

Summary
Immune checkpoint blockade has resulted in durable responses in patients with metastatic melanoma, but only in a fraction of treated patients. For immune checkpoint inhibitors (ICI) to be effective, sufficient infiltration with tumor‐reactive T cells is essential. Oncolytic viruses (OV) selectively replicate in and lyse tumor cells and so induce an immunogenic form of cell death, providing at once a source of tumor‐associated (neo)antigens and of danger signals that together induce effective T cell immunity and tumor infiltration. Melanoma‐associated suppression of dendritic cell (DC) differentiation effectively hampers OV‐ or immune checkpoint inhibitor (ICI)‐induced anti‐tumor immunity, due to a consequent inability to prime and attract anti‐tumor effector T cells. Here, we set out to study the effect of ORCA‐010, a clinical stage oncolytic adenovirus, on DC differentiation and functionality in the context of human melanoma. In melanoma and monocyte co‐cultures, employing a panel of five melanoma cell lines with varying origins and oncogenic mutation status, we observed clear suppression of DC development with apparent skewing of monocyte differentiation to a more M2‐macrophage‐like state. We established the ability of ORCA‐010 to productively infect and lyse the melanoma cells. Moreover, although ORCA‐010 was unable to restore DC differentiation, it induced activation and an increased co‐stimulatory capacity of monocyte‐derived antigen‐presenting cells. Their subsequent ability to prime effector T cells with a type I cytokine profile was significantly increased in an allogeneic mixed leukocyte reaction. Our findings suggest that ORCA‐010 is a valuable immunotherapeutic agent for melanoma.
Keywords: dendritic cell, immune suppression, immunotherapy, melanoma, oncolytic virus

Introduction
The incidence of melanoma, the most aggressive form of skin cancer, is rising worldwide [1, 2]. Immune checkpoint blockade has resulted in durable responses in patients with metastatic melanoma, but only in a fraction of treated patients [2, 3, 4]. There is a growing awareness that for immune checkpoint inhibitors (ICI) to be effective, sufficient infiltration with tumor‐reactive T cells is an absolute requirement. Much effort is therefore now ongoing to develop therapies that will render unresponsive tumors ‘hot’, i.e. with high T cell infiltration rates. One of the ways in which to achieve this is by oncolytic virotherapy. Oncolytic viruses (OV) selectively replicate in, and lyse, tumor cells and so induce an immunogenic form of cell death, providing at once a source of tumor‐associated (neo)antigens and of immunological danger signals that together induce effective T cell immunity and tumor infiltration, in effect achieving ‘in vivo vaccination’ [5, 6]. Indeed, intratumoral delivery of talimogene laherparepvec (T‐VEC), an oncolytic herpesvirus encoding the immune stimulatory cytokine granulocyte–macrophage colony‐stimulating factor (GM‐CSF), in melanoma patients has been shown to overcome resistance to programmed cell death (PD)‐1 blockade, resulting in regression of both injected and non‐injected metastases, both of which were shown to present with increased immune infiltration after T‐VEC administration [7].
An immune suppressed myeloid compartment in the tumor microenvironment (TME) has been identified as one of the dominant mechanisms that will stand in the way of T cell activation, either by OV or by ICI, due to the absence of properly developed and activated dendritic cells (DCs) [8]. DCs are professional antigen‐presenting cells in charge of taking up antigens and processing and presenting them through major histocompatibility complex (MHC)‐I or ‐II to naive T cells in the lymph nodes. These T cells are then licensed to mediate tumor elimination [9]. Moreover, DCs are involved in recruiting T cells to the tumor microenvironment, which enables an anti‐tumor effector response. The importance of DCs in facilitating an anti‐tumor immune response has been demonstrated by several reports, showing that a lack of DCs in the tumor bed is one of the main reasons for immunotherapy resistance [10, 11, 12, 13]. Recent reports from Spranger and colleagues have demonstrated that melanoma infiltration by DCs ensures sufficient T cell recruitment to the tumor site for PD‐1 blockade to be effective [14]. Unfortunately, melanomas exploit myeloid plasticity to skew DC differentiation towards immune‐suppressive macrophage‐like subsets that interfere negatively with the immune response [10, 15]. Reports have shown that melanoma‐derived suppressive factors such as interleukin (IL)‐10, IL‐6 or prostaglandin E2 (PGE2) up‐regulate both signal transducer and activator of transcription 3 (STAT‐3) and P38 mitogen‐activated protein kinase (MAPK) activity in DC precursors in murine models and patients [16, 17]. As a result, monocyte‐derived DC (moDC) differentiation is blocked and M2‐like macrophages are induced, which suppress tumor‐specific T cells and promote endothelial cell migration and proliferation, tumor growth and invasion [18, 19]. During cancer development, circulating monocytes are recruited to the tumor site due to the release of monocyte chemoattractant protein‐1 (MCP‐1) and vascular endothelial growth factor C (VEGF‐C) by melanoma cells [20, 21]. Once they are recruited to the tumor, they differentiate into CD163+ tumor‐associated macrophages (TAMs) [22]. CD163+ TAMs produce diverse immune suppressive chemokines that recruit regulatory T cells (Tregs) to the tumor site, thus hampering anti‐tumor responses [23]. The immune‐suppressive cytokine IL‐10, produced by TAMs, induces programmed cell death ligand 1 (PD‐L1) expression in an autocrine fashion, thus hampering T cell activity further [24]. Tumor‐associated dendritic cells (TADCs) have been described to express low levels of CD1a, a marker for moDC differentiation, intermediate levels of CD14 and high levels of CD163, markers linked to non‐differentiated monocytes and M2‐like suppressive macrophages, respectively [15, 25]. Moreover, these TADCs have been described as tolerogenic antigen‐presenting cells with a poor capacity to induce tumor‐specific T cell proliferation and activation, but rather with an increased capacity for IL‐10 release and expansion of Tregs [26, 27].
Through both pathogen‐ and damage‐associated molecular patterns, derived from the OV and resulting from oncolysis, respectively, TADCs may be activated upon OV administration, thus facilitating anti‐tumor T cell (cross‐)priming and increased lymphocyte infiltration at the tumor site [28]. The latter would, in turn, increase the likelihood of patients responding to immune checkpoint blockade.
The use of adenoviruses for virotherapy is attractive in view of their previously reported intrinsic DC‐activating and proinflammatory anti‐tumor properties, which were suggested to be mediated by both Toll‐like receptor (TLR)‐dependent and ‐independent mechanisms [29, 30, 31, 32]. However, an extensive description of the effect of adenoviral oncolysis on DC differentiation and functionality in the context of human melanoma has not been addressed to date. To study the effect of a recently described oncolytic adenovirus with potent anti‐tumor activity, ORCA‐010 [33], on melanoma‐conditioned moDC differentiation and activation, we established an in‐vitro co‐culture model, using a panel of five different melanoma cell lines from varying origins (primary versus metastatic and skin versus lymph node) and with diverging v‐raf murine sarcoma viral oncogene homolog B (BRAF), neuroblastoma rat sarcoma oncogene (NRAS) and phosphatase and tensin homolog (PTEN) mutational status. Our data reveal the ability of ORCA‐010 to both lyse melanoma cells and activate melanoma‐exposed moDC, thus increasing their ability to prime type‐1 effector T cells. These findings indicate that ORCA‐010 would be an attractive candidate to apply in the treatment of melanoma and to enhance the efficacy of immune checkpoint blockade.
Materials and methods
Cell lines and cell culture
The established human melanoma cell lines JUSO, BRO, Mel57, WM9 and SK‐MEL‐28 and their origins were all described previously [34, 35, 36, 37]. Their identities were confirmed by short tandem repeat (STR) analysis, using the human cell line authentication service provided by Eurofins based on 21 independent polymerase chain reaction (PCR) single‐locus technology, following the ISO 17025 standard guidelines (https://www.eurofinsgenomics.eu/en/genotyping‐gene‐expression/applied‐genomics‐services/cell‐lineauthentication/). All cell lines were cultured in Iscove’s modified Dulbecco’s medium (IMDM) (BioWhittaker Lonza, Basel, Switzerland) supplemented with 10% heat‐inactivated fetal calf serum (FSC) (HyClone, Logan, UT, USA), 100 IU/ml sodium–penicillin, 100 μg/ml streptomycin, 2 mM L‐glutamine and 50 μM β‐mercaptoethanol (2ME) (referred to as complete IMDM) at 37°C and 5% CO2. Cells were passaged twice a week, avoiding complete confluence. CD40‐ligand (CD40L)‐expressing J558 mouse myeloma cells were cultured in complete IMDM at 37°C and 5% CO2, and were passaged twice a week.
Peripheral blood mononuclear cell and monocyte isolation
Monocytes were isolated from heparinized blood obtained from healthy donors under informed consent (Sanquin Blood Supply Services, Amsterdam, the Netherlands). Peripheral blood mononuclear cells (PBMCs) were isolated using LymphoprepTM (Axis‐Shield, Dundee, UK) gradient centrifugation (Fresenius 114547), as previously described [24]. CD14+ cells were isolated from PBMCs via magnetic activated cell sorting (MACS) by the use of CD14 magnetic beads (MACS; Milteny Biotec, Bergisch Gladbach, Germany), according to the manufacturer’s protocol. After sorting, the purity of the monocytes was assessed by flow cytometry using a fluorescence‐activated cell sorter (FACS)Calibur (BD Biosciences, San Jose, CA, USA) or LSR FortessaTM (BD Biosciences) and found to routinely exceed 98%.
Viruses
ORCA‐010 was derived from wild‐type adenovirus serotype 5 (Ad5) through genetic modification, including a 24 nt deletion in the E1A gene, a mutation in the gene encoding for the E3/19K protein and an insertion of a cyclic RGD motif in the fiber protein, resulting in the Ad5‐Δ24‐RGD‐T1 (ORCA‐010) virus [33].
Determination of optimal ORCA‐010 multiplicity of infection (MOI) on melanoma cells
Melanoma cells were infected with ORCA‐010 oncolytic virus with different multiplicities of infection (MOIs). A total of 1 × 104 melanoma cells were plated in 400 μl of complete IMDM in flat‐bottomed 48‐well plates and incubated for 24 h at 37°C and 5% CO2. Next, the melanoma cells were infected with ORCA‐010 virus, at MOIs of 0, 25, 100 and 400. Cells were then incubated for 6 days at 37°C and 5% CO2. Cells were harvested and collected in FACS tubes. Collected cells were subsequently used for FACS analysis to determine viability on a FACS CaliburTM, after staining with 7‐aminoactinomycin D (7‐AAD; Sigma, St Louis, MO, USA).
Melanoma and monocyte co‐culture for monocyte‐derived DC (moDC) phenotypical read‐out in the presence or absence of ORCA‐010
Monocytes were co‐cultured for 6 days with the melanoma cell lines either in the presence or absence of ORCA‐010. A total of 1 × 104 melanoma cells were plated in 300 μl of complete IMDM in flat‐bottomed 48‐well plates and incubated for 24 h at 37°C and 5% CO2. Next, the melanoma cells were infected with ORCA‐010 virus at an MOI of 25, 100 or 400 and incubated for 1 h at 37°C and 5% CO2. Subsequently, 2 × 105 monocytes, supplemented with 1000 IU/ml recombinant human (rh)GM‐CSF (Genzyme/Bayer HealthCare Pharmaceuticals, Seattle, WA, USA) and 20 ng/ml rhIL‐4 (R&D Systems, Minneapolis, MN, USA), were plated on top of the melanoma monolayer in 300 μl of complete RPMI. Without further washing, the cells were then incubated for 6 days at 37°C and 5% CO2, after which the cells were harvested and collected in FACS tubes. Collected cells were used for phenotype readout by flow cytometry, using an LSR FortessaTM (BD Biosciences). Melanoma cells were similarly cultured without monocytes and GM‐CSF/IL‐4 to determine the effect of ORCA‐010 infection on PD‐L1 expression on the melanoma cell surface.
MoDC cytokine release
After 6 days of melanoma/monocyte co‐culture as described above, 4 × 104 J558 cells expressing CD40L were added to the co‐culture in the presence of 1000 IU/ml recombinant human interferon gamma (rhIFN‐γ) (e‐Bioscience, San Diego, CA, USA) and incubated for 24 h at 37°C and 5% CO2. After 24 h, supernatants were collected and stored at −20°C. IL‐10 and IL‐12‐p70 were determined by cytometric bead array (CBA), according to the manufacturer’s protocol (BD Biosciences; 551811).
Mixed leukocyte reaction
Melanoma cells (1 × 104) from cell lines were plated in 24‐well plates suitable for Transwell inserts in complete IMDM and incubated for 24 h at 37°C and 5% CO2. Next, ORCA‐010 virus was added at MOI 25, 100 or 400. After 1 h at 37°C and 5% CO2, 4 × 104 CD14+ monocytes, supplemented with 1000 IU/ml GM‐CSF and 20 ng/ml rhIL‐4, were added in complete RPMI to the upper compartments of the Transwell system with a pore size of 0·4 μM (Life Sciences, New York, NY, USA), to avoid migration of the monocytes; and cells were incubated for 6 days at 37°C and 5% CO2. Next, moDCs were harvested from the top compartment and analyzed by flow cytometry or assessed for their T cell priming capacity in an allogeneic mixed lymphocyte reaction (MLR). Before use in the MLR, maturation was induced using 2400 U/ml tumor necrosis factor (TNF)‐α (MACS; Miltenyi Biotec), 100 ng/ml IL‐6 (R&D Systems, Minneapolis, MN, USA), 25 ng/ml IL‐1β (MACS; Miltenyi Biotec) and 1 μg/ml PGE2 (Sigma Aldrich) for 48 h at 37°C and 5% CO2; 104 matured moDCs were used as stimulator cells and were plated in round‐bottomed 96‐well tissue‐culture plates (Greiner, Kremsmünster, Austria). As responder cells, 105 allogeneic peripheral blood lymphocytes (PBL), obtained from PBMCs after CD14+ monocyte depletion, were labeled with 3 μM carboxyfluorescein succinimidyl ester (CFSE) (Sigma Aldrich) and added to the wells. PBL stimulation was performed in duplicate. Cells were cultured in IMDM supplemented with 10% human pooled serum (HPS), 100 IU/ml sodium–penicillin, 100 μg/ml streptomycin, 2 mM L‐glutamine and 50 μM 2ME for 6 days at 37°C and 5% CO2. After 6 days, supernatants were collected and PBL were harvested and T cell proliferation was determined by flow cytometry using a FACSCalibur. The supernatants, obtained at day 6 from the MLR, were used for CBA analysis to measure T cell cytokines including IL‐2, IL‐10, TNF‐α and IFN‐γ, according to the manufacturer’s protocol (BD Biosciences; 560484).
Flow cytometry
Antibody staining was performed by washing moDCs and/or melanoma cells with PBS supplemented with 0·1% bovine serum albumin (BSA) and 0·02% sodium azide (NaN3) (FACS buffer) and centrifuged for 5 min. Supernatants were discarded and cells were stained for surface marker expression in the remaining FACS buffer volume for 30 min at 4°C. After incubation, the excess of antibodies was removed by washing with FACS buffer. Analysis was performed on FACSCalibur or LSR FortessaTM.
Fluorescein isothiocyanate (FITC)‐, phycoerythrin (PE)‐, peridinin chlorophyll cyanin 5·5 (PerCPCy5.5)‐, allophycocyanin (APC)‐, AF700‐, PECy7‐, BV711‐, BV421 and BV650‐ labeled antibodies directed against human CD14, CD4, CD8, CD3, human leukocyte antigen R‐related (HLA‐DR), CD16, PD‐L1 (BD Biosciences), CD80, CD86, CD1a, HLA‐DR (BD PharMingen, San Diego, CA, USA), BDCA3 (Miltenyi Biotec), HLA‐ABC, CD1c, CD45 (Biolegend, San Diego, CA, USA), PD‐L1 (eBioscience), CD40, CD163, PD‐L2 (BD Horizon, BD Biosciences) and CD40 (Beckman Coulter, Brea, CA, USA) were used for flow cytometric analyses.
Obtained data were analyzed using CellQuest analysis software (BD Biosciences) or Kaluza analysis software version 1.3 (Beckman Coulter).
IncuCyte live cell imaging
Melanoma cell lines were plated at 1 × 104 cells in complete IMDM in flat‐bottomed 48‐well plates and incubated for 24 h at 37°C and 5% CO2. Next day, the melanoma cells were incubated with ORCA‐010, at an MOI of 25, 100 or 400 for 1 h at 37°C and 5% CO2. Subsequently, 2 × 105 monocytes, supplemented with 1000 IU/ml rhGM‐CSF and 20 ng/ml rhIL‐4, were plated on top of the melanoma monolayer in complete RPMI. Cells were then incubated for 6 days at 37°C and 5% CO2 in the IncuCyte® ZOOM live cell analysis system (Essen Bioscience, Ann Arbor, MI, USA), imaging the cultures every 4 h. Controls included conditions without ORCA‐010 and/or without monocytes added. To establish growth kinetics, melanoma cell lines were plated at 1 × 104 cells in complete IMDM in flat‐bottomed 48‐well plates and incubated for 9 days in the IncuCyte® ZOOM live cell analysis system (Essen Bioscience). The images were processed with IncuCyte ZOOM software.
Caspase 3/7 apoptosis assay
A total of 2000 BRO cells were plated in 50 μl complete IMDM, supplemented with 1 : 1000 diluted caspase‐3/7 green apoptosis assay reagent (IncuCyte®, cat. no. 4440), in a flat‐bottomed 384‐well plate. Next, the cells were infected with ORCA‐010 virus at an MOI of 400 or left untreated. Cells were then incubated for 9 days at 37°C and 5% CO2 in the IncuCyte® ZOOM (Essen Bioscience), with live imaging every 4 h. The images were analysed with IncuCyte ZOOM software, measuring the confluence and green apoptotic marker signal.
Measurement of cytokine release
A total of 1 × 104 melanoma cells were plated in 300 μl of complete IMDM in flat‐bottomed 48‐well plates and incubated for 24 h at 37°C and 5% CO2. Protein secretion into culture supernatants after 24 h was assessed by enzyme‐linked immunosorbent assay (ELISA), according to the manufacturer’s instructions. Transforming growth factor beta 1 (TGF‐β1) and macrophage–colony‐stimulating factor (M‐CSF) were detected using DuoSet ELISA kits (R&D Systems). VEGF, IL‐12p70, IL‐10, IL‐6, IL‐8, TNF‐α and IL‐1β levels were determined by CBA (BD Biosciences), according to the manufacturer’s protocol (VEGF, BD protocol 558336; others, BD protocol 551811).
Statistical analysis
Statistical analysis was performed using GraphPad Prism version 7.03. The statistical tests used were an ordinary two‐way analysis of variance (anova) followed by Sidak’s post‐hoc test; a matched repeated‐measures (RM) one‐way anova followed by Fisher’s least significance difference (LSD) post‐hoc test; or a paired two‐sided Student’s t‐test, as indicated in the Figure legends. A P‐value was considered significant when P < 0·05.
Results
Melanoma cell lines suppress moDC differentiation
To determine the suppressive effect of melanoma on moDC differentiation, we used a panel of five melanoma cell lines, i.e. BRO, SK‐MEL‐28, WM9‐ MEL57 and JUSO. The origin and BRAF, NRAS and PTEN mutational status of these cell lines is summarized in Table 1. Their release profiles of potentially immune suppressive cytokines are shown in Table 2. The melanoma cells were co‐cultured with healthy donor PBL for 6 days in the presence of the stimulatory cytokines GM‐CSF and IL‐4 (GM4), which induce DC differentiation. As controls, we included monocytes that were cultured in the presence of GM4 alone (i.e. optimal moDC differentiation) or with GM4 + IL‐10 (i.e. maximally blocked DC differentiation) [38]. Figure 1a shows FACS dot‐plots of CD1a and CD14 expression on the CD45+ populations. Our data demonstrate that melanoma cell lines are able, to different degrees, to inhibit moDC differentiation, as evidenced by maintained CD14 expression and, most notably, reduced CD1a levels. This is further demonstrated in Fig. 1b, showing the collective data of three independent experiments with monocytes from different donors. We observed significant differences in CD1a and CD14 expression levels upon co‐culture with all five melanoma cell lines, except for CD14 levels upon co‐culture with WM9, compared to the normal GM4‐induced moDC cultures. A melanoma‐related block in DC differentiation was further supported by significantly reduced expression levels of the moDC‐related marker CD1c (clearly absent from the monocytes at baseline) upon co‐culture with all cell lines, with WM9 again being the exception (Fig. 1c). A melanoma‐induced reduction in co‐stimulatory capacity was evidenced by significant reductions in CD80 (Fig. 1d) and, to a lesser extent, CD40 (Supporting information, Fig. S1a) levels. CD163 expression was maintained in comparison with the GM4 condition upon most melanoma co‐cultures, indicating differentiation towards a more M2‐like suppressive macrophage state (Fig. 1e). In keeping with this, CD16 was also increased (Supporting information, Fig. S1b), similarly to our previous observations for melanoma‐associated M2‐like macrophages [15, 39]. Of note, all cell lines displayed similar growth curves and reached confluence around day 4 (Supporting information, Fig. S1c).
Table 1.
Panel of used melanoma cell lines, their origins, and their oncogene mutational status
| Melanoma cell line (reference) | BRAF | RAS | PTEN | Tumor |
|---|---|---|---|---|
| BRO (37) | WT | Q61R | Unknown | Skin primary |
| SK‐MEL‐28 (36) | V600E | WT | A499G | Skin metastasis |
| WM9 (34) | V600E | WT | Deletion exon 3‐9 | Lymph node metastasis |
| MEL57 (35) | V600E | WT | Unknown | Skin metastasis |
| JUSO (36) | WT | Q61L and G13D | WT | Skin primary |
Table 2.
Melanoma cell lines release profile of (potentially immune suppressive) cytokines
| Cytokines (pg/ml) | BRO | SK‐MEL‐28 | WM9 | MEL57 | JUSO |
|---|---|---|---|---|---|
| IL‐12p70 | 0 | 0 | 0 | 0 | 0 |
| IL‐10 | 0 | 0 | 5·96 | 0 | 0 |
| TNF | 0 | 0 | 0 | 0 | 0 |
| IL‐6 | 3·65 | 0·57 | 1·33 | 0 | 0 |
| IL‐1β | 0 | 0 | 0 | 0 | 0 |
| IL‐8 | 2778·2 | 9004 | 354·12 | 4209·86 | 686·63 |
| M‐CSF | 0 | 0 | 4·059 | 4·059 | 0 |
| TGF‐β | 1643·86 | 1966·63 | 1685·09 | 1210·07 | 1446·78 |
| VEGF | 590·87 | 217·75 | 0 | 271·85 | 34·55 |
IL = interleukin; TNF = tumor necrosis factor; M‐CSF = macrophage–colony‐stimulating factor; TGF = transforming growth factor; VEGF = vascular endothelial growth factor.
Fig. 1.
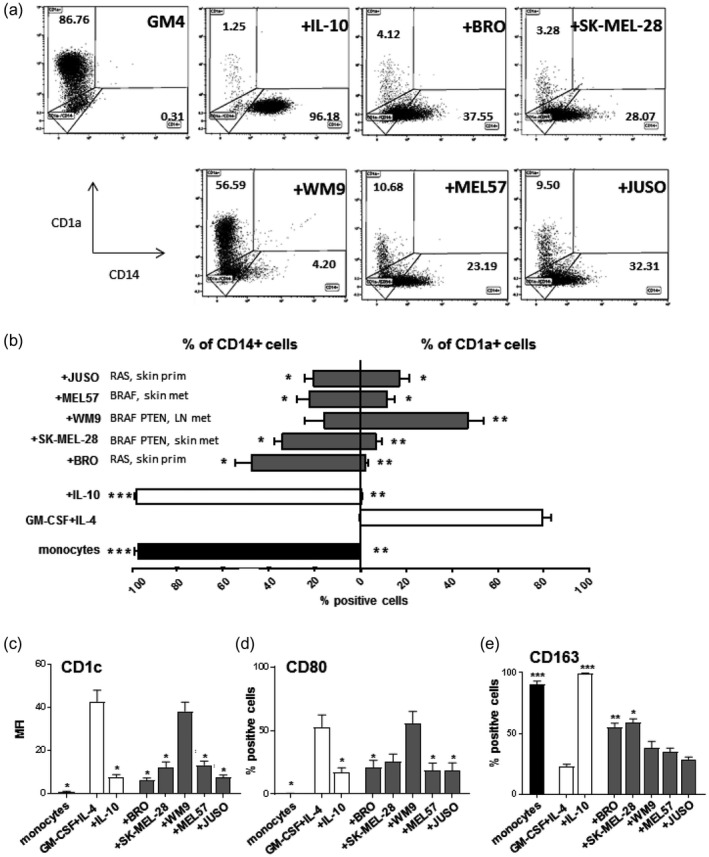
Melanoma cell lines suppress monocyte‐derived dendritic cell (moDC) differentiation. Monocytes were phenotyped at day 0 or cultured for 6 days under different conditions and phenotyped after culture. (a) Representative fluorescence activated cell sorter (FACS) dot‐plots showing CD1a versus CD14 expression on moDC under different culture conditions. (b) Average percentages of CD1a+ and CD14+ expressing monocytes or moDC under different culture conditions, n = 3. The conditions shown are: day 0 initial monocytes (monocytes), granulocyte–macrophage colony‐stimulating factor (GM‐CSF)+interleukin (IL)‐4 (GM4), GM‐CSF+IL‐4+IL‐10 (+IL‐10), GM‐CSF+IL‐4+BRO (+BRO), GM‐CSF+IL‐4+SK‐MEL‐28 (+SK‐MEL‐28), GM‐CSF+IL‐4+WM9 (+WM9), GM‐CSF+IL‐4+MEL57 (+MEL57) and GM‐CSF+IL‐4+ JUSO (+JUSO). (c) Average geometric mean intensity of CD1c staining, (d) average percentage of CD80+ cells and (e) average percentage CD163+ cells (M2‐like macrophages) in the different monocyte or conditioned moDC conditions, n = 3. Control day 0 uncultured monocytes are shown in black bars; GM4 and +IL‐10 conditions are represented by white bars; and melanoma‐conditioned moDCs by gray bars. Means ± standard error of the mean (s.e.m.) are shown; statistical significances are shown compared to the GM4 condition, applying a matched RM 1‐way analysis of variance (ANOVA) test with Fisher’s least significant difference (LSD) post‐hoc test. Significance shown as *P < 0·05, **P < 0·01 and ***P < 0·001.
Oncolytic effect of ORCA‐010 on melanoma cell lines
We determined the oncolytic potential of ORCA‐010 for melanoma in the established cell line panel. As shown in Fig. 2a, up to 80% of melanoma cells were killed (as measured by flow cytometric analysis with 7‐AAD, 6 days after infection). For each of the tested cell lines we selected an MOI resulting in suboptimal lysis (50–70%, indicated by arrows in Fig. 2a), which we used in subsequent melanoma/monocyte co‐cultures. To visualize and assess the oncolytic effect of ORCA‐010 at these selected MOIs, the melanoma cell lines were plated together with caspase‐3/7 green apoptosis assay reagent in the IncuCyte ZOOM for a culture period of 8 days. In Fig. 2b, as a representative result, live cell imaging of BRO cultured in the presence or absence of ORCA‐010 is shown over time, revealing an increase in apoptotic cells (positive for caspase 3/7, shown in green) becoming apparent after day 4. Of note, ORCA‐010 infection at these MOIs resulted in only marginal increases in PD‐L1, while no effects were observed in PD‐L2 expression on the melanoma cells (Fig. 2c), whereas MHC‐I and ‐II expression levels were not affected at all (data not shown).
Fig. 2.

Oncolytic effect of ORCA‐010 on melanoma cell lines. The melanoma cell lines BRO, SK‐MEL‐28, WM9, MEL57 and JUSO were infected with the oncolytic virus ORCA‐010 at different multiplicities of infection (MOI = 0, 25, 100 and 400). (a) Percentage of viable cells measured with 7‐aminoactinomycin D (7‐AAD) staining by flow cytometric analysis 6 days after infection with ORCA‐010 at the indicated MOIs. Data are means ± standard error of the mean (s.e.m.) of three independent experiments. The MOI chosen for further experiments is indicated with a black arrow. (b) Representative pictures taken by IncuCyte imaging of the melanoma cell line BRO with or without ORCA‐010 at different time‐points of culture with the caspase‐3/7 green apoptosis assay reagent. Cells marked in green are apoptotic cells. (c) Percentages of programmed cell death ligand 1 (PD‐L1) and PD‐L2 expressing melanoma cells with or without ORCA‐010 infection. Means ± s.e.m. (n = 3) are shown. An ordinary two‐way analysis of variance (anova) test with Sidak’s post‐hoc test was applied. **P < 0·01.
ORCA‐010 is not able to restore melanoma‐mediated inhibition of moDC differentiation, but induces moDC activation
To model the effect of ORCA‐010 on moDC differentiation in the context of melanoma, the adherent melanoma cell lines were pre‐incubated for 1 h with ORCA‐010 at a per cell line selected MOI of 25, 100 or 400 (Fig. 2a), after which monocytes were added to the cultures together with the GM4 DC differentiation‐inducing cytokine cocktail. After 6 days, morphology and phenotype of the moDCs were assessed. As shown in Fig. 3, live imaging of ORCA‐010‐infected (co‐)cultures over time clearly showed oncolysis to coincide with enhanced dendritic morphology of the moDCs (examples indicated with black arrow), while their simultaneous clustering was indicative of activation. These morphological changes were not reflected in a clear phenotypical shift towards a more DC‐like phenotype; only small, non‐significant changes in CD1a and CD14 were observed (Fig. 4a) and no decreases in CD163 levels were achieved (Fig. 4b). Although hampered DC differentiation was not restored by ORCA‐010, the resulting monocyte‐derived antigen‐presenting cells (APCs) were more activated, as evidenced by significantly elevated expression levels of the co‐stimulatory molecules CD80, CD86 and CD40; of the immune checkpoints PD‐L1 and ‐L2; and of MHC‐I and ‐II (Fig. 4c). Although CD163 levels were unaffected, decreased levels of CD16 were consistent with a diminished tumor‐associated M2‐like phenotype which, in turn, was consistent with the observed increases in the DC‐ and M1‐associated marker CD80 (Fig. 4c). Supporting information, Fig. S2a illustrates that the up‐regulated levels of CD80 and CD86 on moDC in the presence of ORCA‐010‐induced lysis of melanoma cells (in this Figure exemplified by the BRO cell line) even exceeded levels on control GM4‐differentiated moDC, indicating bona fide activation. This activation translated into consistently increased percentages of CD80‐ and CD86‐positive cells across all tested melanoma cell lines (Supporting information, Fig. S2b). Of note, these stimulatory effects were not observed in the GM4 or IL‐10 control cultures without melanoma cells, to which ORCA‐010 was added at MOI 400 (Fig. 4c and Supporting information, Fig. S2b). This suggests that the observed activation may have been the result of either the release of danger‐associated molecular motifs or the reduced release of suppressive cytokines, or a combination of both, resulting from ORCA‐010‐induced oncolysis.
Fig. 3.
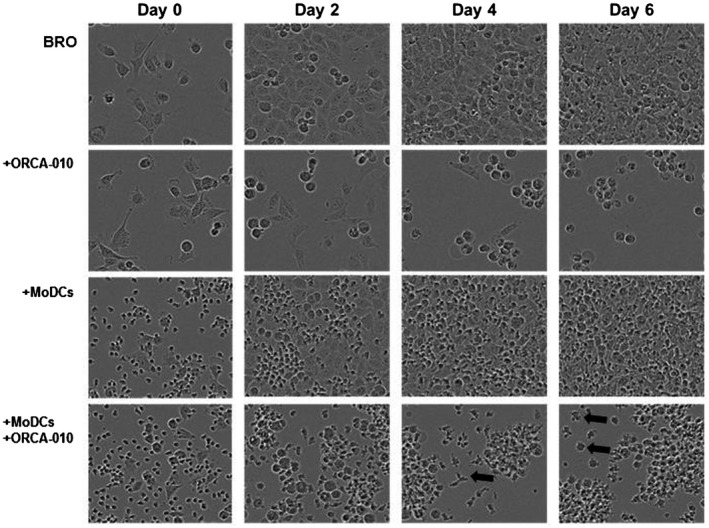
Live cell imaging of melanoma/monocyte co‐cultures. Representative pictures of BRO cells, with or without ORCA‐010; and with or without monocytes and a monocyte‐derived dendritic cell (moDC)‐inducing growth factor cocktail were taken with the IncuCyte ZOOM on different days of culture. Arrows indicate a differentiated moDC.
Fig. 4.
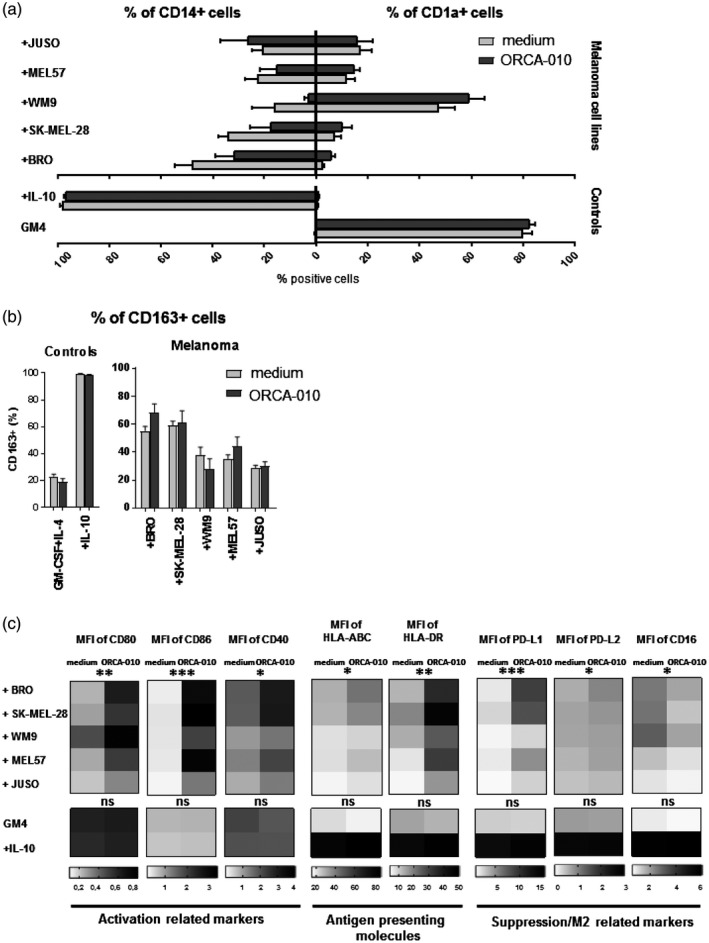
ORCA‐010 is not able to undo melanoma‐mediated inhibition of the differentiation of monocyte‐derived dendritic cells (moDCs) but induces their activation. MoDCs were characterized after 6 days in monoculture [GM4 and +interleukin (IL)‐10] or in co‐culture with melanoma cell lines in the presence or absence of ORCA‐010 virus. Statistical significances are shown when comparing presence or absence of the oncolytic virus within each condition, applying a paired t‐test. (a) Percentages of CD1a+ and CD14+ cells (b) percentage CD163+ cells (M2‐like macrophages). Data in (a) and (b) are means ± standard error of the mean (s.e.m.) of three independent experiments. (c) Heat‐maps of the geometric mean intensity expression of the activation‐related markers CD80, CD86 and CD40; antigen‐presenting molecules human leukocyte antigen (HLA)‐ABC and HLA‐DR; and the suppression/M2‐related markers programmed cell death ligand 1 (PD‐L1), PD‐L2 and CD16, n = 3. Ordinary two‐way analysis of variance (anova) results are displayed, *P < 0·05, **P < 0·01 and ***P < 0·001.
We subsequently tested the ability of the melanoma‐conditioned monocyte‐derived APCs to release IL‐10 or IL‐12p70. As shown in Supporting information, Fig. S3, melanoma exposure generally severely hampered the CD40L‐ and IFN‐γ‐induced release of both IL‐10 and IL‐12p70, except for the WM9 and MEL57 conditioned IL‐12p70 release, which was unaffected. Whereas ORCA‐010 did not affect IL‐10 release (Supporting information, Fig. S3a), for two out of three cell lines with inhibited IL‐12p70 release, this release could be partially restored by ORCA‐010, although not significantly so (Supporting information, Fig. S3b).
Of note, no significant effect of the viral infection was found on the phenotypical activation of already differentiated DC co‐cultured with tumor cells for 2 days (data not shown).
Hampered induction of type 1 effector T cells by melanoma‐conditioned moDCs is counteracted by ORCA‐010
To assess the effect of melanoma exposure in the presence or absence of ORCA‐010 on the moDCs’ capacity to prime T cells, we set up an allogeneic MLR. To this end, differentiating moDCs were cultured and exposed to melanoma cells with or without ORCA‐010 for 6 days in a Transwell system. This allowed harvesting of the moDCs without contaminating melanoma cells, which would otherwise have confounded interpretation of the subsequent MLR. Figure 5a shows that the suppressive effects of the melanoma cell lines were similar for direct co‐culture and co‐culture in the Transwell system. This indicates that the observed suppression was mediated by soluble factors. The melanoma‐conditioned moDCs were cultured with fresh CFSE‐labeled allogeneic PBLs for another 6 days, after which supernatants were harvested for T cell cytokine profiling, and the cells were stained for CD4 or CD8 and analyzed by FACS for CFSE dilution as a measure of T cell proliferation.
Fig. 5.
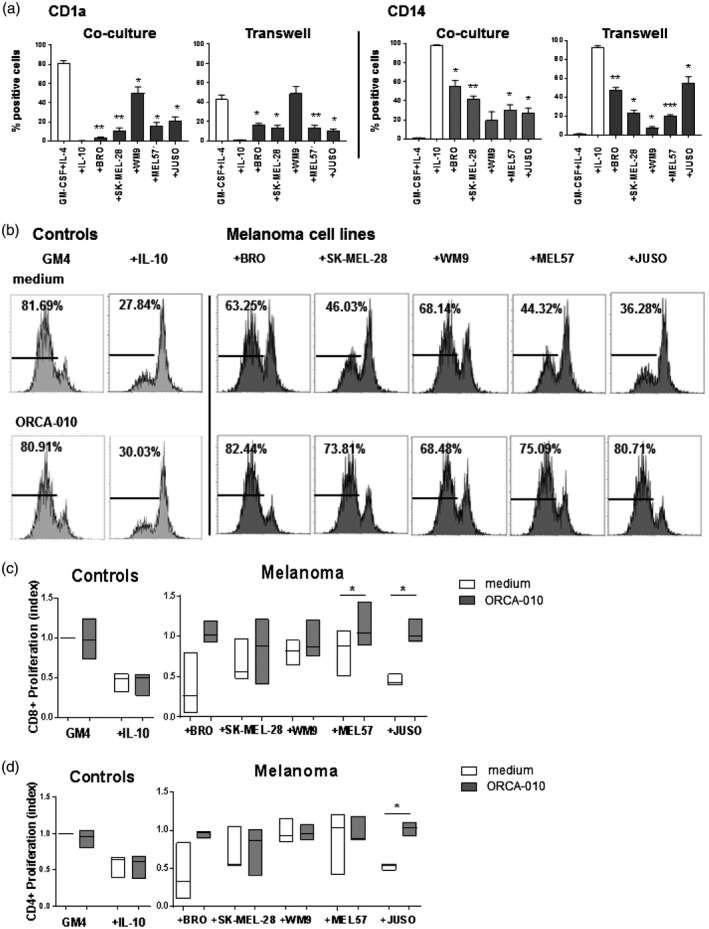
Hampered proliferation of type‐1 effector T cells by melanoma‐conditioned monocyte‐derived dendritic cells (moDCs) is counteracted by ORCA‐010. Using a Transwell system, the 6‐day monocultured [GM4 and +(IL)‐10] or melanoma‐conditioned moDCs (+BRO, +SK‐MEL‐28, +WM9, +MEL57 and +JUSO), with or without infection with the ORCA‐010 virus, were matured and cultured with carboxyfluorescein succinimidyl ester (CFSE)‐stained allogeneic T cells for an extra 6 days. (a) Percentage of CD1a+ and CD14+ conditioned monocytes or moDC under different culture conditions. Statistical significances are shown compared to the GM4 condition. (b) Representative histograms of CD8+ T cell proliferation induced by the different moDC conditions; percentages of proliferated CD8+ T cells are shown per condition. (c,d) Box‐plots of the relative CD8+ (c) and CD4+ (d) T cell proliferation (GM4 was set at 1·0), induced by the differently conditioned moDCs. Box‐plots show the means ± standard error of the mean (s.e.m.) of three independent experiments. A matched repeated‐measures one‐way analysis of variance (anova) with Fisher’s least significant difference (LSD) post‐hoc test was performed to test the significance of oncolytic virus effects per condition. *P < 0·05, **P < 0·01 and ***P < 0·001.
Figure 5b shows representative histograms of the induced CD8+ T cell proliferation measured by the CFSE stain and Fig. 5c the mean relative proliferation of CD8+ T cells from three different donors. Consistent with the phenotypical activation data, melanoma exposure generally resulted in a reduced T cell priming capacity of the moDCs, which was restored by inclusion of ORCA‐010 in the co‐cultures. Again, this stimulatory effect of ORCA‐010 was dependent upon the presence of melanoma cells as it left the cell priming by GM4 moDCs or hampered T cell priming by IL‐10 control moDCs unaffected. Although the strength of the inhibitory effects of the cell lines varied, ORCA‐010 consistently enhanced the conditioned moDCs’ priming capacity for both CD8+ (Fig. 5c) and CD4+ T cells (Fig. 5d).
Cytokine profiling of the MLR supernatants revealed decreased IFN‐γ (Fig. 6a) and IL‐2 (Fig. 6b) release upon priming by the melanoma‐conditioned moDCs, which was restored by the presence of ORCA‐010 in the melanoma/moDC co‐cultures. Although less pronounced, a similar trend was observed for TNF‐α (Fig. 6c). Again, hampered release of these cytokines in the MLR with IL‐10‐conditioned moDC was not overcome by prior differentiation of the moDCs in the presence of ORCA‐010. Finally, IL‐10 levels in the MLR supernatants were not consistently affected by either melanoma conditioning or ORCA‐010 inclusion in the moDC differentiation cultures (Fig. 6d).
Fig. 6.
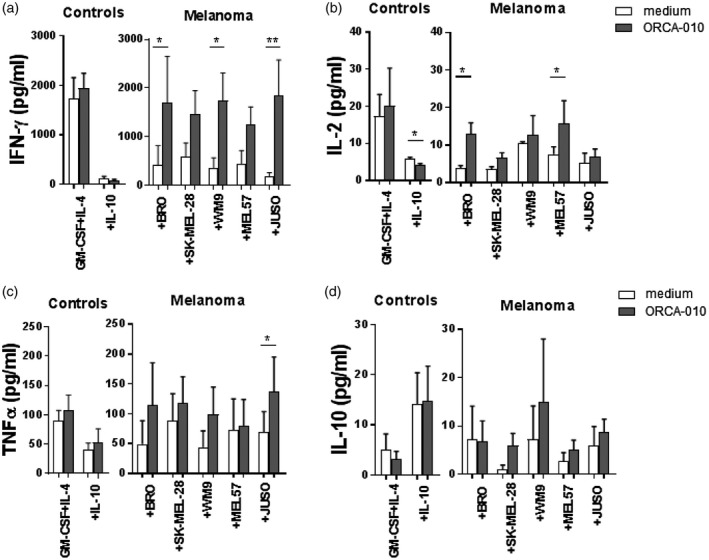
ORCA‐010 counteracts melanoma‐mediated inhibition of the release of type‐1 effector cytokines by monocyte‐derived dendritic cells (moDCs). Day 6 supernatants from the mixed lymphocyte reactions from three independent experiments were collected and cytokines measured. (a) interferon (IFN)‐γ, (b) interleukin (IL)‐2, (c) tumor necrosis factor (TNF)‐α and (d) IL‐10 levels secreted by T cells under the different conditions. Data shown are means ± standard error of the mean (s.e.m.). Statistical significances between cultures with or without oncolytic virus were tested using a matched repeated‐measures one‐way analysis of variance (anova) with Fisher’s least significant difference (LSD) post‐hoc test. *P < 0·05 and **P < 0·01.
Overall, these data demonstrate that ORCA‐010 helps to overcome the immune suppression exerted by the melanoma cells, by promoting activation and functionality of moDCs, which in turn are then better equipped to induce a type‐1 effector response which may contribute to anti‐tumor immunity.
Discussion
DCs represent the first line of defense of our immune system against invaders, and are essential for the induction of a tumor‐specific immune response [9]. Unfortunately, tumors are able to exploit DC plasticity and skew their differentiation towards tolerogenic myeloid regulatory subsets that impact anti‐tumor immunity negatively. In fact, several reports have shown that a lack of DC‐like subsets in the TME can lead to immunotherapy resistance [11, 14]. Under tumor conditions, the main source of DCs does not arise from committed DC precursors, but rather from monocytes recruited to the TME [10]. Melanoma cells release a wide array of molecules that recruit monocytes to the TME where they differentiate into M2‐like suppressive macrophages, mediated by different tumor‐derived suppressive factors and which, in turn, suppress tumor‐specific T cells, while expanding Tregs [18, 40, 41, 42]. Therefore, it is important to develop new therapeutic approaches that will boost the differentiation of monocytes into DCs. Oncolytic virotherapy represents a new attractive therapy approach for cancer treatment, thanks to its ability to induce specific tumor lysis and its capability to prompt an immune response through the in‐situ release of danger signals and tumor (neo‐)antigens [6, 28, 43, 44].
Here, similarly to what had previously been observed by us and others in patient‐derived melanoma samples [15, 45], we showed that melanoma cell lines with varying oncogenic mutations were all able to block the normal monocyte‐to‐dendritic cell differentiation towards a more M2‐like macrophage‐like state. This was clear from a maintained CD14 expression on the monocytes and a failure to up‐regulate CD1a [25, 39, 46], and was further confirmed by lower expression levels of the DC marker CD1c compared to moDC that were unimpeded in their differentiation [47]. In order to establish whether co‐culture with the melanoma cell lines merely resulted in a maintained undifferentiated monocytic state or actually induced skewing of differentiation towards an M2‐like state (suggested by expression of CD163), we also assessed CD16 and CD40 expression. Monocytes are CD14high and CD16low, whereas macrophages are known to co‐express CD14 and CD16 at intermediate levels [48, 49]. Our results showed elevated levels of CD16 and modestly reduced CD40 levels compared to control moDCs, but both up‐regulated compared to levels on baseline monocytes, indicative of macrophage‐like differentiation. Finally, the balance in expression between CD80 and CD163 is generally regarded as a measure for M1 versus M2 macrophage skewing. Clearly, in the melanoma co‐cultures, ORCA‐010 did not impact CD163 expression, but induced up‐regulation of the co‐stimulatory machinery, including CD80, suggestive of a more M1‐like differentiation [50]. Although modest and non‐significant, observed ORCA‐010‐induced changes in IL‐12p70 release by the monocyte‐derived APCs upon exposure to the BRO or SK‐MEL‐28 cell lines supported this notion [51, 52]. Certainly, inclusion of ORCA‐010 in the co‐cultures translated into a generally improved allogeneic T cell priming by the resulting APCs, accompanied by a clear shift to a type‐1 T cell cytokine release profile. In vivo, this should translate into proinflammatory conditions in the melanoma TME that would favor the induction of cell‐mediated anti‐tumor immunity.
The observed phenotype of CD1alow/CD1clow/CD80low/CD14+/CD16+/CD163+ macrophage‐like cells is consistent with previous observations in in‐vitro cultures and clinical melanoma specimens [53, 54, 55, 56]. Several reports have shown that the main players in this impairment of DC differentiation are soluble factors such as IL‐10 or IL‐6 [15, 57]; indeed, Transwell experiments showed soluble factors in the melanoma/moDC co‐cultures to be responsible for the observed inhibition of moDC differentiation. However, neither IL‐6 nor IL‐10 were secreted by the established melanoma cell lines (Table 2) used in the monocyte co‐cultures, indicating other factors to be involved. For instance, IL‐8, found at high concentrations in all the melanoma cell line supernatants (Table 2), was recently shown to participate in the skewing of monocytes to M2‐like macrophages [58]. Additionally, the melanoma cell lines were able to secrete relatively high levels of VEGF (Table 2) which has been described to interfere with the ability to generate functional mature DCs through the inhibition of nuclear factor kappa B (NF‐κB) pathway [59]. Notably, WM9 was the only cell line that did not secrete VEGF, corresponding to its lower capacity to inhibit moDC differentiation. Alternatively, cross‐talk between melanoma and the monocytes may have resulted in monocytes secreting the factors suppressing their differentiation to DCs in an autocrine fashion. Indeed, tumor‐infiltrating myeloid cells have been identified as major mediators of immune suppression [60, 61, 62, 63]. Of note, we were unable to relate the observed DC suppression to a particular activated oncogenic signaling pathway or to the origins of the tested melanoma cell lines.
The oncolytic adenovirus ORCA‐010 was able to induce tumor cell death and to activate the suppressed moDCs, evidenced by the up‐regulation of CD80, CD86 and CD40 co‐stimulatory markers as well as of HLA‐DR and HLA‐ABC antigen‐presenting molecules. Theoretically, this could have been achieved by binding to pattern recognition receptors by virus‐derived motifs or by cell damage‐associated motifs resulting from oncolysis. The fact that ORCA‐010 alone could not activate moDCs (Fig. 4c) strongly suggests the latter to be the case. Indeed, it has previously been shown that adenoviral infection of tumor cells is able to activate autophagy together with necroptotic and pyroptotic cell death, leading to the release of DAMPs such as ATP and HMGB1, with the ability to directly trigger TLR‐mediated maturation of DC [64]. This DC activation can occur relatively quickly [65]. Although both TLR‐ and STING‐L have been implicated in adenovirus‐mediated immune activation [30, 31, 32], at present the actual underlying mechanism for ORCA‐010‐mediated activation remains obscure. Rather than restoring DC differentiation, ORCA‐010 activated the monocyte‐derived APCs and the apparent shift in balance in CD80 and CD163 suggested that they adopted an M1‐like macrophage phenotype. Indeed, treatment with an oncolytic adenovirus in glioma resulted in a dense accumulation of M1 macrophages, which proved conducive to anti‐tumor immunity [66]. This is in keeping with our own in‐vitro findings of enhanced priming of type‐1 effector T cells.
The observed proinflammatory immunity bolstering capacity of ORCA‐010, together with its intrinsic ability to induce specific tumor cell lysis, makes it a promising therapeutic tool for patients with melanoma. Intratumoral administration of ORCA‐010 could conceivably lead to in‐vivo vaccination at the tumor site, priming or boosting anti‐tumor immunity that could, in the end, provide tumor control not only at the site of injection, but also at distal metastatic sites. Moreover, ORCA‐010 may be armed to produce immune modulatory proteins specifically at the tumor site, which could promote further DC development and recruitment or activate and mobilize the host immune system in other ways [67, 68]. As such, ORCA‐010 may prove to be a valuable and versatile addition to the therapeutic arsenal in the fight against melanoma, and cancer in general.
Disclosures
W. D. is employed by ORCA Therapeutics; V. W. v. B. serves as CSO for ORCA Therapeutics.
Author contributions
M. L. G. designed and performed research, analyzed and interpreted data, and wrote the manuscript; H. d. H. and J. v. E. v. S. performed research and collected and analyzed data; W. D. provided materials and designed research; R. v. d. V. and V. W. v. B. designed research and interpreted data; T. D. d. G. designed research, analyzed and interpreted data and wrote the manuscript. All authors read, edited and approved the manuscript.
Supporting information
Fig. S1. Melanoma cell lines suppress monocyte‐to‐dendritic cell differentiation. Monocytes were phenotyped at day 0 or cultured for 6 days in different culture conditions and phenotyped after culture. The conditions shown are: day 0 uncultured monocytes (monocytes), monocytes cultured in the presence of GM‐CSF+IL‐4 (GM4), GM‐CSF+IL‐4+IL‐10 (+IL‐10), GM‐CSF+IL‐4+BRO (+BRO), GM‐CSF+IL‐4+SK‐MEL‐28 (+SK‐MEL‐28), GM‐CSF+IL‐4+WM9 (+WM9), GM‐CSF+IL‐4+MEL57 (+MEL57), and GM‐CSF+IL‐4+ JUSO (+JUSO). Monocytes are shown in black bars; GM4 and +IL‐10 conditions are represented by white bars; and melanoma‐conditioned moDCs by gray bars. Statistical significances are shown compared to the GM4 condition applying a matched RM 1‐way ANOVA test with Fisher’s LSD post‐hoc test. (a) Average geometrical mean intensity of the surface marker CD40, and (b) average percentage of CD16+ monocytes or conditioned moDCs, n = 3. * = P < 0.05, and ** = P < 0.01. (c) Average percentage of 3 independent experiments of confluent cells for the 5 different melanoma cell lines (SK‐MEL‐28, JUSO, MEL57, WM9 and BRO) over a growth period of 9 days.
Fig. S2. ORCA‐010 induces activation of melanoma‐inhibited MoDCs. MoDCs were phenotyped after 6 days in mono‐culture (GM4 and +IL‐10) or in co‐culture with melanoma cell lines, in the presence or absence of ORCA‐010. (a) Representative histograms of the geometric mean intensity expression of CD80 and CD86 in the GM4, BRO or BRO plus ORCA‐010 conditions. (b) Percentage of CD80+ and CD86+ expressed on the differently conditioned MoDCs. Individual data from three independent experiments are shown. Statistical significances between conditions with and without oncolytic virus were tested using a paired T‐test. * = P < 0.05, and ** = P < 0.01.
Fig. S3. Effects of ORCA‐010 on cytokine release by melanoma‐conditioned monocyte‐derived APCs. MoDCs that had been cultured for 6 days with or without melanoma cells, with or without ORCA‐010 infection, were freshly stimulated with J558 (expressing CD40L)‐conditioned medium and INF‐γ. After 24h, supernatants were collected and the released cytokines were analyzed. (a) IL‐10, and (b) IL‐12‐p70 released by the differently conditioned MoDCs; data shown are means ± SEM of three independent experiments. No significant differences were observed when comparing the presence or absence of the oncolytic virus within each condition using a paired T‐test.
Acknowledgements
The authors thank Elisabetta Michielon and Sinead M. Lougheed for technical assistance. This study was supported by the European Union’s Horizon 2020 research and innovation program under grant agreement no. 643130.
References
- 1. Hollestein LM, van den Akker SA, Nijsten T, Karim‐Kos HE, Coebergh JW, de Vries E. Trends of cutaneous melanoma in The Netherlands: increasing incidence rates among all Breslow thickness categories and rising mortality rates since 1989. Ann Oncol 2012; 23:524–30. [DOI] [PubMed] [Google Scholar]
- 2. Kaufman HL, Kirkwood JM, Hodi FS et al The Society for Immunotherapy of Cancer consensus statement on tumour immunotherapy for the treatment of cutaneous melanoma. Nat Rev Clin Oncol 2013; 10:588–98. [DOI] [PubMed] [Google Scholar]
- 3. Chapman PB, Hauschild A, Robert C et al Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011; 364:2507–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Flaherty KT, Infante JR, Daud A et al Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med 2012; 367:1694–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Uusi‐Kerttula H, Hulin‐Curtis S, Davies J, Parker AL. Oncolytic adenovirus: strategies and insights for vector design and immuno‐oncolytic applications. Viruses 2015; 7:6009–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Larson C, Oronsky B, Scicinski J et al Going viral: a review of replication‐selective oncolytic adenoviruses. Oncotarget 2015; 6:19976–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ribas A, Dummer R, Puzanov I et al Oncolytic virotherapy promotes intratumoral T cell infiltration and improves anti‐PD‐1 immunotherapy. Cell 2017; 170:1109–19 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Spranger S, Luke JJ, Bao R et al Density of immunogenic antigens does not explain the presence or absence of the T‐cell‐inflamed tumor microenvironment in melanoma. Proc Natl Acad Sci U S A 2016; 113:E7759–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lanzavecchia A, Sallusto F. Regulation of T cell immunity by dendritic cells. Cell 2001; 106:263–6. [DOI] [PubMed] [Google Scholar]
- 10. Gabrilovich D. Mechanisms and functional significance of tumour‐induced dendritic‐cell defects. Nat Rev Immunol 2004; 4:941–52. [DOI] [PubMed] [Google Scholar]
- 11. Spranger S, Bao R, Gajewski TF. Melanoma‐intrinsic beta‐catenin signalling prevents anti‐tumour immunity. Nature 2015; 523:231–5. [DOI] [PubMed] [Google Scholar]
- 12. Bottcher JP, Bonavita E, Chakravarty P et al Cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune Control. Cell 2018; 172:1022–37 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Luke JJ, Bao R, Sweis RF, Spranger S, Gajewski TF. WNT/β‐catenin pathway activation correlates with immune exclusion across human cancers. Clin Cancer Res 2019; 25:3074–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Spranger S, Dai D, Horton B, Gajewski TF. Tumor‐residing Batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer Cell 2017; 31:711–23 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. van de Ven R, Lindenberg JJ, Oosterhoff D, de Gruijl TD. Dendritic cell plasticity in tumor‐conditioned skin: CD14(+) cells at the cross‐roads of immune activation and suppression. Front Immunol 2013; 4:403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang S, Yang J, Qian J, Wezeman M, Kwak LW, Yi Q. Tumor evasion of the immune system: inhibiting p38 MAPK signaling restores the function of dendritic cells in multiple myeloma. Blood 2006; 107:2432–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Trempolec N, Dave‐Coll N, Nebreda AR. SnapShot: p38 MAPK signaling. Cell 2013; 152:656–e1. [DOI] [PubMed] [Google Scholar]
- 18. He J, Liu Z, Zheng Y et al p38 MAPK in myeloma cells regulates osteoclast and osteoblast activity and induces bone destruction. Cancer Res 2012; 72:6393–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Murray PJ, Allen JE, Biswas SK et al Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 2014; 41:14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Duff SE, Li C, Jeziorska M et al Vascular endothelial growth factors C and D and lymphangiogenesis in gastrointestinal tract malignancy. Br J Cancer 2003; 89:426–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Koga M, Kai H, Egami K et al Mutant MCP‐1 therapy inhibits tumor angiogenesis and growth of malignant melanoma in mice. Biochem Biophys Res Commun 2008; 365:279–84. [DOI] [PubMed] [Google Scholar]
- 22. Fujimura T, Kakizaki A, Furudate S, Kambayashi Y, Aiba S. Tumor‐associated macrophages in skin: How to treat their heterogeneity and plasticity. J Dermatol Sci 2016; 83:167–73. [DOI] [PubMed] [Google Scholar]
- 23. Furudate S, Fujimura T, Kambayashi Y, Kakizaki A, Hidaka T, Aiba S. Immunomodulatory effect of imiquimod through CCL22 produced by tumor‐associated macrophages in B16F10 melanomas. Anticancer Res 2017; 37:3461–71. [DOI] [PubMed] [Google Scholar]
- 24. Getts DR, Turley DM, Smith CE et al Tolerance induced by apoptotic antigen‐coupled leukocytes is induced by PD‐L1+ and IL‐10‐producing splenic macrophages and maintained by T regulatory cells. J Immunol 2011; 187:2405–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lindenberg JJ, van de Ven R, Lougheed SM et al Functional characterization of a STAT3‐dependent dendritic cell‐derived CD14(+) cell population arising upon IL‐10‐driven maturation. Oncoimmunology 2013; 2:e23837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ma Y, Shurin GV, Gutkin DW, Shurin MR. Tumor associated regulatory dendritic cells. Semin Cancer Biol 2012; 22:298–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shurin MR, Naiditch H, Zhong H, Shurin GV. Regulatory dendritic cells: new targets for cancer immunotherapy. Cancer Biol Ther 2011; 11:988–92. [DOI] [PubMed] [Google Scholar]
- 28. Loskog A. Immunostimulatory gene therapy using oncolytic viruses as vehicles. Viruses 2015; 7:5780–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tormo D, Ferrer A, Bosch P et al Therapeutic efficacy of antigen‐specific vaccination and Toll‐like receptor stimulation against established transplanted and autochthonous melanoma in mice. Cancer Res 2006; 66:5427–35. [DOI] [PubMed] [Google Scholar]
- 30. Appledorn DM, Patial S, McBride A et al Adenovirus vector‐induced innate inflammatory mediators, MAPK signaling, as well as adaptive immune responses are dependent upon both TLR2 and TLR9 in vivo . J Immunol 2008; 181:2134–44. [DOI] [PubMed] [Google Scholar]
- 31. Zhu J, Huang X, Yang Y. Innate immune response to adenoviral vectors is mediated by both Toll‐like receptor‐dependent and ‐independent pathways. J Virol 2007; 81:3170–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nociari M, Ocheretina O, Schoggins JW, Falck‐Pedersen E. Sensing infection by adenovirus: Toll‐like receptor‐independent viral DNA recognition signals activation of the interferon regulatory factor 3 master regulator. J Virol 2007; 81:4145–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dong W, van Ginkel JW, Au KY, Alemany R, Meulenberg JJ, van Beusechem VW. ORCA‐010, a novel potency‐enhanced oncolytic adenovirus, exerts strong antitumor activity in preclinical models. Hum Gene Ther 2014; 25:897–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Herlyn D, Iliopoulos D, Jensen PJ et al In vitro properties of human melanoma cells metastatic in nude mice. Cancer Res 1990; 50:2296–302. [PubMed] [Google Scholar]
- 35. van Leeuwen A, Schrier PI, Giphart MJ et al TCA: a polymorphic genetic marker in leukemias and melanoma cell lines. Blood 1986; 67:1139–42. [PubMed] [Google Scholar]
- 36. Wach F, Eyrich AM, Wustrow T, Krieg T, Hein R. Comparison of migration and invasiveness of epithelial tumor and melanoma cells in vitro . J Dermatol Sci 1996; 12:118–26. [DOI] [PubMed] [Google Scholar]
- 37. Lockshin A, Giovanella BC, De Ipolyi PD et al Exceptional lethality for nude mice of cells derived from a primary human melanoma. Cancer Res 1985; 45:345–50. [PubMed] [Google Scholar]
- 38. Buelens C, Verhasselt V, De Groote D, Thielemans K, Goldman M, Willems F. Interleukin‐10 prevents the generation of dendritic cells from human peripheral blood mononuclear cells cultured with interleukin‐4 and granulocyte/macrophage‐colony‐stimulating factor. Eur J Immunol 1997; 27:756–62. [DOI] [PubMed] [Google Scholar]
- 39. Oosterhoff D, Lougheed S, van de Ven R et al Tumor‐mediated inhibition of human dendritic cell differentiation and function is consistently counteracted by combined p38 MAPK and STAT3 inhibition. Oncoimmunology 2012; 1:649–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jakubzick CV, Randolph GJ, Henson PM. Monocyte differentiation and antigen‐presenting functions. Nat Rev Immunol 2017; 17:349–62. [DOI] [PubMed] [Google Scholar]
- 41. Ma Y, Adjemian S, Mattarollo SR et al Anticancer chemotherapy‐induced intratumoral recruitment and differentiation of antigen‐presenting cells. Immunity 2013; 38:729–41. [DOI] [PubMed] [Google Scholar]
- 42. Anzai A, Choi JL, He S et al The infarcted myocardium solicits GM‐CSF for the detrimental oversupply of inflammatory leukocytes. J Exp Med 2017; 214:3293–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vassilev L, Ranki T, Joensuu T et al Repeated intratumoral administration of ONCOS‐102 leads to systemic antitumor CD8(+) T‐cell response and robust cellular and transcriptional immune activation at tumor site in a patient with ovarian cancer. Oncoimmunology 2015; 4:e1017702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Oh DS, Kim TH, Lee HK. Differential role of anti‐viral sensing pathway for the production of type I interferon beta in dendritic cells and macrophages against respiratory syncytial virus A2 strain infection. Viruses 2019; 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Biswas SK, Allavena P, Mantovani A. Tumor‐associated macrophages: functional diversity, clinical significance, and open questions. Semin Immunopathol 2013; 35:585–600. [DOI] [PubMed] [Google Scholar]
- 46. Lindenberg JJ, van de Ven R, Oosterhoff D et al Induction of dendritic cell maturation in the skin microenvironment by soluble factors derived from colon carcinoma. Hum Vaccin Immunother 2014; 10:1622–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dyduch G, Tyrak KE, Glajcar A, Szpor J, Ulatowska‐Bialas M, Okon K. Melanomas and dysplastic nevi differ in epidermal CD1c+ dendritic cell count. Biomed Res Int 2017; 2017:6803756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Aldo PB, Racicot K, Craviero V, Guller S, Romero R, Mor G. Trophoblast induces monocyte differentiation into CD14+/CD16+ macrophages. Am J Reprod Immunol 2014; 72:270–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Grimm M, Feyen O, Coy JF, Hofmann H, Teriete P, Reinert S. Analysis of circulating CD14+/CD16+ monocyte‐derived macrophages (MDMs) in the peripheral blood of patients with oral squamous cell carcinoma. Oral Surg Oral Med Oral Pathol Oral Radiol 2016; 121:301–6. [DOI] [PubMed] [Google Scholar]
- 50. Jaguin M, Houlbert N, Fardel O, Lecureur V. Polarization profiles of human M‐CSF‐generated macrophages and comparison of M1‐markers in classically activated macrophages from GM‐CSF and M‐CSF origin. Cell Immunol 2013; 281:51–61. [DOI] [PubMed] [Google Scholar]
- 51. Lichtenegger FS, Mueller K, Otte B et al CD86 and IL‐12p70 are key players for T helper 1 polarization and natural killer cell activation by Toll‐like receptor‐induced dendritic cells. PLOS ONE 2012; 7:e44266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Dannenmann SR, Thielicke J, Stockli M et al Tumor‐associated macrophages subvert T‐cell function and correlate with reduced survival in clear cell renal cell carcinoma. Oncoimmunology 2013; 2:e23562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell 2010; 141:39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Beyer M, Mallmann MR, Xue J et al High‐resolution transcriptome of human macrophages. PLOS ONE 2012; 7:e45466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Vogel DY, Glim JE, Stavenuiter AW et al Human macrophage polarization in vitro: maturation and activation methods compared. Immunobiology 2014; 219:695–703. [DOI] [PubMed] [Google Scholar]
- 56. Gerlini G, Tun‐Kyi A, Dudli C, Burg G, Pimpinelli N, Nestle FO. Metastatic melanoma secreted IL‐10 down‐regulates CD1 molecules on dendritic cells in metastatic tumor lesions. Am J Pathol 2004; 165:1853–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chen J, Ye Y, Liu P et al Suppression of T cells by myeloid‐derived suppressor cells in cancer. Hum Immunol 2017; 78:113–9. [DOI] [PubMed] [Google Scholar]
- 58. Krawczyk KM, Nilsson H, Allaoui R et al Papillary renal cell carcinoma‐derived chemerin, IL‐8, and CXCL16 promote monocyte recruitment and differentiation into foam‐cell macrophages. Lab Invest 2017; 97:1296–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Oyama T, Ran S, Ishida T et al Vascular endothelial growth factor affects dendritic cell maturation through the inhibition of nuclear factor‐kappa B activation in hemopoietic progenitor cells. J Immunol 1998; 160:1224–32. [PubMed] [Google Scholar]
- 60. Ostrand‐Rosenberg S, Sinha P, Beury DW, Clements VK. Cross‐talk between myeloid‐derived suppressor cells (MDSC), macrophages, and dendritic cells enhances tumor‐induced immune suppression. Semin Cancer Biol 2012; 22:275–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Thuwajit C, Ferraresi A, Titone R, Thuwajit P, Isidoro C. The metabolic cross‐talk between epithelial cancer cells and stromal fibroblasts in ovarian cancer progression: Autophagy plays a role. Med Res Rev 2018; 38:1235–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ye J, Wu D, Wu P, Chen Z, Huang J. The cancer stem cell niche: cross talk between cancer stem cells and their microenvironment. Tumour Biol 2014; 35:3945–51. [DOI] [PubMed] [Google Scholar]
- 63. Hui L, Chen Y. Tumor microenvironment: sanctuary of the devil. Cancer Lett 2015; 368:7–13. [DOI] [PubMed] [Google Scholar]
- 64. Ma J, Ramachandran M, Jin C et al Characterization of virus‐mediated immunogenic cancer cell death and the consequences for oncolytic virus‐based immunotherapy of cancer. Cell Death Dis 2020; 11:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Beck B, Dorfel D, Lichtenegger FS et al Effects of TLR agonists on maturation and function of 3‐day dendritic cells from AML patients in complete remission. J Transl Med 2011; 9:151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. van den Bossche WBL, Kleijn A, Teunissen CE et al Oncolytic virotherapy in glioblastoma patients induces a tumor macrophage phenotypic shift leading to an altered glioblastoma microenvironment. Neuro Oncol 2018; 20:1494–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. de Gruijl TD, Janssen AB, van Beusechem VW. Arming oncolytic viruses to leverage antitumor immunity. Expert Opin Biol Ther 2015; 15:959–71. [DOI] [PubMed] [Google Scholar]
- 68. de Graaff P, Govers C, Wichers HJ, Debets R. Consumption of beta‐glucans to spice up T cell treatment of tumors: a review. Expert Opin Biol Ther 2018; 18:1023–40. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Melanoma cell lines suppress monocyte‐to‐dendritic cell differentiation. Monocytes were phenotyped at day 0 or cultured for 6 days in different culture conditions and phenotyped after culture. The conditions shown are: day 0 uncultured monocytes (monocytes), monocytes cultured in the presence of GM‐CSF+IL‐4 (GM4), GM‐CSF+IL‐4+IL‐10 (+IL‐10), GM‐CSF+IL‐4+BRO (+BRO), GM‐CSF+IL‐4+SK‐MEL‐28 (+SK‐MEL‐28), GM‐CSF+IL‐4+WM9 (+WM9), GM‐CSF+IL‐4+MEL57 (+MEL57), and GM‐CSF+IL‐4+ JUSO (+JUSO). Monocytes are shown in black bars; GM4 and +IL‐10 conditions are represented by white bars; and melanoma‐conditioned moDCs by gray bars. Statistical significances are shown compared to the GM4 condition applying a matched RM 1‐way ANOVA test with Fisher’s LSD post‐hoc test. (a) Average geometrical mean intensity of the surface marker CD40, and (b) average percentage of CD16+ monocytes or conditioned moDCs, n = 3. * = P < 0.05, and ** = P < 0.01. (c) Average percentage of 3 independent experiments of confluent cells for the 5 different melanoma cell lines (SK‐MEL‐28, JUSO, MEL57, WM9 and BRO) over a growth period of 9 days.
Fig. S2. ORCA‐010 induces activation of melanoma‐inhibited MoDCs. MoDCs were phenotyped after 6 days in mono‐culture (GM4 and +IL‐10) or in co‐culture with melanoma cell lines, in the presence or absence of ORCA‐010. (a) Representative histograms of the geometric mean intensity expression of CD80 and CD86 in the GM4, BRO or BRO plus ORCA‐010 conditions. (b) Percentage of CD80+ and CD86+ expressed on the differently conditioned MoDCs. Individual data from three independent experiments are shown. Statistical significances between conditions with and without oncolytic virus were tested using a paired T‐test. * = P < 0.05, and ** = P < 0.01.
Fig. S3. Effects of ORCA‐010 on cytokine release by melanoma‐conditioned monocyte‐derived APCs. MoDCs that had been cultured for 6 days with or without melanoma cells, with or without ORCA‐010 infection, were freshly stimulated with J558 (expressing CD40L)‐conditioned medium and INF‐γ. After 24h, supernatants were collected and the released cytokines were analyzed. (a) IL‐10, and (b) IL‐12‐p70 released by the differently conditioned MoDCs; data shown are means ± SEM of three independent experiments. No significant differences were observed when comparing the presence or absence of the oncolytic virus within each condition using a paired T‐test.