
Figure 8. Signaling mechanisms of BCR-induced CSR.
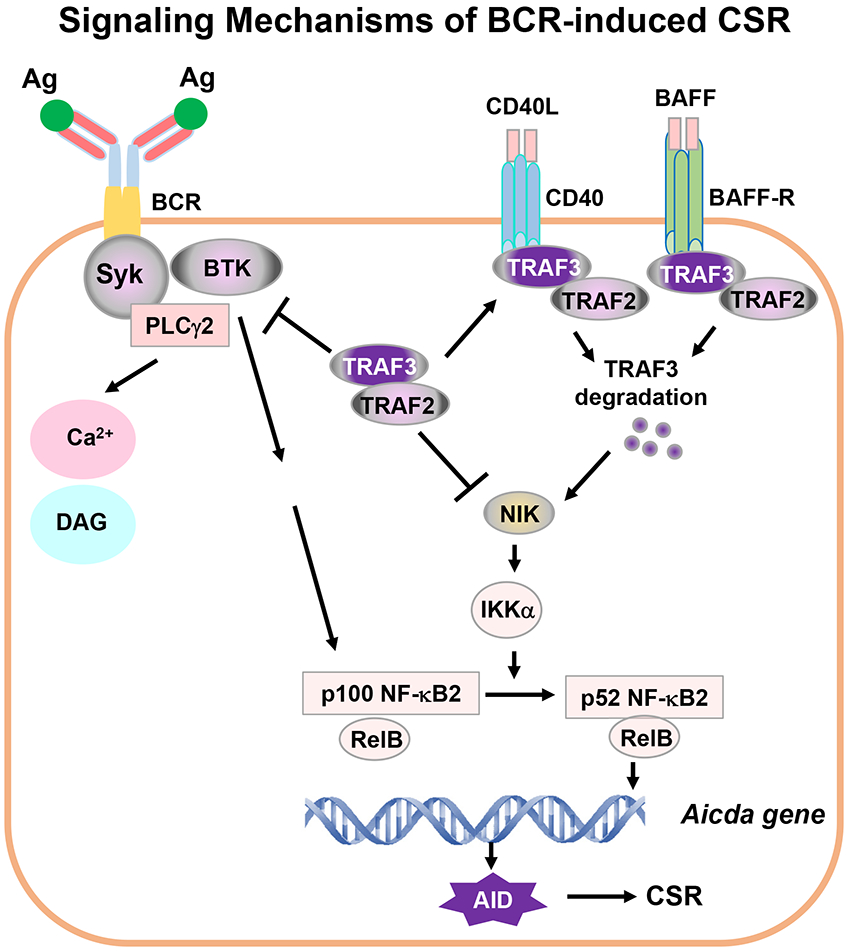
Ag stimulation of BCR activates proximal signaling elements, Syk, BTK and PLCγ2, whose activation triggers Ca2+ flux. Syk/BTK/PLCγ2 complex may activate additional unknown signaling pathway(s) to promote NF-κB2 precursor p100 synthesis. TRAF2 and TRAF3 block NIK activity. Thus, Syk/BTK/PLCγ2 complex cannot activate signaling components downstream of NF-κB2 p100, i.e., cannot activate NF-κB2 transcription factor to initiate AID expression. Removal of TRAF3 and/or TRAF2 leads to constitutively active NF-κB2 p52 via NIK and IKKα pathway. Active NF-κB2 complexes act together with additional factors to initiate AID transcription. AID protein targets Igh locus to induce CSR. During infection or immunization, CD40/CD40L or BAFF/BAFFR interaction recruits TRAF3/TRAF2 to membrane lipid rafts, which eventually causes TRAF3 degradation, a physiological situation of TRAF3-deficiency. TRAF3 degradation results in NIK and NF-κB2 complex activation. NF-κB2 activation is specifically required for the BCR to induce CSR. We propose that one critical function of co-stimulatory signals is to degrade TRAF3 to allow NF-κB2-dependent BCR-induced CSR that is essential for in vivo antibody responses. Of note, TRAF3 not only restricts Syk, BTK and PLCγ2 hyperactivation upon Ag stimulation but also blocks NF-κB2 activation, which may be especially important for maintaining autoreactive B-cell anergy.