab does not appear to affect ligand binding, indicating that SRP2070Fab can be used for SBDD. Considering the versatility of SRP2070Fab, we anticipate that this antibody will be beneficial for performing SBDD applications. Because SRP2070Fab recognises only BRIL, it is thought that it has no effect on the GPCR; this would suggest that BRIL may absorb any structure fluctuations caused by binding of SRP2070Fab. While 5HT1B/ERG and BRIL were linearly aligned through TM5 and TM6 and SRP2070Fab was vertically bound to BRIL, AT2R/s-Ang II and BRIL were unaligned by ~ 30°, and SRP2070Fab was vertically bound to BRIL (Fig. 1). This result suggests that there is some structural flexibility between GPCR and BRIL when SRP2070Fab functions as a crystallisation enhancer. Because of this property and the fact that SRP2070Fab is applicable to structure solution of 5HT1B in intermediate active state, which is rather similar to inactive state structure, as well as active AT2R, it is reasonable to hypothesize that SRP2070Fab can be used to study both active and inactive states. This is another important feature of SRP2070 as is applicable to both of agonist and antagonist drug developments.
In recent years, the resolution of cryo-electron microscopy has improved, and several structures of GPCRs have been determined by this technique27. We anticipate that SRP2070Fab could also be used for cryo-EM analyses. By attaching SRP2070Fab to BRIL-fused GPCRs, the molecular weight can be increased and the SRP2070Fab shape may be useful as a marker for 2D image classification and averaging.
To our knowledge, the use of an antibody like SRP2070Fab to promote crystallisation has not been previously reported. This method may facilitate the structural study of many types of GPCRs. Considering the results described herein, SRP2070Fab is a promising structural biology tool for studying GPCRs and could be useful for developing drugs that target GPCRs.
Methods
Ethics statement
Mice experiments in this research were conducted in accordance with the Fundamental Guidelines for Proper Conduct of Animal Experiment and Related Activities in Academic Research Institutions under the jurisdiction of the Ministry of Education, Culture, Sports, Science and Technology of Japan. The protocols conducted in this study were reviewed and approved by Kyoto University institutional review board.
Complete amino acid sequences and constructs for crystallisation
The sequence of 5HT1B residues 33-390 (UniProt accession: P28222) with BRIL insertion (underlined)15 is provided in Table 2. The additional N-terminal residue retained after cleavage with tobacco etch virus (TEV) is italicised.
Table 2.
The amino acids sequences.
| 5HT1B-BRIL | GCSAKDYIYQDSISLPWKVLLVMLLALITLATTLSNAFVIATVYRTRKLHTPANYLIASLAVTDLLVSILVMPISTMYTVTGRWTLGQVVCDFWLSSDITCCTASIWHLCVIALDRYWAITDAVEYSAKRTPKRAAVMIALVWVFSISISLPPFFWRQAKAEEEVSECVVNTDHILYTVYSTVGAFYFPTLLLIALYGRIYVEARSRIADLEDNWETLNDNLKVIEKADNAAQVKDALTKMRAAALDAQKATPPKLEDKSPDSPEMKDFRHGFDILVGQIDDALKLANEGKVKEAQAAAEQLKTTRNAYIQKYLAARERKATKTLGIILGAFIVCWLPFFIISLVMPICKDACWFHLAIFDFFTWLGYLNSLINPIIYTMSNEDFKQAFHKLIRFKCTS |
| AT2R-BRIL | GCSQKPSDKHLDAIPILYYIIFVIGFLVNIVVVTLFCCQKGPKKVSSIYIFNLAVADLLVLATLPLWATYYSYRYDWLFGPVMCKVFGSFLTLNMFASIWFITCMSVDRYQSVIYPFLSQRRNPWQASYIVPLVWCMACLSSLPTFYFRDVRTIEYLGVNACIMAFPPEKYAQWSAGIALMKNILGFIIPLIFIATCYFGIRKHLLKTNADLEDNWETLNDNLKVIEKADNAAQVKDALTKMRAAALDAQKATPPKLEDKSPDSPEMKDFRHGFDILVGQIDDALKLANEGKVKEAQAAAEQLKTTRNAYIQKYLKNRITRDQVLKMAAAVVLAFIICWLPFHVLTFLDALAWMGVINSCEVIAVIDLALPFAILLGFTNSCVNPFLYCFVGNRFQQKLRSVFRVPITWLQGKRESENLYFQ |
| The variable region of the SRP2070 light chain | DIVLTQSPATLSVTPGDRVSLSCRASQSVSNYLHWYQQKSHESPRLLIKYASQSISGIPSRFSGSGSGTDFTLSINSVETEDFGMYFCQQSNSWPLTFGAGTKLELR |
| The variable region of the SRP2070 heavy chain | QIQLQQSGPELVKPGASVKISCKASGYTFTDFYINWMKQRPGQGLEWIGWIFPGSGNTHYNEKFKGKATLIVDTSSSTAFMQLNSLTSEDSAVYFCTRPVSYYYDFDYWGQGTTLTVSS |
The sequence of AT2R residues 35-346 (UniProt accession: P50052) with the BRIL insertion15 (underlined) is provided in Table 2. The mutations, L93V and F133W, were added to improve stabilisation. The additional N- and C-terminal residues retained after cleavage with tobacco etch virus (TEV) cleavage are italicised.
The subclass of the SRP2070 was a mouse IgG2a. The sequence of the variable region of the light chain is provided in Table 2; CDR-L1, CDR-L2, and CDR-L3 are underlined.
The sequence of the variable region of the heavy chain is provided in Table 2; CDR-H1, CDR-H2, and CDR-H3 are underlined.
Codon-optimised cDNA encoding H. sapiens AT2R-BRIL and 5HT1B-BRIL was cloned into pFastBac1 (Thermo Fisher Scientific, Waltham, MA, USA). For AT2R-BRIL, the sequences encoding haemagglutinin (HA) and FLAG tags followed by a TEV cleavage site were added to the N-terminus, and a TEV cleavage site followed by a His8 tag were added to the C-terminus. For 5HT1B-BRIL, haemagglutinin (HA), FLAG, and His8 tags followed by a TEV cleavage site were added to the N-terminus.
Culture and membrane preparation
AT2R-BRIL was expressed in Spodoptera frugiperda (Sf9) cells using the baculovirus-based expression system (cell density = 2.0–4.0 × 106 cells/mL; MOI = 0.5; expression time = 60 h). The culture was harvested by centrifugation (7,000 × g, 10 min, 4 °C), and cell pellets were flash frozen at − 80 °C. Frozen cells were thawed and suspended in the hypotonic buffer (10 mM HEPES pH 7.5, 20 mM KCl, 10 mM MgCl2, 1 × protein inhibitor cocktail tablet). The suspension was centrifuged, and the pellet was resuspended in high osmotic buffer [10 mM HEPES pH 7.5, 10 mM MgCl2, 20 mM KCl, 1 M NaCl, 1 × protease inhibitor cocktail (Sigma Aldrich, Saint Louis, MO, USA)]. The cells were lysed using a dounce homogeniser, and the lysate was centrifuged (100,000 × g, 30 min, 4 °C). The pellet was resuspended in high osmotic buffer containing 40% glycerol and stored at − 80 °C. Plasma membranes expressing 5HT1B-BRIL were prepared using the same method as for AT2R-BRIL.
Production of monoclonal antibody fragment (SRP2070Fab)
Purified GPCR-BRIL proteins were reconstituted into liposomes consisting of 20:1 (w/w) egg phosphatidylcholine (Sigma-Aldrich)/monophosphoryl lipid A (Sigma-Aldrich). MRL/lpr mice were immunised with liposomal GPCR-BRIL. After immunisation, the mouse spleens were removed, and spleen cells were fused with myeloma cells. Hybridoma cells were screened by liposome-ELISA, denatured dot blot analysis, and fluorescence size exclusion chromatography (FSEC). To screen antibodies that specifically bind to the antigen with correct structure, but not the denatured one, liposome-ELISA positive and denatured dot blot clones were pooled, and then we confirmed the binding capacity in aqueous solution by FSEC. Hybridoma cells producing SRP2070 were intraperitoneally administered to mice, and ascites were collected. SRP2070 was purified by ammonium sulphate precipitation and protein G column chromatography. The subclass of SRP2070 was mouse IgG2a. After cleaving SRP2070 with papain, SRP2070Fab was purified by protein A column chromatography to remove the Fc portion and further purified by size exclusion chromatography (Superdex200 10/300 GL, GE Healthcare) by the changing buffer into PBS.
Purification of 5HT1B-BRIL/ERG/SRP2070Fab
The construct for structural analysis was expressed in accordance with a previous study17. The amino acids from Leu240 to Met305 were replaced with BRIL. In addition, the glycosylation site was removed by deleting 32 amino acid residues at the N-terminus. The HA signal sequence, FLAG tag, His10-tag, and TEV protease recognition site were added. Furthermore, the L138W mutation was introduced to increase thermal stability. The DNA coding sequence was inserted into pFastBac1 using BamHI and HindIII.
Purified plasma membranes were thawed on ice and resuspended in solubilisation buffer (50 mM HEPES pH 7.5, 500 mM NaCl, 10% glycerol, 20 mM imidazole, 1% n-dodecyl-β-d-maltoside (DDM), 0.2% cholesteryl hemisuccinate (CHS), 1 × protein inhibitor cocktail) supplemented with 100 mg/mL iodoacetamide (Wako Pure Chemical Industries, Ltd.) and 50 μM ERG (Sigma Aldrich, Saint Louis, MO, USA), and then solubilised by rotating gently for 3 h at 4 °C. The supernatant was isolated by centrifugation at 235,000 × g for 45 min and incubated with TALON Superflow Metal Affinity Resin (Clontech; 1 mL of resin per 500 mL of original culture volume was used) for 2 h at 4 °C. After incubation, the resin was washed with five column volumes of wash buffer (50 mM HEPES pH 7.5, 500 mM NaCl, 10% glycerol, 0.1% DDM, 0.02% CHS, 20 mM imidazole, 50 μM ERG, and 10 mM ATP), followed by five column volumes of wash buffer containing 800 mM NaCl. 5HT1B-BRIL protein was eluted with three column volumes of elution buffer (50 mM HEPES pH 7.5, 500 mM NaCl, 10% glycerol, 0.1% DDM, 0.02% CHS, 500 mM imidazole, and 50 μM ERG), concentrated, and then buffer exchanged into wash buffer without imidazole. The sample was treated with His-tagged TEV protease (expressed in-house) at 4 °C overnight to remove the N-terminal tags. The His-tagged TEV protease and cleaved N-terminal fragment were removed using Ni–NTA fast flow resin (GE Healthcare). 5HT1B-BRIL/ERG complex was collected as the Ni–NTA column flow-through fraction and concentrated to ~ 30 mg/mL using a 100-kDa MWCO Amicon concentrator.
The 5HT1B-BRIL/ERG/SRP2070Fab complex was prepared by mixing 5HT1B-BRIL/ERG and SRP2070Fab at a molar ration of 1:1.5 for 1 h on ice. The complex was purified by SEC (Superdex 200 increase 200, GE healthcare). Peak fractions containing 5HT1B-BRIL/ERG/SRP2070Fab were collected and concentrated to 30 mg/mL using a 100-kDa MWCO Amicon concentrator and then used for crystallisation experiments.
Crystallisation and data collection of 5HT1B-BRIL/ERG/SRP2070Fab complex
5HT1B-BRIL/ERG/SRP2070Fab were reconstituted into LCP by mixing with host lipids (monoolein:cholesterol = 9:1) at a protein:lipid ratio (v:w) of 2:3 using a mixer consisting of two 100 μL gas tight syringes (Hamilton Company). Crystal trays (96 well glass sandwich plates), containing 30 nL LCP sample covered by 800 nL mother liquor per well, were set up using a Gryphon LCP crystallisation robot (Art Robbins Instruments) and then incubated at 20 °C. Droplets were imaged chronologically by RockImager 1,000 (FORMULATRIX, Bedford, MA). Crystals were obtained from precipitant conditions containing 0.4 M KSCN, 0.1 M NaOAc pH 5.5, and 30% PEG 400. Plate-shaped crystals grew to a maximum size of 30 × 20 × 5 μm3 in 3‒4 days, were harvested using MiTeGen MicroMounts (MiTeGen), and flash frozen in liquid nitrogen. X-ray data were collected at the BL32XU beamline at SPring-8 (Japan Synchrotron Radiation Research Institute). Most crystals diffracted to ~ 3.0 Å resolution (1.0 s exposure, 1.0° oscillation, 10 μm beam attenuated by a 500‒1,200 μm aluminium sheet). To reduce radiation damage, crystals were exchanged after every 5°–10° of data collection. A complete data set at 3.0 Å resolution was obtained by merging the individual data sets collected from 144 crystals using KAMO28.
The initial structures were solved by molecular replacement as follows: as MR search models, the BRIL/SRP2070 complex (solved in-house) structure was separated into BRIL/Fv region and Fc region, and the receptor region of 5HT1B was clipped from 5HT1B-BRIL structure (PDB ID: 4IAR). The MR search was performed with MOLREP29 using three search models. Rigid-body and restrained refinements were performed with REFMAC530. The ligand was placed in the electron density and the model was corrected with COOT31. The polder ligand omit map 26 was calculated with PHENIX32.
Purification of AT2R-BRIL/s-Ang II/SRP2070Fab
Residues 1–34 and 347–363 of human AT2R were deleted, and BRIL was inserted between N242 and K246 of AT2R. The HA signal sequence, FLAG tag, and TEV protease recognition site were added to the N-terminus, and TEV protease recognition site and His8 tag were added to the C-terminus of AT2R. The membrane fraction purified from the Sf9 cell pellet was solubilised and incubated (1 h, 4 °C) with solubilisation buffer (50 mM HEPES pH 7.5, 800 mM NaCl, 10% glycerol, 1% DDM, 0.2% CHS, and 15 mM imidazole) supplemented with 100 mg/mL iodoacetamide (Wako Pure Chemical Industries, Ltd.) and 200 μM s-Ang II (Peptide Institute Inc.). After centrifugation (100,000 × g for 30 min, 4 °C), the supernatant was incubated with TALON Superflow Metal Affinity Resin (Clontech, 1–2 mL of resin per 500 mL of original culture volume was used) overnight at 4 °C with gentle agitation. After incubation, the resin was washed with twenty column volumes of wash buffer (50 mM HEPES pH 7.5, 200 mM NaCl, 10% glycerol, 0.1% DDM, 0.02% CHS, 15 mM imidazole, and 200 μM s-Ang II), then eluted by five column volumes of elution buffer (50 mM HEPES pH 7.5, 200 mM NaCl, 10% glycerol, 0.03% DDM, 0.006% CHS, 250 mM imidazole, and 200 μM s-Ang II). The eluted protein sample was concentrated using a 100-kDa MWCO Amicon concentrator, and the buffer was exchanged into wash buffer without imidazole.
The AT2R-BRIL/s-Ang II/SRP2070Fab complex was prepared by mixing AT2R-BRIL/s-Ang II and SRP2070Fab at a molar ratio of 1:1.5 for 1 h on ice. His-tagged TEV protease was added to the sample and incubated 4 °C overnight to remove the N-terminal FLAG tag and C-terminal His8-tag. The His8-tagged TEV protease and cleaved C-terminal fragment were removed by incubating with 2 mL of Ni–NTA fast flow resin (GE Healthcare) at 4 °C for 1 h. AT2R-BRIL/s-Ang II/SRP2070Fab complex was collected as the Ni–NTA column flow-through fraction and concentrated using a 100-kDa MWCO Amicon concentrator. The final complex sample was purified by SEC (Superdex 200 10/300, GE Healthcare). Peak fractions containing AT2R-BRIL/s-Ang II/SRP2070Fab were collected and concentrated to ~ 30 mg/mL using a 100-kDa MWCO Amicon concentrator.
Crystallisation and data collection of AT2R-BRIL/s-Ang II/SRP2070Fab complex
AT2R-BRIL/s-Ang II/SRP2070Fab and host lipids (monoolein:cholesterol = 9:1) were mixed at a protein:lipid ratio (v:w) of 2:3 using a mixer consisting of two 100 μL gas tight syringes (Hamilton Company). Crystal trays (96-well glass sandwich plates), containing 30 nL LCP sample overlaid by 800 nL mother liquor per well, were set up using a NT-8 crystallisation robot (FORMULATRIX), incubated at 20 °C, and imaged chronologically with a RockImager 1,000.
The crystals were obtained from crystallisation conditions containing 50 mM KOAc, 0.1 mM MES pH 6.5, 26–36% PEG 300, 100 μM s-Ang II. Rod-shaped crystals grew to a maximum size of 30 × 5 × 5 μm3 within a week, were harvested using MiTeGen MicroMounts (MiTeGen), and flash frozen in liquid nitrogen. X-ray data were collected at the BL32XU beamline at SPring-8 (Japan Synchrotron Radiation Research Institute). Most crystals diffracted to ~ 3.5 Å resolution (1.0 s exposure, 1.0° oscillation, 10 μm beam attenuated by a 500–1,200 μm aluminium sheet). To reduce radiation damage, crystals were exchanged after every 5°–10° of data collection. Finally, a complete data set at 3.4 Å resolution was obtained by merging the individual data sets collected from 118 crystals using the KAMO system28.
The initial structure was determined by molecular replacement with PHASER using BRIL/SRP2070Fv, SRP2070Fv, and the receptor region of AT1R-BRIL (PDB ID: 4ZUD) as search models. Rigid-body and restrained refinements were performed with REFMAC530. The ligand was placed in the electron density and the model was corrected with COOT31. The polder ligand omit map 26 was calculated with PHENIX32.
The detailed crystal packing calculations for 5HT1B-BRIL/ERG/SRP2070Fab and AT2R-BRIL/s-Ang II/SRP2070Fab crystal structures were calculated with PISA33 and AREAIMOL in CCP434,35 and shown in Supplementary Table 1S and Supplementary Fig. 4S. Structural figures were prepared using the CCP4mg molecular-graphics software36 except for Fig. 4C,D. Figure 4C,D were prepared using CueMol (https://www.cuemol.org/).
Supplementary information
Supplementary Information 1 (PDF 2608 kb)
Acknowledgements
This work was performed jointly by Kyoto University and Kyowa Kirin Co. Ltd. DNA sequencing was performed at the Medical Research Support Center, Graduate School of Medicine, Kyoto University. The X-ray crystallographic data were collected at SPring-8. (Proposal No. 2013B1092, 2014B1355, and 2015A1044) and Photon Factory (Proposal No. 2015R-62). We would like to thank Editage (https://www.editage.jp) for English language editing.
Author contributions
J.S. and S.I. designed the project. H.M. and H.A. generated the hybridoma cells expressing SRP2070. H.M. purified 5HT1B/ERG/SRP2070Fab, and M.S. crystallised it. M.S. collected and processed the synchrotron data of 5HT1B/ERG/SRP2070Fab. H.A., M.Y., and M.S. purified, crystallised, collected, and processed the synchrotron data of AT2R-BRIL/s-Ang II/SRP2070Fab. C.S. suppoted the purification of AT2R-BRIL/s-Ang II/SRP2070Fab. M.S. and H.A. solved and refined the structures. Y.T. supported the data collection and analysis. H.M., H.A., M.S., J.S., and S.I. analysed the data, compiled the figures, and wrote the manuscript. H.M. and H.A. are equally contributed.
Data availability
Atomic coordinates and structure factors are deposited in the Protein Data Bank under accession codes 7C61 for the 5HT1B-BRIL/ERG/SRP2070Fab and 7C6A for the AT2R-BRIL/s-Ang II/SRP2070Fab. All of the data and information about constructs used in this study are available upon reasonable request from the corresponding author.
Competing interests
This research has been funded by Kyowa Kirin Co., Ltd. which is a Japanese pharmaceutical company. H.M., M.S., Y.T., M.Y., and J.S. have been employees of Kyowa Kirin Co., Ltd. and M.Y. owns stock in the company.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Hikaru Miyagi and Hidetsugu Asada.
Supplementary information
is available for this paper at 10.1038/s41598-020-68355-x.
References
- 1.Conn PM, Ulloa-Aguirre A, Ito J, Janovick JA. G protein-coupled receptor trafficking in health and disease: Lessons learned to prepare for therapeutic mutant rescue in vivo. Pharmacol. Rev. 2007;59:225–250. doi: 10.1124/pr.59.3.2. [DOI] [PubMed] [Google Scholar]
- 2.Thomsen W, Frazer J, Unett D. Functional assays for screening GPCR targets. Curr. Opin. Biotechnol. 2005;16:655–665. doi: 10.1016/j.copbio.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 3.Robertson N, et al. The properties of thermostabilised G protein-coupled receptors (StaRs) and their use in drug discovery. Neuropharmacol. 2011;60:36–44. doi: 10.1016/j.neuropharm.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 4.Lee Y, Basith S, Choi S. Recent advances in structure-based drug design targeting class A G protein-coupled receptors utilizing crystal structures and computational simulations. J. Med. Chem. 2018;61:1–46. doi: 10.1021/acs.jmedchem.6b01453. [DOI] [PubMed] [Google Scholar]
- 5.Shoichet BK, Kobilka BK. Structure-based drug screening for G-protein-coupled receptors. Trends Pharmacol. Sci. 2012;33:268–272. doi: 10.1016/j.tips.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kumari P, Ghosh E, Shukla AK. Emerging approaches to GPCR ligand screening for drug discovery. Trends Mol. Med. 2015;21:687–701. doi: 10.1016/j.molmed.2015.09.002. [DOI] [PubMed] [Google Scholar]
- 7.Zhao Q, Wu BL. Ice breaking in GPCR structural biology. Acta Pharmacol. Sin. 2012;33:324–334. doi: 10.1038/aps.2011.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chae PS, et al. Maltose-neopentyl glycol (MNG) amphiphiles for solubilization, stabilization and crystallization of membrane proteins. Nat. Methods. 2010;7:1003–1008. doi: 10.1038/nmeth.1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maita N, Taniguchi H, Sakuraba H. Crystallization, X-ray diffraction analysis and SIRAS phasing of human alpha-l-iduronidase. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2012;68:1363–1366. doi: 10.1107/S1744309112040432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith JL, Fischetti RF, Yamamoto M. Micro-crystallography comes of age. Curr. Opin. Struct. Biol. 2012;22:602–612. doi: 10.1016/j.sbi.2012.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xiang J, et al. Successful strategies to determine high-resolution structures of GPCRs. Trends Pharmacol. Sci. 2016;37:1055–1069. doi: 10.1016/j.tips.2016.09.009. [DOI] [PubMed] [Google Scholar]
- 12.Johansson LC, Stauch B, Ishchenko A, Cherezov V. A bright future for serial femtosecond crystallography with XFELs. Trends Biochem. Sci. 2017;42:749–762. doi: 10.1016/j.tibs.2017.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Manglik A, Kruse AC. Structural basis for G protein-coupled receptor activation. Biochemistry. 2017;56:5628–5634. doi: 10.1021/acs.biochem.7b00747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rosenbaum DM, et al. GPCR engineering yields high-resolution structural insights into beta2-adrenergic receptor function. Science. 2007;318:1266–1273. doi: 10.1126/science.1150609. [DOI] [PubMed] [Google Scholar]
- 15.Chun E, et al. Fusion partner toolchest for the stabilization and crystallization of G protein-coupled receptors. Structure. 2012;20:967–976. doi: 10.1016/j.str.2012.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Caffrey M, Cherezov V. Crystallizing membrane proteins using lipidic mesophases. Nat. Protoc. 2009;4:706–731. doi: 10.1038/nprot.2009.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wacker D, et al. Structural features for functional selectivity at serotonin receptors. Science. 2013;340:615–619. doi: 10.1126/science.1232808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Asada H, et al. Crystal structure of the human angiotensin II type 2 receptor bound to an angiotensin II analog. Nat. Struct. Mol. Biol. 2018;25:570–576. doi: 10.1038/s41594-018-0079-8. [DOI] [PubMed] [Google Scholar]
- 19.Valentin-Hansen L, Holst B, Frimurer TM, Schwartz TW. PheVI:09 (Phe6.44) as a sliding microswitch in seven-transmembrane (7TM) G protein-coupled receptor activation. J. Biol. Chem. 2012;287:43516–43526. doi: 10.1074/jbc.M112.395137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Katritch V, Cherezov V, Stevens RC. Structure-function of the G protein-coupled receptor superfamily. Annu. Rev. Pharmacol. Toxicol. 2013;53:531–556. doi: 10.1146/annurev-pharmtox-032112-135923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wingler LM, McMahon C, Staus DP, Lefkowitz RJ, Kruse AC. Distinctive activation mechanism for angiotensin receptor revealed by a synthetic nanobody. Cell. 2019;176:479–490 e412. doi: 10.1016/j.cell.2018.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ishchenko A, et al. Structural insights into the extracellular recognition of the human serotonin 2B receptor by an antibody. Proc. Natl. Acad. Sci. USA. 2017;114:8223–8228. doi: 10.1073/pnas.1700891114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cherezov V, et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rasmussen SG, et al. Crystal structure of the human beta2 adrenergic G-protein-coupled receptor. Nature. 2007;450:383–387. doi: 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]
- 25.Bhat TN, Cohen GH. OMITMAP: An electron density map suitable for the examination of errors in a macromolecular model. J. Appl. Cryst. 1984;17:244–248. doi: 10.1107/S0021889884011456. [DOI] [Google Scholar]
- 26.Liebschner D, et al. Polder maps: Improving OMIT maps by excluding bulk solvent. Acta Crystallogr. D. Struct. Biol. 2017;73:148–157. doi: 10.1107/S2059798316018210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Safdari HA, Pandey S, Shukla AK, Dutta S. Illuminating GPCR Signaling by Cryo-EM. Trends Cell Biol. 2018;28:591–594. doi: 10.1016/j.tcb.2018.06.002. [DOI] [PubMed] [Google Scholar]
- 28.Yamashita K, Hirata K, Yamamoto M. KAMO: Towards automated data processing for microcrystals. Acta Crystallogr. D. Struct. Biol. 2018;74:441–449. doi: 10.1107/S2059798318004576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vagin A, Teplyakov A. Molecular replacement with MOLREP. Acta Crystallogr. D. Biol. Crystallogr. 2010;66:22–25. doi: 10.1107/S0907444909042589. [DOI] [PubMed] [Google Scholar]
- 30.Murshudov GN, et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D. Biol. Crystallogr. 2011;67:355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr. D. Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Adams PD, et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D. Struct. Biol. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 34.Collaborative Computational Project N The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 35.Winn MD, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D. Biol. Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mcnicholas S, Potterton E, Wilson KS, Noble ME. Presenting your structures: The CCP4mg molecular-graphics software. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011;67:386–394. doi: 10.1107/S0907444911007281. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information 1 (PDF 2608 kb)
Data Availability Statement
Atomic coordinates and structure factors are deposited in the Protein Data Bank under accession codes 7C61 for the 5HT1B-BRIL/ERG/SRP2070Fab and 7C6A for the AT2R-BRIL/s-Ang II/SRP2070Fab. All of the data and information about constructs used in this study are available upon reasonable request from the corresponding author.