

Supplemental digital content is available in the text.
Key Words/Abbreviations: medullary pancreatic carcinoma, POLE mutation, immunotherapy, long-term survival, tumor mutational burden, CRC - colorectal cancer, EC - endometrial carcinoma, MMR - mismatch repair, MPC - medullary pancreatic carcinoma, MSI - microsatellite instability/instable, MSS - microsatellite stability/stable, PC - pancreatic cancer, PDAC - pancreatic ductal adenocarcinoma, SPN - solid-pseudopapillary neoplasm, TIL - tumor-infiltrating lymphocyte, TMB - tumor mutational burden, POLE - polymerase epsilon
Abstract
Medullary pancreatic carcinoma (MPC) is a rare histological variant of pancreatic ductal adenocarcinoma (PDAC). Because of its rarity, data on the molecular background of MPC are limited. Previous studies have shown that a subset of MPCs is microsatellite instable due to mismatch repair deficiency. Here, we present a unique case of a female patient in her 60s who is a long-term survivor after surgery for pancreatic cancer. The patient had a microsatellite stable MPC with a somatic mutation of the polymerase epsilon gene (POLE). Both microsatellite instable and POLE-mutated cancers are usually associated with high tumor mutational burden and antigen load, resulting in a prominent antitumor immune response and overall better survival. The current case illustrates that, in addition to mismatch repair deficiency, MPC can develop because of a somatic POLE mutation, resulting in a tumor with a high tumor mutational burden and leading to a better prognosis compared with conventional PDAC. This new finding may have important implications in the management of patients with MPC and calls for further studies on the role of POLE in PDAC.
Pancreatic ductal adenocarcinoma (PDAC) is an aggressive malignancy with an exceptionally poor prognosis.1 The overall 5-year survival is <9% for unresected PDACs and improves up to ~15% to 25% after surgical removal of the tumor.1 Other than surgery with radical intent, current treatment options are limited.1
Targeted “precision” therapies have resulted in improved survival for several cancer types, including lung cancer.2 Despite extensive sequencing studies,3–7 molecular targeted therapies have not been successful in the majority of patients with pancreatic cancer (PC).8,9 There are, however, exceptions. For example, a fraction of patients with PC harboring inactivating mutations in homologous recombination repair genes may benefit from targeted therapies.10 The challenge moving forward is to identify additional subgroups of patients with PC who will similarly benefit from therapies selected based on molecular characteristics in their specific cancer.
Medullary pancreatic carcinoma (MPC) is a rare subtype of PDAC with distinctive morphological and molecular features. Goggins et al11 initially described this variant in 1998 as pancreatic adenocarcinoma associated with DNA replication errors, wild-type KRAS, and distinct histopathological hallmarks including poor differentiation, expanding invasion, extensive necrosis, and a syncytial growth pattern. Because of its rarity, very little is known about the molecular pathology of MPC. The largest study to date, describing 18 MPCs, revealed several prominent characteristics significantly associated with this rare tumor type.12 First, microsatellite instability (MSI) was detected in 22% (4/18) of MPCs, whereas the remaining 78% (14/18) were microsatellite stable (MSS). All 4 MSI cases demonstrated loss of MLH1 expression at the protein level. Second, activating mutations in the KRAS oncogene, observed in >90% of conventional PDACs, were detected in only 33% of the MPCs.1,8,12 Third, a medullary phenotype was significantly associated with family history of any cancer type in first-degree relatives.12 Furthermore, MPC has been reported in a Lynch syndrome patient with a germline MSH2 mutation.13
Overall, mismatch repair (MMR) deficiency, both due to germline and somatic MMR gene inactivation, is strongly associated with a medullary phenotype in PC.11–14 In a recent systematic review, medullary histology of PDAC was shown to be strongly associated with MSI and deficient DNA MMR.15 However, the fact that most MPCs reported in the literature are MSS indicates that other unknown molecular mechanisms play a role in the pathogenesis of this distinctive tumor.
Here, we present a unique case of a patient with an MSS MPC. Sequencing revealed a somatic polymerase epsilon gene (POLE) mutation and a high tumor mutational burden (TMB). We hypothesize that this POLE mutation and the resulting hypermutation are responsible for the medullary phenotype in this MPC. In view of the improved prognosis and potential responsiveness to immunotherapy of POLE-mutated cancers, this new finding has important implications for treatment and prognostication of patients and for our understanding of the pathogenesis of MPC.
CASE REPORT
A female patient in her 60s initially presented with nausea, especially progressive after the consumption of a fat-enriched meal, and feelings of discomfort in the epigastric region. The patient had no prior oncological history, and her family history of cancer was negative. Further investigation by magnetic resonance imaging and positron emission tomography scan revealed a positron emission tomography–positive mass in the pancreatic head (Figs. 1A–C). Fine-needle aspiration cytology was positive for malignancy. The tumor was surgically resected by a Whipple procedure, which showed a well-circumscribed 7-cm tumor with extensive central necrosis located in the pancreatic head. No macroscopic involvement of the duodenum was observed. No lymphovascular or perineural invasion was detected, and all 22 isolated pancreaticoduodenal lymph nodes were free of tumor. Based on the final histopathological assessment, the tumor was classified as stage pT3N0M0R0 (American Joint Committee on Cancer eighth edition) MPC. No adjuvant therapy was given. The most recent abdominal magnetic resonance imaging, performed 5 years after surgery, demonstrated no signs of recurrence or metastasis.
FIGURE 1.
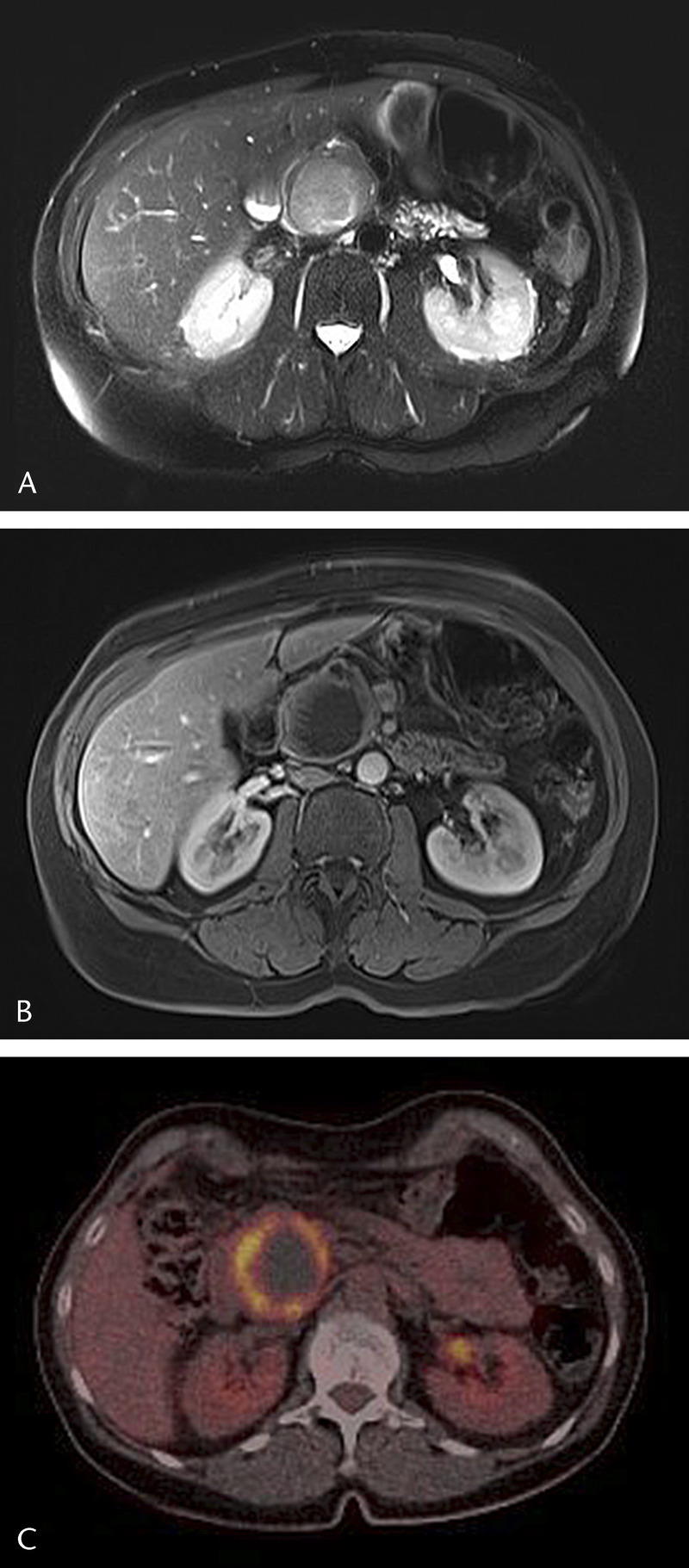
Axial T2-weighted MR image (half-Fourier acquisition single-shot turbo spin echo [HASTE]) with a sharply demarked high signal intensity mass (45 mm) in the pancreatic head (A). Axial T1-weighted image (T1 volumetric interpolated breath-hold examination [T1-VIBE] with fat suppression) after intravenous injection of a contrast agent (gadoterate meglumine) shows enhancement of the wall with some linear papillary projections and a large nonenhancing center, possibly because of necrosis or mucinous component (B). On fluorine-18 fluorodeoxyglucose positron emission tomography (18F-FDG-PET), the wall of the tumor is avid with a large photopenic center (C).
Pathological Findings
Microscopically, the neoplasm was well-demarcated with a pushing border growth pattern and extensive necrosis in the center. A prominent lymphocytic infiltrate surrounded and infiltrated the tumor (Fig. 2A). The tumor was composed of a proliferation of loosely cohesive and solitary cells, sometimes organized around pseudopapillae, reminiscent of a solid-pseudopapillary neoplasm (SPN) (Fig. 2B). The neoplastic cells demonstrated marked cytonuclear atypia and frank mitotic activity (Fig. 2C). In the more solid areas, the tumor showed a syncytial growth pattern with loss of cell boundaries.
FIGURE 2.
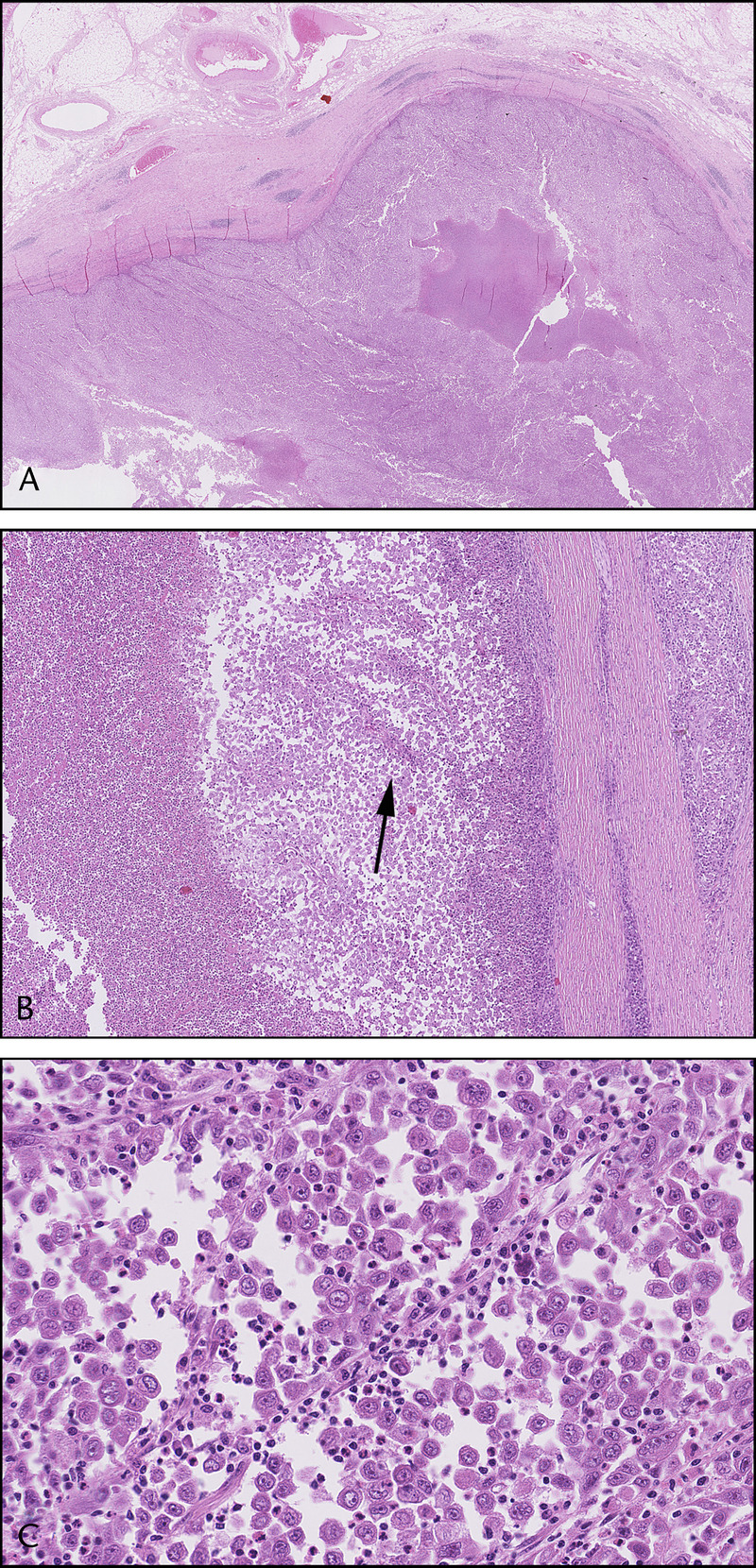
Microscopically, the tumor was characterized by a pushing border growth pattern, extensive central necrosis, and a prominent lymphoid infiltrate surrounding the tumor (A). The tumor was composed of a proliferation of loosely cohesive and solitary cells, sometimes organized around pseudopapillae (arrow), reminiscent of an SPN (B). The tumor cells demonstrated marked cytonuclear atypia and frank mitotic activity (C).
Immunolabeling revealed that the neoplastic cells expressed cytokeratin 7, whereas markers for acinar differentiation (BCL10) and SPN (CD10 and nuclear β-catenin) were negative. Moreover, complete loss of p53 and SMAD4 expression was detected, suggesting genetic inactivation of both genes. All 4 MMR proteins, MLH1, MSH2, MSH6, and PMS2, were expressed, consistent with an MSS tumor.
Immunohistochemical analysis further revealed marked infiltration of CD4+ and CD8+ T lymphocytes, both at the leading edge of the tumor as well as in between of neoplastic cells (Figs. 3A, B). Furthermore, a substantial portion of the tumor-infiltrating lymphocytes (TILs) showed granular cytoplasmatic staining for granzyme B, further confirming their cytotoxic phenotype (Fig. 3C). Immunohistochemical analysis and list of antibodies are described in Supplemental Digital Content (Supplemental Table 1, http://links.lww.com/MPA/A792).
FIGURE 3.
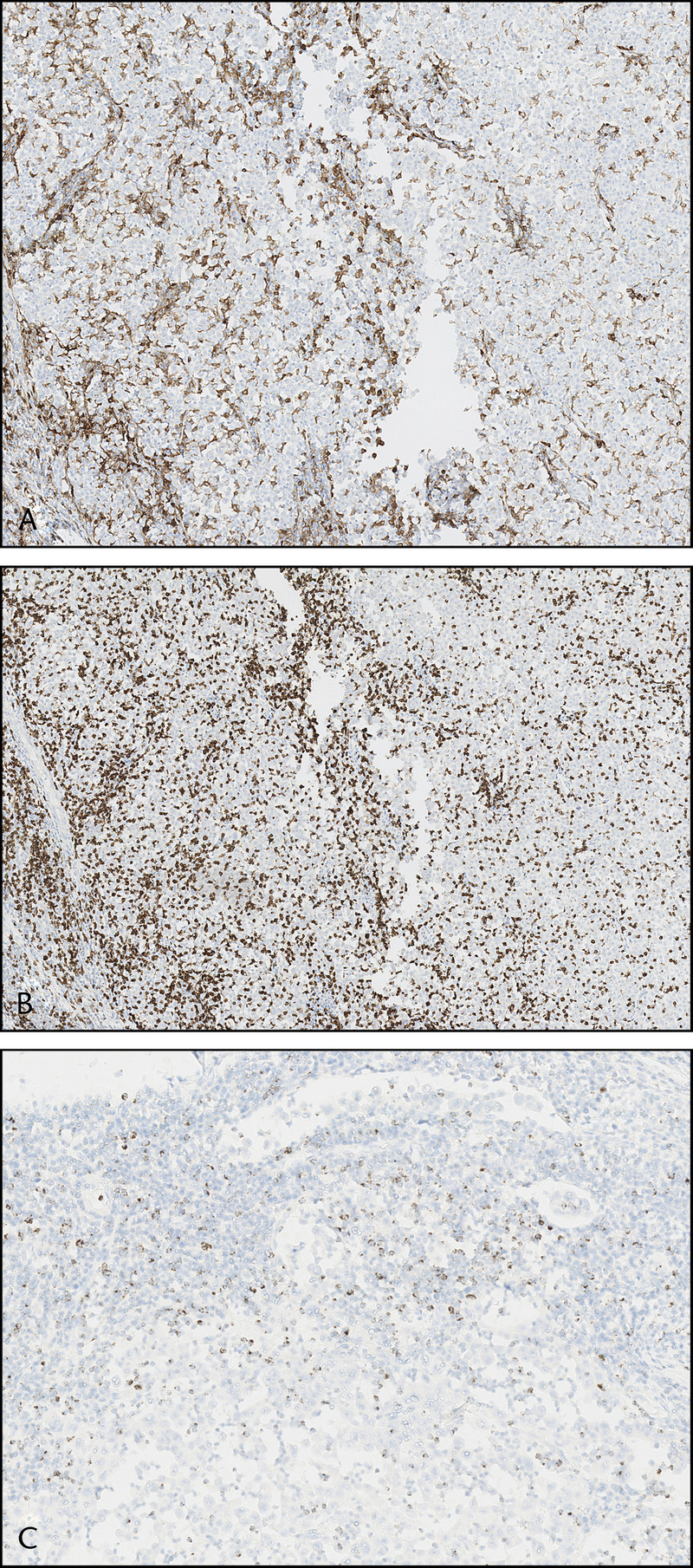
The tumor showed marked infiltration of CD4+ (A) and CD8+ (B) T lymphocytes both at the tumor border and in between of tumor cells. Multiple TILs stained positively for granzyme B (C), indicative of their cytotoxic phenotype.
Despite the lack of MSI, based on morphological grounds and World Health Organization classification, the tumor was signed out as an MPC.
Molecular Findings
Because of the MSS phenotype, targeted next-generation sequencing was performed to unravel the molecular basis of the MPC in our patient, using a custom panel for frequently mutated regions of 30 cancer-related genes, the entire SMAD4 coding region, and 5 mononucleotide MSI markers (Supplemental Digital Content, Supplementary Table 2, http://links.lww.com/MPA/A792).16
All 5 mononucleotide MSI markers included in the next-generation sequencing panel, BAT25, BAT26, NR21, NR24, and NR27, were stable, confirming the MSS phenotype of the tumor. A tumor-specific somatic POLE mutation (NM_006231.3: c.1231G > T or p.Val411Leu; VAF 17%) was detected that is annotated as likely pathogenic by ClinVar.17 This is a known hotspot mutation in the exonuclease proofreading domain of POLE polymerase, resulting in a protein with compromised proofreading activity during DNA replication. In addition, somatic mutations were detected in the following genes: ERBB2 c.929C>T (p.Ser310Phe; VAF 15%), GNAS c.601C>T (p.Arg201Cys; VAF 16%), KRAS c.183A>C (p.Gln61His; VAF 17%), MAP2K1 c.316G>A (p.Ala106Thr; VAF 17%), and TP53 c.637C>T (p.Arg213*; VAF 18%), suggestive of a hypermutation phenotype. No mutations were detected in the hotspot positions of CTNNB1, excluding the differential diagnosis of SPN.18
Tumor mutational burden was assessed using TruSight Oncology 500 (Illumina, Inc, San Diego, Calif) and showed 111 mutations/Mb in the tumor tissue, compared with 1.6 mutations/Mb in the normal nonneoplastic control tissue. This is consistent with a high TMB caused by the POLE mutation. Further analysis revealed a mutational signature (signature 10, COSMIC), known to be associated with POLE mutations (for details, see Fig. 5 in Kroeze et al19; current MPC is represented as UPN40). For details on TMB analysis, see Supplemental Methods, http://links.lww.com/MPA/A792. For the complete list of somatic variants identified in the tumor, see Supplemental Digital Content (Supplementary Table 3, http://links.lww.com/MPA/A792).
DISCUSSION
Here, we present a case of an MSS MPC with a pathogenic somatic POLE mutation leading to a high TMB. Based on the findings in this case report, we hypothesize that a somatic POLE mutation and the resulting hypermutation can be an alternative molecular mechanism, instead of MSI, underlying MPC, resulting in exceptionally improved overall survival.
The current case is the first description of a medullary phenotype observed in a PC with a somatic POLE mutation. In a recent study, Guenther et al,20 examined 115 unselected PDACs but did not identify any hotspot POLE mutations. Moreover, they checked 741 PDAC samples from the publicly available sequencing platform, cbioportal.org, and identified only 1 case with a pathogenic POLE mutation and 2 with possibly damaging variants.20 Histology of the few reported POLE-mutated tumors has not been specified, and POLE mutation is very rare in unselected PC.3 The current case indicates that the medullary phenotype is likely to be a marker for a genetic defect that leads to hypermutation, either by MMR deficiency or by POLE mutation. Therefore, further research is necessary to investigate the role of POLE mutations in MPCs.
The POLE gene encodes the DNA polymerase ε catalytic subunit (Polε), which is a large polymerase involved in the synthesis of the DNA leading strand during replication.21 Mutations in the proofreading domain of POLE lead to DNA repair deficiency characterized by MSS and ultramutated phenotype.22 Interestingly, in contrast to the “two-hit” paradigm of tumor suppressor genes inactivation, similar to other studies,22,23 no second hit by either somatic mutation or loss of heterozygosity has been detected in this MPC. This indicates that a single affected allele is sufficient to hinder the proofreading activity of POLE polymerase and promote POLE-mediated tumorigenesis and high TMB.24 Moreover, a major contribution of mutational signature 10a and 10b, associated with POLE mutations, indicates that somatic monoallelic mutation in the exonuclease domain of POLE is a main mutational process driving the tumorigenesis and responsible for high TMB in this MPC.19 Germline mutations in POLE and POLD1 polymerases predispose to rare polymerase proofreading-associated polyposis, imposing increased risk to develop polyposis, early-onset colorectal cancer (CRC), and endometrial carcinomas (ECs).22 Recently, a large family harboring a novel germline POLE variant was described to have multiple cancers including 3 early-onset PC cases, potentially indicating that PC belongs to the tumor spectrum of polymerase proofreading-associated polyposis.24 Germline POLE mutations were also reported in familial PC patients, and their frequency positively correlated with family history of breast, ovarian, or PC.25
Because of deficient proofreading capacity, POLE-mutated tumors are characterized by an ultramutated phenotype and exceptionally high TMB.26,27 A distinct ultramutated subgroup of CRCs and ECs was indeed shown to harbor somatic POLE mutations.28,29 Consistently, high TMB was detected in addition to the somatic POLE mutation in the current case. Furthermore, it is well established that high TMB results in increased presentation of neoantigens on tumor cells, facilitating the activation of immune cells.30,31 Because of the enhanced activation of the immune system and consequent antitumor immune response, POLE-mutated ECs have excellent prognosis and better survival rates.32–35 Moreover, POLE-deficient CRCs demonstrating increased infiltration with CD8+ lymphocytes exhibited a decreased recurrence risk.36 Indeed, in the current case, a high number of TILs was demonstrated by immunohistochemistry.
Although it is known that patients with MPC have prolonged survival compared with the extremely poor prognosis of conventional PDAC,14,37 a recent review of MPC described that most patients (15/21; 71%) died from their disease, often within 1 year of diagnosis (11/21; 52%).38 Our patient showed a remarkable 5-year disease-free survival, more consistent with the previously stated better prognosis of POLE-mutated tumors. This may indicate that, in the heterogeneous group of MPC, POLE mutation could segregate a unique type of PDAC patients with long-term survival.
Finally, high TMB alone is a known predictor of response to immunotherapies in multiple cancer types.27,39 Interestingly, POLE-mutated cancers were reported to carry an average of 15 times more neoantigens than MSI tumors and 100 times more than MSS tumors.40,41 Therefore, we anticipate that POLE-mutated PC may be very promising target for immunotherapies.
To conclude, we describe an MSS MPC with a somatic POLE mutation. This case indicates that POLE mutations represent a novel molecular mechanism underlying medullary histology in PC that might be particularly sensitive to immunotherapies and could be associated with a better prognosis. This case further highlights that histopathology can provide a clue to the underlying genetic drivers of a neoplasm.
Supplementary Material
ACKNOWLEDGMENTS
For determination of the TMB, TruSight Oncology 500 reagents were made available by Illumina, Inc. We are grateful to Johan Offerhaus for his intellectual contribution and mentorship. We thank Folkert Morsink for his help in preparing the figures.
Footnotes
V.K. and E.t.L. contributed equally to this work.
This study was supported by a grant from the Dutch Cancer Society (KWF) 2016 10289 and the National Institutes of Health CA 62924.
The authors declare no conflict of interest.
This report was written with an approval from the patient.
Supplemental digital contents are available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal’s Web site (www.pancreasjournal.com).
REFERENCES
- 1.Kleeff J, Korc M, Apte M, et al. Pancreatic cancer. Nat Rev Dis Primers. 2016;2:16022. [DOI] [PubMed] [Google Scholar]
- 2.Reck M, Rabe KF. Precision diagnosis and treatment for advanced non–small-cell lung cancer. N Engl J Med. 2017;377:849–861. [DOI] [PubMed] [Google Scholar]
- 3.Cancer Genome Atlas Research Network. Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell. 2017;32:185–203.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Waddell N, Pajic M, Patch AM, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518:495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Witkiewicz AK, McMillan EA, Balaji U, et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun. 2015;6:6744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bailey P, Chang DK, Nones K, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016;531:47–52. [DOI] [PubMed] [Google Scholar]
- 7.Biankin AV, Waddell N, Kassahn KS, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491:399–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dreyer SB, Chang DK, Bailey P, et al. Pancreatic cancer genomes: implications for clinical management and therapeutic development. Clin Cancer Res. 2017;23:1638–1646. [DOI] [PubMed] [Google Scholar]
- 9.Chang DK, Grimmond SM, Biankin AV. Pancreatic cancer genomics. Curr Opin Genet Dev. 2014;24:74–81. [DOI] [PubMed] [Google Scholar]
- 10.Golan T, Hammel P, Reni M, et al. Maintenance olaparib for germline BRCA-mutated metastatic pancreatic Cancer. N Engl J Med. 2019;381:317–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goggins M, Offerhaus GJ, Hilgers W, et al. Pancreatic adenocarcinomas with DNA replication errors (RER+) are associated with wild-type K-ras and characteristic histopathology. Poor differentiation, a syncytial growth pattern, and pushing borders suggest RER+. Am J Pathol. 1998;152:1501–1507. [PMC free article] [PubMed] [Google Scholar]
- 12.Wilentz RE, Goggins M, Redston M, et al. Genetic, immunohistochemical, and clinical features of medullary carcinoma of the pancreas: a newly described and characterized entity. Am J Pathol. 2000;156:1641–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Banville N, Geraghty R, Fox E, et al. Medullary carcinoma of the pancreas in a man with hereditary nonpolyposis colorectal cancer due to a mutation of the MSH2 mismatch repair gene. Hum Pathol. 2006;37:1498–1502. [DOI] [PubMed] [Google Scholar]
- 14.Maple JT, Smyrk TC, Boardman LA, et al. Defective DNA mismatch repair in long-term (≥ 3 years) survivors with pancreatic cancer. Pancreatology. 2005;5:220–228. [DOI] [PubMed] [Google Scholar]
- 15.Luchini C, Brosens LAA, Wood LD, et al. Comprehensive characterisation of pancreatic ductal adenocarcinoma with microsatellite instability: histology, molecular pathology and clinical implications. Gut. 2020 Apr 29[Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eijkelenboom A, Kamping EJ, Kastner-van Raaij AW, et al. Reliable next-generation sequencing of formalin-fixed, paraffin-embedded tissue using single molecule tags. J Mol Diagn. 2016;18:851–863. [DOI] [PubMed] [Google Scholar]
- 17.Landrum MJ, Lee JM, Benson M, et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018;46:D1062–D1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guo M, Luo G, Jin K, et al. Somatic genetic variation in solid pseudopapillary tumor of the pancreas by whole exome sequencing. Int J Mol Sci. 2017;18:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kroeze LI, de Voer RM, Kamping EJ, et al. Evaluation of a hybrid capture-based pan-cancer panel for analysis of treatment stratifying oncogenic aberrations and processes. J Mol Diagn. 2020;22:757–769. [DOI] [PubMed] [Google Scholar]
- 20.Guenther M, Veninga V, Kumbrink J, et al. POLE gene hotspot mutations in advanced pancreatic cancer. J Cancer Res Clin Oncol. 2018;144:2161–2166. [DOI] [PubMed] [Google Scholar]
- 21.Castellucci E, He T, Goldstein DY, et al. DNA polymerase ɛ deficiency leading to an ultramutator phenotype: a novel clinically relevant entity. Oncologist. 2017;22:497–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Palles C, Cazier JB, Howarth KM, et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat Genet. 2013;45:136–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rohlin A, Zagoras T, Nilsson S, et al. A mutation in POLE predisposing to a multi-tumour phenotype. Int J Oncol. 2014;45:77–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hansen MF, Johansen J, Bjørnevoll I, et al. A novel POLE mutation associated with cancers of colon, pancreas, ovaries and small intestine. Fam Cancer. 2015;14:437–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goldstein JB, Zhao L, Javle MM, et al. Characterization of germline genomic alterations in familial pancreas cancer. J Clin Oncol. 2017;35(15 suppl):abstract 4116. [Google Scholar]
- 26.Roberts SA, Gordenin DA. Hypermutation in human cancer genomes: footprints and mechanisms. Nat Rev Cancer. 2014;14:786–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luchini C, Bibeau F, Ligtenberg MJL, et al. ESMO recommendations on microsatellite instability testing for immunotherapy in cancer, and its relationship with PD-1/PD-L1 expression and tumour mutational burden: a systematic review-based approach. Ann Oncol. 2019;30:1232–1243. [DOI] [PubMed] [Google Scholar]
- 28.Kandoth C, Schultz N, Cancer Genome Atlas Research Network et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497:67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Efremova M, Finotello F, Rieder D, et al. Neoantigens generated by individual mutations and their role in cancer immunity and immunotherapy. Front Immunol. 2017;8:1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chabanon RM, Pedrero M, Lefebvre C, et al. Mutational landscape and sensitivity to immune checkpoint blockers. Clin Cancer Res. 2016;22:4309–4321. [DOI] [PubMed] [Google Scholar]
- 32.Bosse T, Nout RA, McAlpine JN, et al. Molecular classification of grade 3 endometrioid endometrial cancers identifies distinct prognostic subgroups. Am J Surg Pathol. 2018;42:561–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Espinosa I, Lee CH, D'Angelo E, et al. Undifferentiated and dedifferentiated endometrial carcinomas with POLE exonuclease domain mutations have a favorable prognosis. Am J Surg Pathol. 2017;41:1121–1128. [DOI] [PubMed] [Google Scholar]
- 34.van Gool IC, Bosse T, Church DN. POLE proofreading mutation, immune response and prognosis in endometrial cancer. Oncoimmunology. 2015;5:e1072675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bellone S, Centritto F, Black J, et al. Polymerase ε (POLE) ultra-mutated tumors induce robust tumor-specific CD4+ T cell responses in endometrial cancer patients. Gynecol Oncol. 2015;138:11–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Domingo E, Freeman-Mills L, Rayner E, et al. Somatic POLE proofreading domain mutation, immune response, and prognosis in colorectal cancer: a retrospective, pooled biomarker study. Lancet Gastroenterol Hepatol. 2016;1:207–216. [DOI] [PubMed] [Google Scholar]
- 37.Schlitter AM, Segler A, Steiger K, et al. Molecular, morphological and survival analysis of 177 resected pancreatic ductal adenocarcinomas (PDACs): identification of prognostic subtypes. Sci Rep. 2017;7:41064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yago A, Furuya M, Mori R, et al. Medullary carcinoma of the pancreas radiologically followed up as a cystic lesion for 9 years: a case report and review of the literature. Surg Case Rep. 2018;4:80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Samstein RM, Lee CH, Shoushtari AN, et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet. 2019;51:202–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Howitt BE, Shukla SA, Sholl LM, et al. Association of Polymerase e-mutated and microsatellite-instable endometrial cancers with neoantigen load, number of tumor-infiltrating lymphocytes, and expression of PD-1 and PD-L1. JAMA Oncol. 2015;1:1319–1323. [DOI] [PubMed] [Google Scholar]
- 41.Shinbrot E, Henninger EE, Weinhold N, et al. Exonuclease mutations in DNA polymerase epsilon reveal replication strand specific mutation patterns and human origins of replication. Genome Res. 2014;24:1740–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.