

SUMMARY
The RNA N6-methyladenosine (m6A) methylation is installed by the METTL3-METTL14 methyltransferase complex. This modification has critical regulatory roles in various biological processes. Here, we report that deletion of Mettl14 dramatically reduces mRNA m6A methylation in developing B cells and severely blocks B cell development in mice. Deletion of Mettl14 impairs interleukin-7 (IL-7)-induced pro-B cell proliferation and the large-pre-B-to-small-pre-B transition and causes dramatic abnormalities in gene expression programs important for B cell development. Suppression of a group of transcripts by cytoplasmic m6A reader YTHDF2 is critical to the IL-7-induced pro-B cell proliferation. In contrast, the block in the large-pre-B-to-small-pre-B transition is independent of YTHDF1 or YTHDF2 but is associated with a failure to properly upregulate key transcription factors regulating this transition. Our data highlight the important regulatory roles of the RNA m6A methylation and its reader proteins in early B cell development.
Graphical Abstract
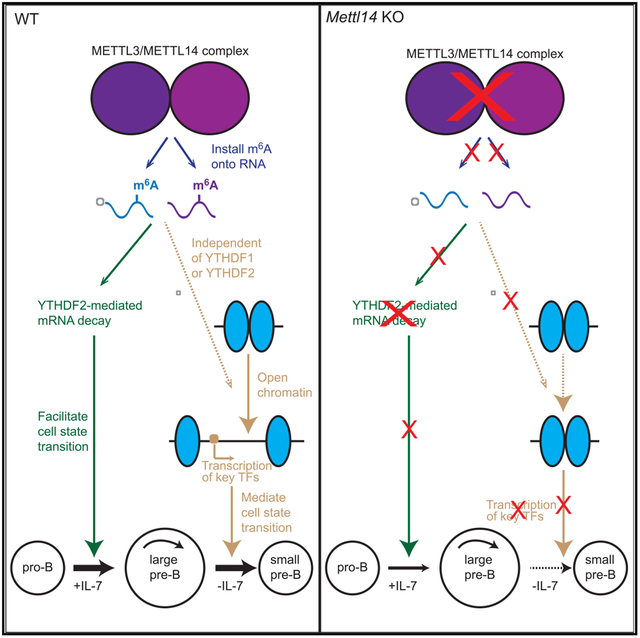
In Brief
Zheng et al. show that B-cell-specific deletion of RNA m6A methylation writer protein METTL14 results in severe defects in B cell development. RNA m6A plays critical roles in regulating gene expressions and controls two major transitions of early B cell development via different mechanisms.
INTRODUCTION
B cell development represents a cellular model system that in-corporates a series of cell fate determination and cell differentiation processes. It is hallmarked by sequential DNA rearrangements of the immunoglobulin heavy (IgH) and light (IgL) chain loci as well as rapid transitions between proliferation and recombination, resulting in a diverse repertoire of peripheral B cells that recognize foreign antigens but are tolerant of self (Clark et al., 2014; Mårtensson et al., 2010). The accurate control of cell state transitions in B lymphopoiesis highly depends on the precise regulation of gene expression in multiple layers. At the gene transcription level, a series of transcription factors (TFs) that directly bind to specific DNA sequence have been well known to sequentially control gene expression, providing the first layer of regulation for cell fate determination and transitions (Busslinger, 2004; Hagman and Lukin, 2006). Epigenetic regulatory pathways involving DNA or histone modifications regulate chromatin accessibility, offering an additional layer of regulation at the transcription level (Barneda-Zahonero et al., 2012; Mandal et al., 2018; Wu et al., 2018). Accumulating evidence indicates that post-transcriptional regulation of RNA represents another vital regulatory layer of gene expression, as RNA binding proteins (RBPs) (Galloway et al., 2016; Inoue et al., 2015; Zhou et al., 2015) and microRNAs (Coffre et al., 2016; Lai et al., 2016; Rodriguez et al., 2007) have been shown to fine-tune gene expression by modulating mRNA degradation, splicing, or translation during B lymphopoiesis. Recently, modifications of RNA have emerged as an additional regulation layer of gene expression, but their roles in B cell development remained unexplored.
m6A methylation is the most prevalent internal modification found in mRNA of eukaryotes (Dominissini et al., 2012; Fu et al., 2014). This modification is installed on mRNA by a dedicated writer methyltransferase complex, with METTL3 and METTL14 forming the catalytic core (Liu et al., 2014; Yue et al., 2015). Several RBPs, including the YTH domain family proteins, have been identified as reader proteins that preferentially recognize the m6A-methylated mRNA and modulate the fate of their target transcripts (Shi et al., 2019). Work from our lab showed that YTHDF2 promotes decay of its m6A-methylated targets (Wang et al., 2014b), and YTHDF1 facilitates translation of the methylated targets (Wang et al., 2015). The physiological roles of m6A and its readers have been identified in various biological processes, such as embryonic stem cell differentiation (Geula et al., 2015), hematopoietic stem cell development (Cheng et al., 2019; Lee et al., 2019; Weng et al., 2018; Zhang et al., 2017), and immune responses (Han et al., 2019; Li et al., 2017a; Liu et al., 2019). These studies collectively provided strong support for the proposed role of m6A as an mRNA chemical mark that allows cells to group hundreds to thousands of transcripts for coordinated modulation in translation or degradation in response to cellular and environmental signals (Roundtree et al., 2017; Zhao et al., 2017a).
Here, we report that the RNA m6A methylation plays essential roles during early B cell development. Conditional deletion of Mettl14 in mouse B-lineage cells causes a dramatic reduction of RNA m6A and severely blocks two major transitions during early B cell development. Our studies highlight crucial regulatory roles of RNA m6A modification in cell state transitions during early B cell development.
RESULTS
METTL14 Deficiency Severely Blocks Early B Cell Development in Mice
To investigate the potential role of RNA m6A in B cells, we generated Mb1cre/+Mettl14fl/fl (Mettl14 knockout [KO]) mice. Compared with the Mb1+/+Mettl14fl/fl or Mb1cre/+Mettl14fl/+ (wild-type [WT]) littermates, Mettl14 KO mice had an over 120-fold reduction in the splenic B cell numbers (Figure 1A) and essentially undetectable B cells in the peritoneal cavity (Figure 1B). Analysis of B cell progenitors in the bone marrow showed that Mettl14 KO mice had a nearly 75% reduction in the percentage of B lineage (CD19+) cells (Figure 1C). Within the CD19+ population, the percentages (Figure 1C) and numbers (Figure 1D) of the immature B cells and the mature B cells were both severely decreased. These data indicate that loss of METTL14 dramatically impairs B cell development.
Figure 1. Loss of METTL14 Severely Blocks Early B Cell Development In Vivo.
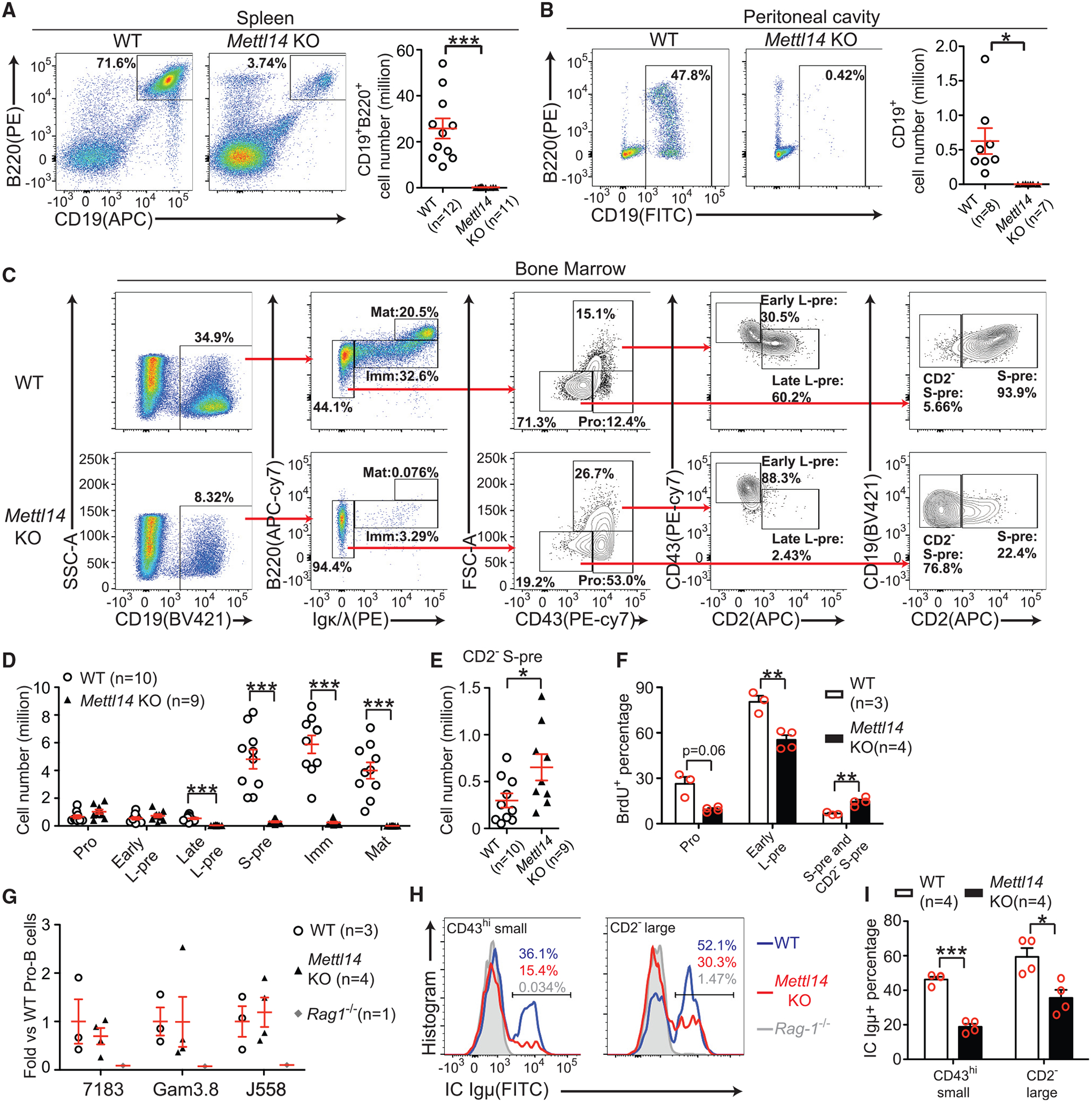
(A and B) Flow cytometry plots and quantifications of B cells in the spleens (A) and the peritoneal cavity (B) of indicated mice. (C andD) Flow cytometry plots (C) and quantifications (D) of indicated populations in the bone marrow of indicated mice. (E) Quantification of the abnormal CD2−small pre-B population of indicated mice. The number of each cell population from two sets of bones (humerus, femur, and tibia) per mouse was calculated (D and E). (F) BrdU (1 mg/mouse) was intraperitoneally injected into mice, and BrdU incorporation in indicated B-lineage cells from indicated mice was analyzed 1 h later. (G) Quantitative PCR of indicated recombined IgH families in the pro-B cells sorted from indicated mice. (H and I) Flow cytometry plots (H) and percentages (I) of intracellular Igμ+ cells in indicated CD19+B220midIgκ/λ− bone marrow subpopulations from indicated mice. SEM or ± SEM is shown; NS, not significant; *, p < 0.05; **, p < 0.01 and ***, p < 0.001.
We further divided the CD19+B220midIgκ/λ− population into pro-B cells, early large pre-B cells, late large pre-B cells, and small pre-B cells (Figure 1C). Our gating scheme is consistent with gating based on other markers (Figures S1A and S1B). Mettl14 KO mice displayed higher portions of CD43hi pro-B cells and large pre-B cells but a much lower portion of CD43lo cells. Whereas WT large pre-B cells contain both early and late populations, Mettl14 KO large pre-B cells lack the late population (Figure 1C). The residual CD43lo cells from the Mettl14 KO mice also downregulated c-Kit (Figure S1C), suggesting that they were a population downstream of the pro-B stage. But the majority of those cells failed to upregulate CD2−or CD25 (Figures 1C and S1C), indicating that they did not reach the small pre-B stage. Quantification of all the subpopulations showed that Mettl14 KO mice had normal numbers of the pro-B cells and the early (CD2−) large pre-B cells, significantly reduced numbers of the late (CD2+) large pre-B cells and the small pre-B cells (CD2+; Figure 1D), and an accumulation of an abnormal CD2−small pre-B population (Figure 1E). To examine how these cellular changes related to the inactivation of the Mettl14 gene, PCR of genomic DNA isolated from various cell populations of Mettl14 KO mice (Figure S1D) showed that, at the earliest pre-pro-B (CD19−B220midIgκ/λ−CD43hi) cell stage when Mb1-cre just starts to be expressed, Mettl14 alleles were still intact. A high proportion of Mettl14 was deleted in the CD43hi pro-B cells, and nearly complete deletion of Mettl14 was seen in the abnormal CD43lo population. The very few remaining immature B cells from the KO mice showed a less efficient deletion, suggesting that “leaky” expression of METTL14 might have allowed them to differentiate to this stage.
Despite the normal cell numbers, the pro-B cells and the early large pre-B cells from the Mettl14 KO mice displayed significantly lower proliferation rates than the respective counterparts from the WT mice (Figure 1F). In contrast, although WT small pre-B cells already exited cell cycle, Mettl14 KO CD43lo small cells remained more proliferative (Figure 1F), further supporting that these cells did not reach the small pre-B stage. Quantitative PCR showed that Mettl14 KO pro-B cells did not have any significant defect in IgH recombination at the DNA level (Figure 1G); however, intracellular staining of Igμ showed that developing B cells from the Mettl14 KO mice had significantly less Igμ+ populations than those from WT mice (Figures 1H and 1I), suggesting that METTL14 deficiency might impair expression of recombined IgH. Altogether, these in vivo data demonstrated that loss of METTL14 caused severe defects during early B cell development.
METTL14 Deficiency Impairs IL-7-Induced Pro-B Cell Proliferation and the Transition to the Large Pre-B Stage
Next, we used well-established in vitro B cell culture system (Kikuchi et al., 2008) to further investigate how METTL14 modulates the major stages of B cell differentiation. WT CD19+Igκ/λ−CD2−cells (representing both pro-B and early large pre-B stages) started to proliferate at day 2 in the presence of interleukin-7 (IL-7), leading to an increase of cell number together with an enlargement of cell size. In contrast, Mettl14 KO group showed an 5- to 10-fold less cell number than WT controls during the expansion, delayed initiation of proliferation, and delayed cell size enlargement (Figures 2A–2C). We confirmed the complete deletion of Mettl14 allele in the KO CD19+Igκ/λ−CD2−cells during the expansion (Figure S2A). The KO cells also displayed a lower proliferation rate during the expansion as measured by bromodeoxyuridine (BrdU) pulse experiment (Figure S2B), which was consistent with the observations in vivo (Figure 1F).
Figure 2. Loss of METTL14 Impairs IL-7-Induced Pro-B Cell Proliferation.
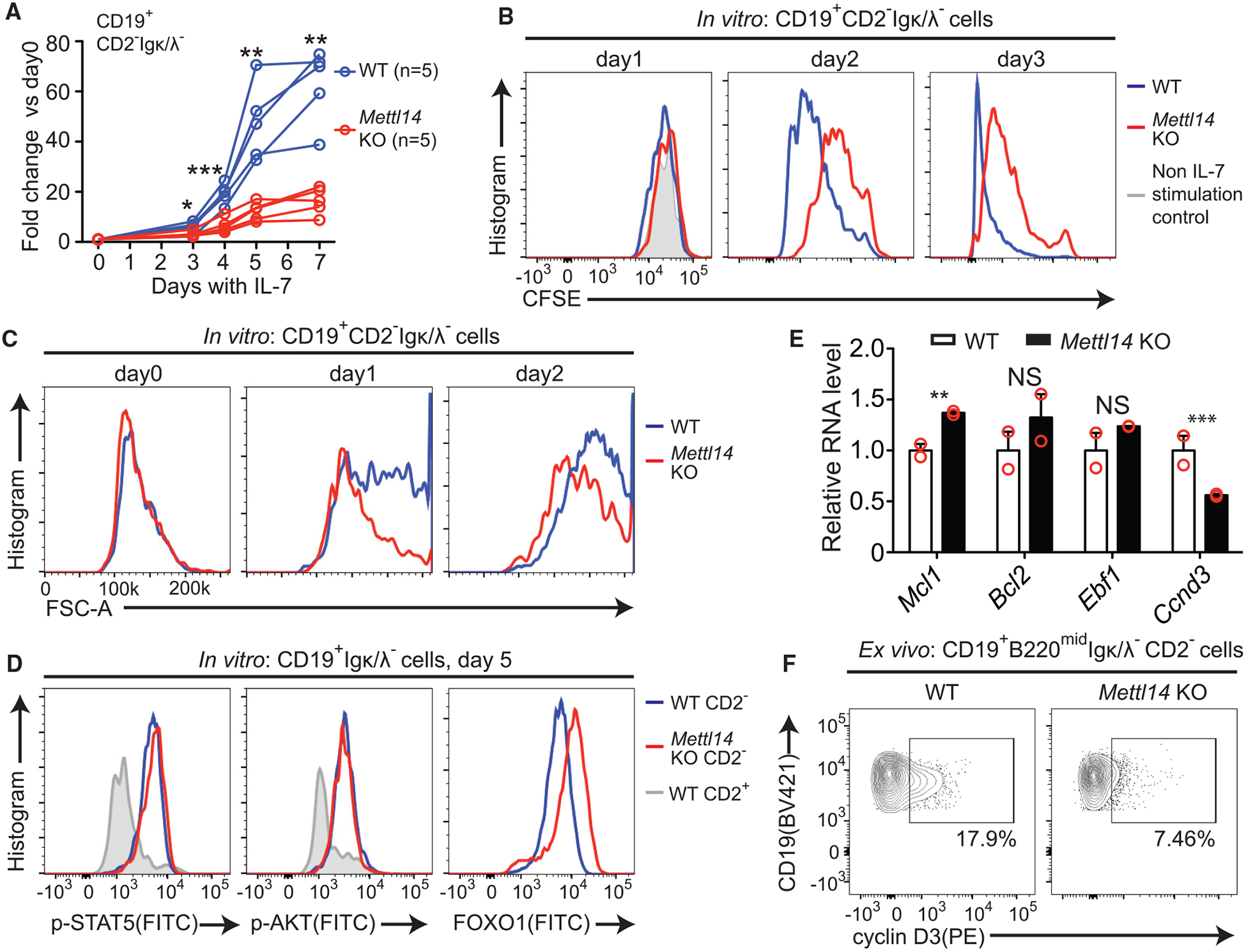
(A) Fold changes of the CD19+CD2−Igκ/λ− cells versus day 0 at indicated times during IL-7-induced expansion are shown. Each line represents an individual mouse. *, p < 0.05; **, p < 0.01 and ***, p < 0.001. (B) CFSE (carboxyfluorescein succinimidyl ester) intensity of the CD19+CD2−Igκ/λ− cells of indicated group at indicated times during IL-7-induced expansion are shown. The same WT cells cultured without IL-7 at day 1 were used as baseline control. Data are representative of two experiments. (C) Forward scatterplots of indicated CD19+CD2−Igκ/λ− cells at indicated times during IL-7-induced expansion. Plots are representative of at least 3 experiments. (D) Intracellular staining of p-STAT5 (left), p-AKT (middle), and FOXO1 in indicated cells after 5-day culture with IL-7. The WT CD19+CD2+ cells from the same culture (gray lines) were used as control for p-STAT5 and p-AKT. Data are representative of at least two experiments. (E) Relative RNA levels of indicated genes in Mettl14 KO large pre-B cells versus WT controls (estimated by RNA-seq; Table S2). NS, not significant; **false discovery rate (FDR) < 0.01; ***FDR < 0.001 (FDRs were calculated as described in STAR Methods). (F) Intracellular staining of cyclin D3 in the CD19+B220midCD2−Igκ/λ− cells from indicated mice. Data are representative of 4 pairs of mice.
Although the pro-B/early large pre-B cells from the KO mice displayed a slight reduction in IL-7 receptor expression (Figure S2C), intracellular staining showed that phosphorylation of STAT5, the key driver of pro-B cell proliferation downstream of IL-7 receptor (Yao et al., 2006), was normal in the Mettl14 KO cells under IL-7 stimulation (Figures 2D and S2D). Phosphorylation of AKT (p-AKT) is also coupled with IL-7 stimulation in the large pre-B cells, which is essential for tuning down B cell receptor (BCR) recombination machinery by suppressing FOXO1 (Ochiai et al., 2012). In vitro expanded Mettl14 KO large pre-B cells had a similar level of p-AKT as WT controls but displayed a higher level of FOXO1 (Figure 2D), suggesting that the IL-7-induced p-AKT is unable to properly downregulate FOXO1 in the absence of METTL14 at the large pre-B stage.
High-throughput RNA sequencing (RNA-seq) showed that, before IL-7 stimulation, ex vivo sorted pro-B cells from Mettl14 KO mice displayed relatively small differences in the transcriptome compared with the WT controls (Figure S3A; Table S1). 6-h post-IL-7 stimulation, more difference in gene expression was detected between the WT and Mettl14 KO pro-B cells, as 4.5% and 8.3% of genes were, respectively, higher or lower in the KO group (Figure S3B; Table S1). Differential expressed genes (DEGs) with lower expression in the Mettl14 KO cells after IL-7 stimulation strongly enrich cell-cycle-related genes (Figure S3C), which is consistent with decreased proliferation observed in these cells (Figure S2B).
As revealed by RNA-seq and proteome quantification by multiplex tandem mass tag labeling with two-dimensional liquid chromatography and tandem mass spectrometry (TMT-LC/LC-MS/MS) (Bai et al., 2017; Tan et al., 2017), Mettl14 KO large B cells that were expanded in the presence of IL-7 for 5 days displayed dramatic alterations in gene expression at both mRNA (Figure S3D; Table S2) and protein levels (Figure S3E; Table S3), with good correlation observed between these changes (Figures S3F and S3G). DEGs with lower expression in the Mettl14 KO cells were also strongly enriched with cell-cycle-related genes (Figure S3H). Both transcriptome and proteome analysis confirmed that the IL-7/STAT5 signaling cascade was intact in the Mettl14 KO large pre-B cells, as the expression induction of the key STAT5-regulated genes Mcl1, Bcl2 (Malin et al., 2010), and Ebf1 (Timblin and Schlissel, 2013) were normal, but the key STAT5-stimulated cyclin essential for the initiation of proliferation in pro-B cells, cyclin D3 (Cooper et al., 2006; Mandal et al., 2009), was significantly lower in the Mettl14 KO cells at both RNA and protein levels (Figures 2E and S3I). We confirmed that the CD2−pro-B/early large pre-B cells from the Mettl14 KO mice had a lower percentage of cyclin-D3-expressing cells in vivo (Figure 2F). Notably, Foxo1 and BCR recombination effectors Rag1, Rag2, and Dntt were higher in the KO group (Figures S3J and S3K). These data suggest that loss of METTL14 results in abnormal regulation of genes involved in both cell cycle and BCR recombination machinery in the large pre-B cells.
METTL14 Deficiency Impairs the Large-Pre-B-to-Small-Pre-B Transition
To further understand how METTL14 deficiency blocks the transition from the CD2−stage to the CD2+ stage, we withdrew IL-7 from the in vitro expanded large pre-B cells (Mandal et al., 2009; Ochiai et al., 2012). After IL-7 removal, a large portion of the WT large pre-B cells differentiated to CD2+ small pre-B cells and Igκ/λ+ immature B cells, but Mettl14 KO large pre-B cells were unable to do so (Figures 3A–3C), which is consistent with the major block observed in vivo (Figures 1C and 1D). After IL-7 removal, 90% of WT pre-B cells dramatically decreased their sizes and downregulated CD43 expression. In contrast, only 60% of Mettl14 KO pre-B cells underwent the same change (Figure S4A). Mettl14 KO pre-B cells also remained more proliferative than WT controls after IL-7 removal (Figure 3D), indicating that METTL14 deficiency impairs the exit from cell cycle in large pre-B cells. Furthermore, Mettl14 KO cells were unable to rearrange Igk (Figures 3E and S4B) or downregulate the pre-BCR component VPREB (Figures 3F and S4C), as their WT counterparts do during this transition. These data indicated that METTL14 deficiency totally blocks the differentiation to the small pre-B stage.
Figure 3. Loss of METTL14 Blocks the Large-Pre-B-to-Small-Pre-B Transition.
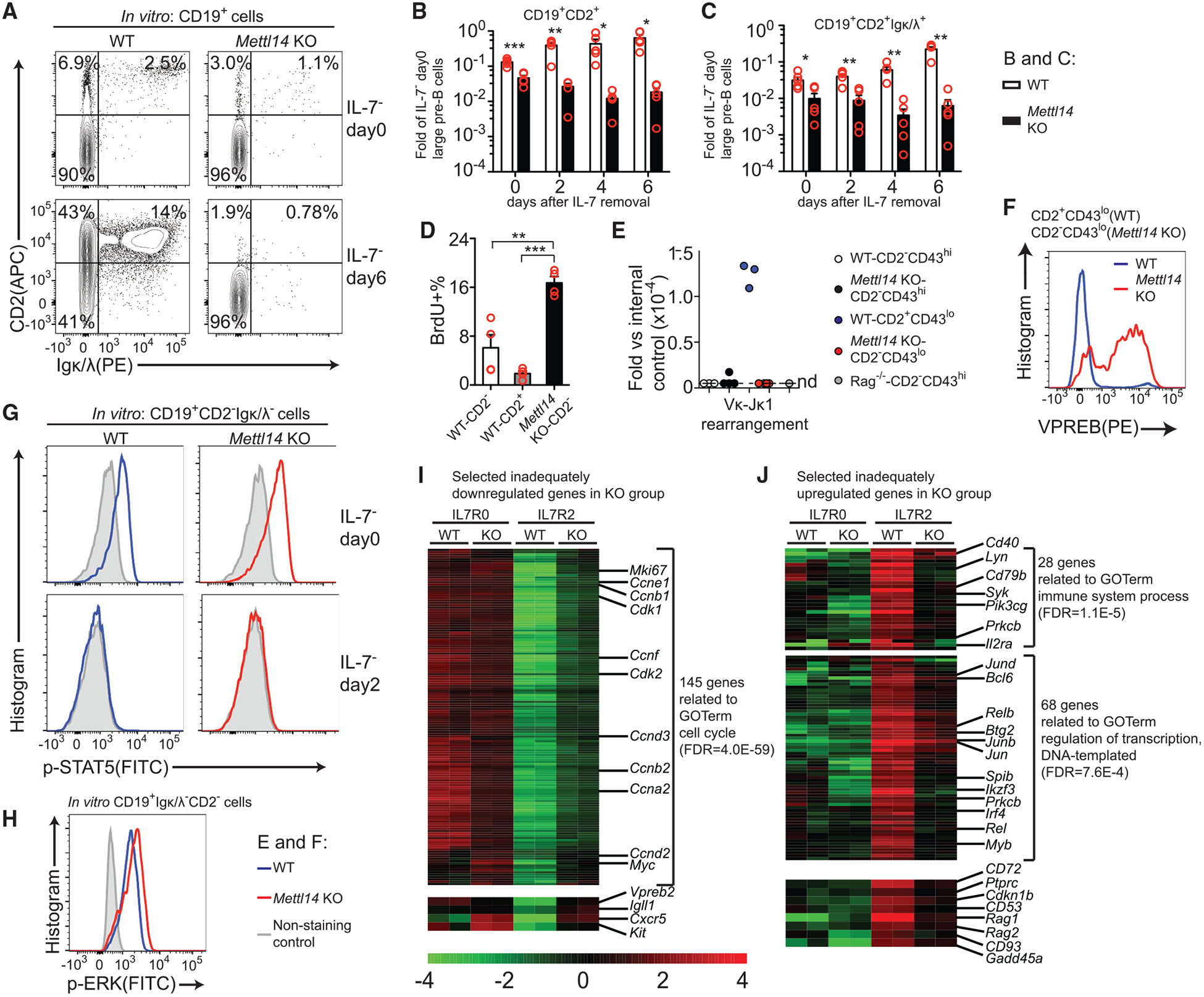
(A) Flow cytometry plots of indicated markers in CD19+ cells of indicated groups before or 6 days after IL-7 removal. Data are representative of at least 5 mice per group. (B and C) Quantification of the CD19+CD2+ cells (B) and the CD19+CD2+Igκ/λ+ cells (C) at indicated times after IL-7 removal (n = 5 per group). Data are normalized with the number of starting CD19+Igκ/λ− CD2−cells before IL-7 removal. (D) BrdU (10 μM) was added to the cell culture for 45 min on day 4 after IL-7 removal. BrdU+ percentages in indicated CD19+Igκ/λ− cells (n = 4 per group) are shown. (E) Quantitative PCR of Vκ1-Jκ1 recombination with the genomic DNAs from indicated CD19+B220midIgκ/λ− subpopulations sorted from indicated mice (n = 3 for WT; n = 4 for Mettl14 KO; n = 1 for Rag1 −/−). Samples below the detection limits are shown on the nd (not detected) line. (F) Intracellular staining of VPREB in indicated CD19+B220midIgκ/λ− subpopulations from indicated mice. Plots are representative of at least 3 mice per group. (G) Intracellular staining of p-STAT5 in indicated in CD19+CD2−Igκ/λ− cells before or 2 days after IL-7 removal. Data are representative of 4 mice per group. (H) Intracellular staining of p-ERK in indicated in vitro expanded large pre-B cells at day 5. Data are representative of 3 mice per group. (I and J) Heatmaps of selected inadequately downregulated (I) and inadequately upregulated (J) genes in the KO large pre-B cells upon IL-7 removal (Table S4). Selected enriched GO terms are used for grouping some genes, and the FDRs are shown after the term names. SEM or ± SEM is shown; NS, not significant; *, p < 0.05; **, p < 0.01 and ***, p < 0.001.
Upon IL-7 removal, Mettl14 KO large pre-B cells showed no defect in p-STAT5 downregulation (Figure 3G), suggesting that loss of METTL14 does not impair the downregulation of the IL-7-induced pathway (Clark et al., 2014). Loss of METTL14 did not cause any detectable defect in the expression of pre-BCR (Figure S4D), and the in vitro cultured Mettl14 KO large pre-B cells constantly showed a higher level of p-ERK than WT controls (Figure 3H), suggesting that the phosphorylation of ERK downstream of the pre-BCR (Mandal et al., 2009) was also intact in the absence of METTL14.
RNA-seq analysis showed that, upon IL-7 removal, although the majority of both upregulated and downregulated genes in the WT cells also followed the same trend in Mettl14 KO cells (Figure S4E), the fold changes in Mettl14 KO group were significantly smaller (median 1.76 versus 2.48 for the activated genes and 1.60 versus 2.20 for the suppressed genes in WT versus KO, respectively; Figure S4F). As a result, after IL-7 removal, 17% (414/2,640) of the upregulated genes showed lower expression in the Mettl14 KO than in WT and 38% (957/2,489) of the downregulated ones showed higher expression in the Mettl14 KO than in WT (Figure S4G; Table S4). Principal-component analysis (PCA) (Figure S4H) also showed how these cell populations are related yet distinct. Parallel results were obtained by comparing the proteomes of WT and Mettl14 KO cells upon IL-7 removal (Figures S4I–S4K).
Gene ontology analysis identified a large group of genes related to cell cycle as well as the pre-BCR components (Vpreb and Igll1) in the inadequately downregulated genes due to METTL14 deficiency (Figure 3I), consistent with that these cells failed to cease proliferation (Figure 3D) or downregulate pre-BCR components (Figures 3F and S4C). The inadequately upregulated genes include key TFs mediating the large-pre-B-to-small-pre-B transition (Ikzf3, Irf4, Spib, and Bcl6), B cell markers (Ptprc, Il2ra, and Cd93), BCR signaling components (Syk, Lyn, and Cd79b), BCR recombination components (Rag1 and Rag2), and proliferation inhibitors (Cdkn1b; Figure 3J). These data demonstrated that Mettl14 KO large pre-B cells are unable to completely switch the gene expression program during the transition from the large pre-B stage to the small pre-B stage.
Pre-arranged IgH or IgL Does Not Rescue B Cell Development in Mettl14 KO Mice
Loss of METTL14 caused a reduction of the Igμ+ pro-B cells (Figures 1H and 1I), leading us to test whether a pre-arranged IgH could rescue the B cell development block in the KO mice by crossing the Mettl14 KO mice with the SWHEL mice (referred to as KO/IgH mice and controls as WT/IgH; Phan et al., 2003). Compared to WT/IgH, KO/IgH mice still had dramatic reduction of B cell numbers in the spleen (Figure 4A). Expression of the pre-arranged IgH did not rescue the block between the CD43hiCD2− early large pre-B cells and the CD43loCD2+ late large pre-B cells (Figure 4C). However, compared to KO, KO/IgH mice showed less severe reductions in the percentages and numbers of the CD19+ cells, the immature and mature B cells, and the CD43lo small “pre-B” population (Figures 4B, 4D, and 4E versus Figures 1C and 1D). By intracellular staining, we confirmed that the pre-arranged IgH corrected the reduction of the Igμ+ population in the pro-B cells of the Mettl14 KO mice (Figure 4F).
Figure 4. Expression of the Pre-arranged IgH Does Not Rescue B Cell Development Defects in Mettl14 KO Mice.
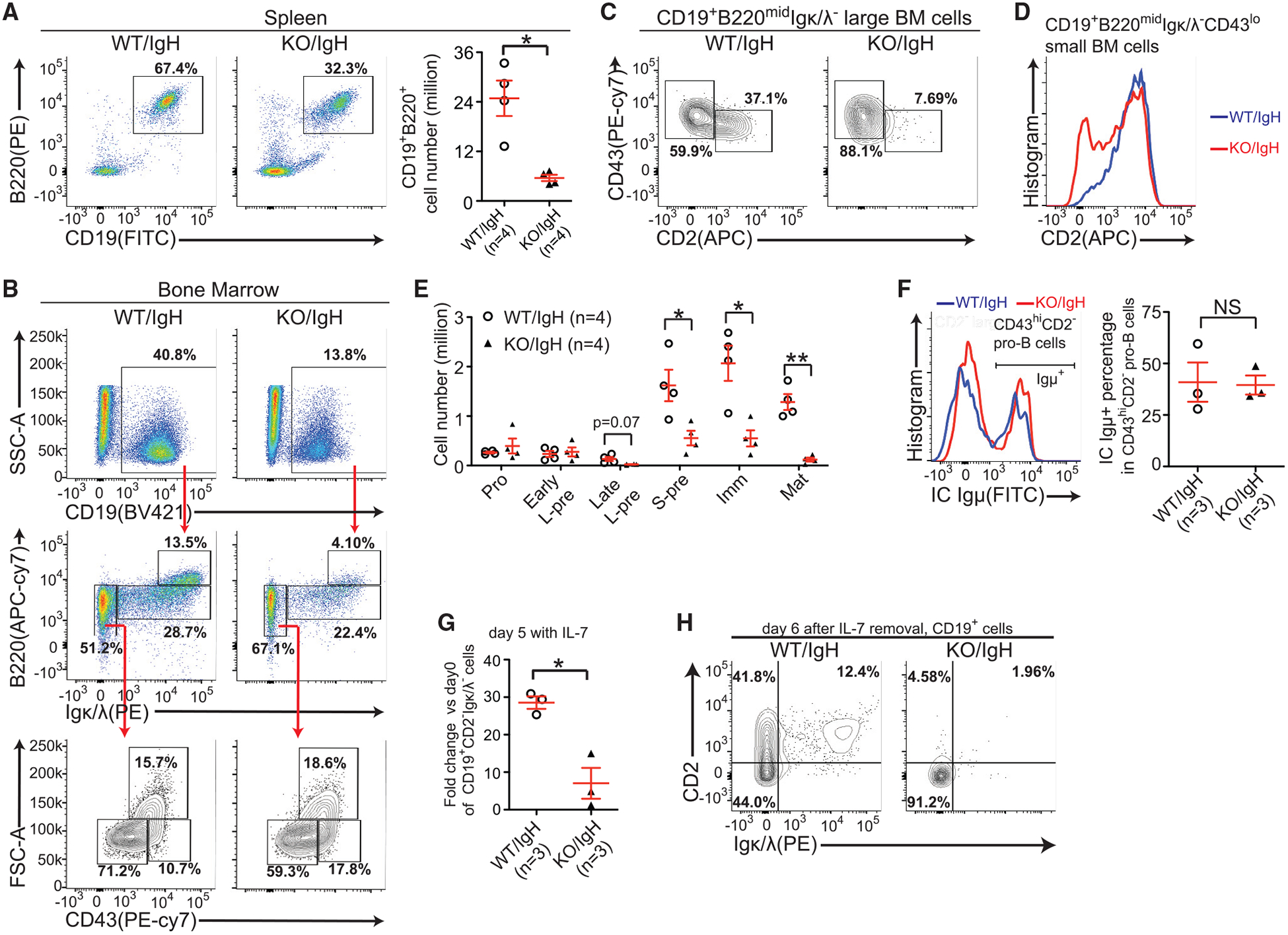
(A) Flow cytometry plots and quantification of splenic B cells of indicated mice. (B) Flow cytometry plots of B lineage cells in the bone marrow of indicated mice. (C) Expression of indicated markers in the large pre-B cells of indicated mice. (D) Expression of CD2−in the small CD43lo population of indicated mice. (B–D) Data are representative of 4 mice per group. (E) Quantification of indicated B-lineage cells (Figure 1C) in indicated mice. The number of each cell population from 2 femurs per mouse was calculated. (F) Flow cytometry plots and quantification of intracellular Igμ+ cells in indicated cells from indicated mice. (G) The fold changes of CD19+Igκ/λ− CD2−cell numbers were compared between indicated groups after 5-day IL-7-induced expansion. (H) Flow cytometry plots of Igκ/λ−/CD2−expression in CD19+ cells 6 days after IL-7 removal. Data are representative of 3 mice per group. SEM or ± SEM is shown; NS, not significant; *, p < 0.05; **, p < 0.01 and ***, p < 0.001.
In parallel, we examined B cell development in vitro. We found that KO/IgH cells displayed as severe defects in IL-7-induced pro-B cell expansion (Figure 4G) and large pre-B to small pre-B transition (Figure 4H) as in Mettl14 KO cells (Figure 2A). Altogether, these data indicate that the pre-arranged IgH does not rescue B cell development blocks due to METTL14 deficiency.
Pre-arranged IgL could not rescue the B cell development block in METTL14 KO mice either. Compared to WT controls expressing the SWHEL transgenic IgL (WT/IgL) (Phan et al., 2003), KO/IgL mice showed an over 100-fold reduction in splenic B cell number (Figure S5A). The presence of the pre-arranged IgL resulted in an increase of the Igκ/λ+ percentage in the bone marrow of mice, but most of Igκ/λ− and Igκ/λ+ cells in the KO/IgL mice exhibited a CD43hiCD2−phenotype, indicating they were still blocked from reaching the CD2+ small pre-B stage even with the expression of IgL (Figure S5B). Consequently, KO/IgL still displayed a dramatic reduction in bone marrow CD2+ small pre-B/immature B cells compared with the WT/IgL controls (Figure S5C).
METTL14 Is Required for Installing m6A onto a Wide Range of Transcripts in Developing B Cells
To link mRNA m6A to B cell development, we mapped the whole transcriptome m6A of developing B cells by m6A-seq (Hsu and He, 2018). Over 7,000 highly confident m6A sites were identified in WT ex vivo sorted pro-B cells and in vitro expanded large pre-B cells, enriched with the m6A consensus motif GGACU(G) identified before (Fu et al., 2014; Figure 5A). m6A is primarily enriched in the CDS (coding sequence) and 3′ UTR surrounding the stop codons in developing B cells (Figure 5B), as the case in cell lines and other tissues (Dominissini et al., 2012; Fu et al., 2014). Roughly 40%–50% of expressed genes are confidently identified as m6A labeled in developing B cells (Table S5), and m6A-labeled genes identified in two stages showed large overlap (Figure 5C). These data indicated that the m6A methylation is widely distributed in the transcriptome of developing B cells.
Figure 5. METTL14 Is Required for Writing m6A onto a Wide Range of mRNAs in Developing B Cells.
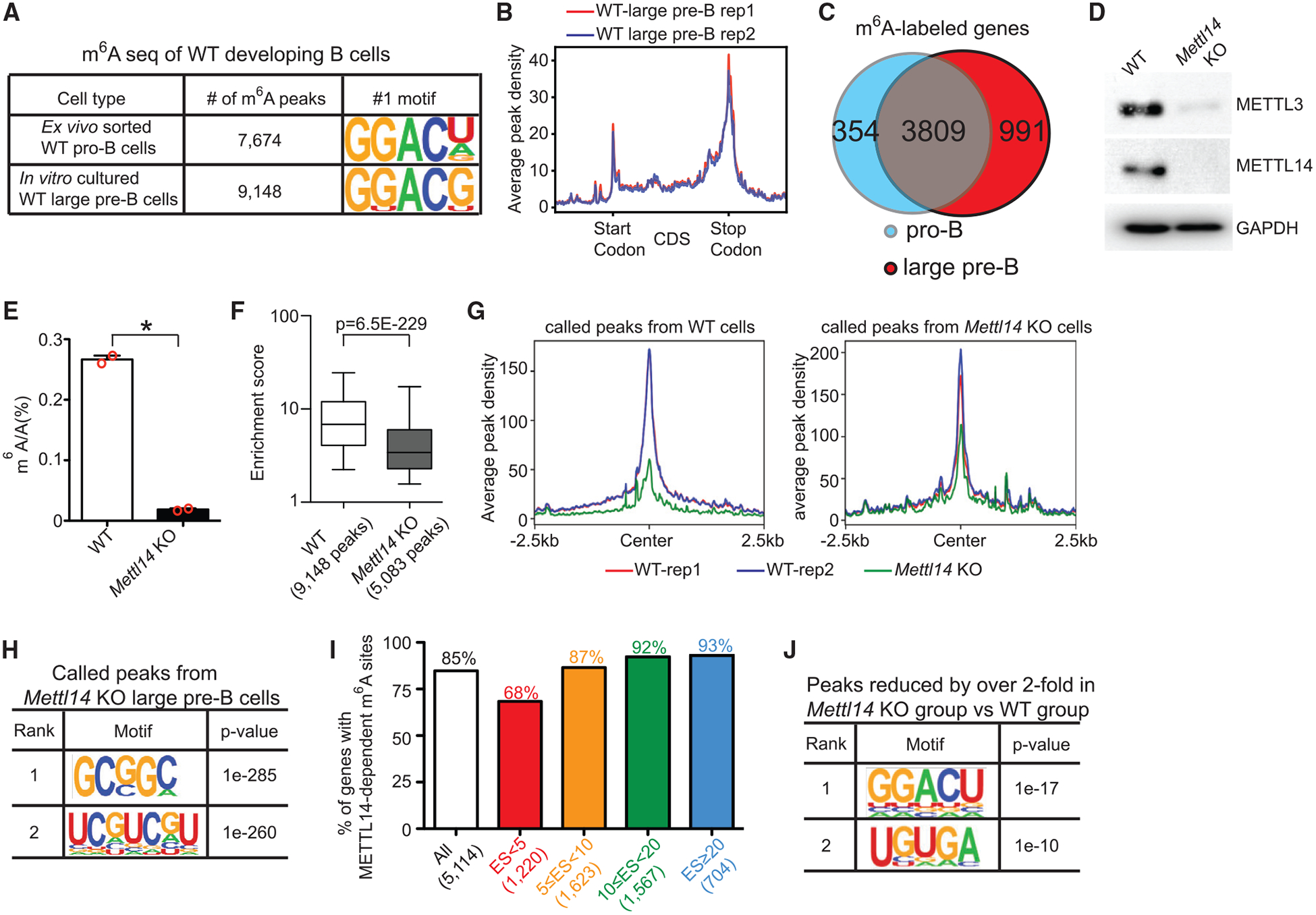
(A) Numbers of m6A peaks identified in indicated cells and their top consensus sequences. (B) Metagene profiles of enrichment of m6A modifications across the whole transcriptome in the two WT large pre-B cell biological replicates. (C) Overlapping of m6A-labeled genes identified in WT pro-B cells and in WT large pre-B cells. Only genes with RPKM (reads per kilobase million) > 1 in both WT pro-B cells and WT large pre-B cells were counted. (D) Western blot against indicated proteins in in vitro expanded large pre-B cells. Data shown are representative of 2 independent experiments. (E) Quantification of m6A methylation by LC-MS/MS in the poly-A enriched mRNAs from indicated in vitro expanded large pre-B cells (n = 2 per group). (F) Fold enrichment scores of called peaks from indicated groups were compared. Box and whisker (5–95 percentile) plots were shown. p value was calculated by Mann-Whitney-Wilcoxon test. (G) Global comparison of the peaks identified in indicated groups between all samples. (H) Consensus sequences and their p values of peaks called by m6A-seq from the Mettl14 KO group. (I) Percentages of genes with significantly reduced m6A peaks in Mettl14 KO group in genes grouped by m6A methylation levels. (J) Top two consensus sequences and their p values of peaks that were significantly reduced in Mettl14 KO group. (H and J) p value was calculated by HOMER Motif discovery tools.
Loss of METTL14 caused disappearance of the whole METTL3/METTL14 core in the large pre-B cells (Figure 5D), which is consistent with observations in T cells (Li et al., 2017a). Quantification of m6A in poly-A-enriched mRNAs by mass spectrometry showed that the Mettl14 KO large pre-B cells had a 93% reduction in the mRNA m6A level compared to the WT controls (Figure 5E). Similarly, by m6A-seq, 44% fewer peaks, with significantly lower enrichment scores (Figure 5F) and peak density (Figure 5G), were identified in Mettl14 KO large pre-B cells (5,083 peaks) than in WT counterparts (9,148 peaks). Importantly, the peaks identified in Mettl14 KO cells are not enriched with the consensus m6A motif GGACU/G (Figure 5H), indicating that our assay is specific and the remaining m6A sites might be of different nature. On average, 85% of m6A-labeled genes in WT large pre-B cells contained m6A peaks that are significantly reduced in the Mettl14 KO large pre-B cells (Figure 5I; Table S5). These significantly reduced peaks were primarily enriched with the consensus motif GGACU (Figure 5J). These data confirm that METTL14 is required for the installation of m6A onto a wide range of transcripts in developing B cells.
In addition to the dramatic reduction in mRNA m6A levels, loss of METTL14 broadly affects the expression of genes involved in writing or reading m6A in large pre-B cells. Nearly all proteins known to be associated with the METTL3/METTL14 complex (Shi et al., 2019) plus another m6A methyltransferase, METTL16 (Mendel et al., 2018; Warda et al., 2017), were elevated at both RNA (Figure S6A) and protein (Figure S6B) levels in the Mettl14 KO large pre-B cells. These increases might reflect a certain compensatory mechanism, as cells attempt to stabilize the remaining METTL3/METTL14 core and compensate its functions. Four of five YTH domain-containing m6A readers were also elevated in the same pattern (Figures S6C and S6D). We did not detect significant changes in m6A erasers or other known m6A binding proteins in the KO cells (Figures S6E and S6F).
mRNA m6A Decreases a Group of YTHDF2-Bound Transcripts in Developing B Cells and Modulates IL-7-Induced Pro-B Cell Proliferation via YTHDF2
Consistent with the well-established notion that m6A inversely correlates with mRNA stability, in large pre-B cells, m6A-labeled genes are more likely to have increased mRNA levels in the absence of METTL14 (Figures 6A and 6B). A strong correlation between higher m6A labeling in transcripts and higher gene expression levels in Mettl14 KO cells was also detected (Figures S7A and S7B), suggesting that m6A directly decreases the mRNA levels of a group of genes.
Figure 6. m6A Modulates IL-7-Induced Pro-B Cell Proliferation by Suppressing a Group of YTHDF2-Bound Transcripts.
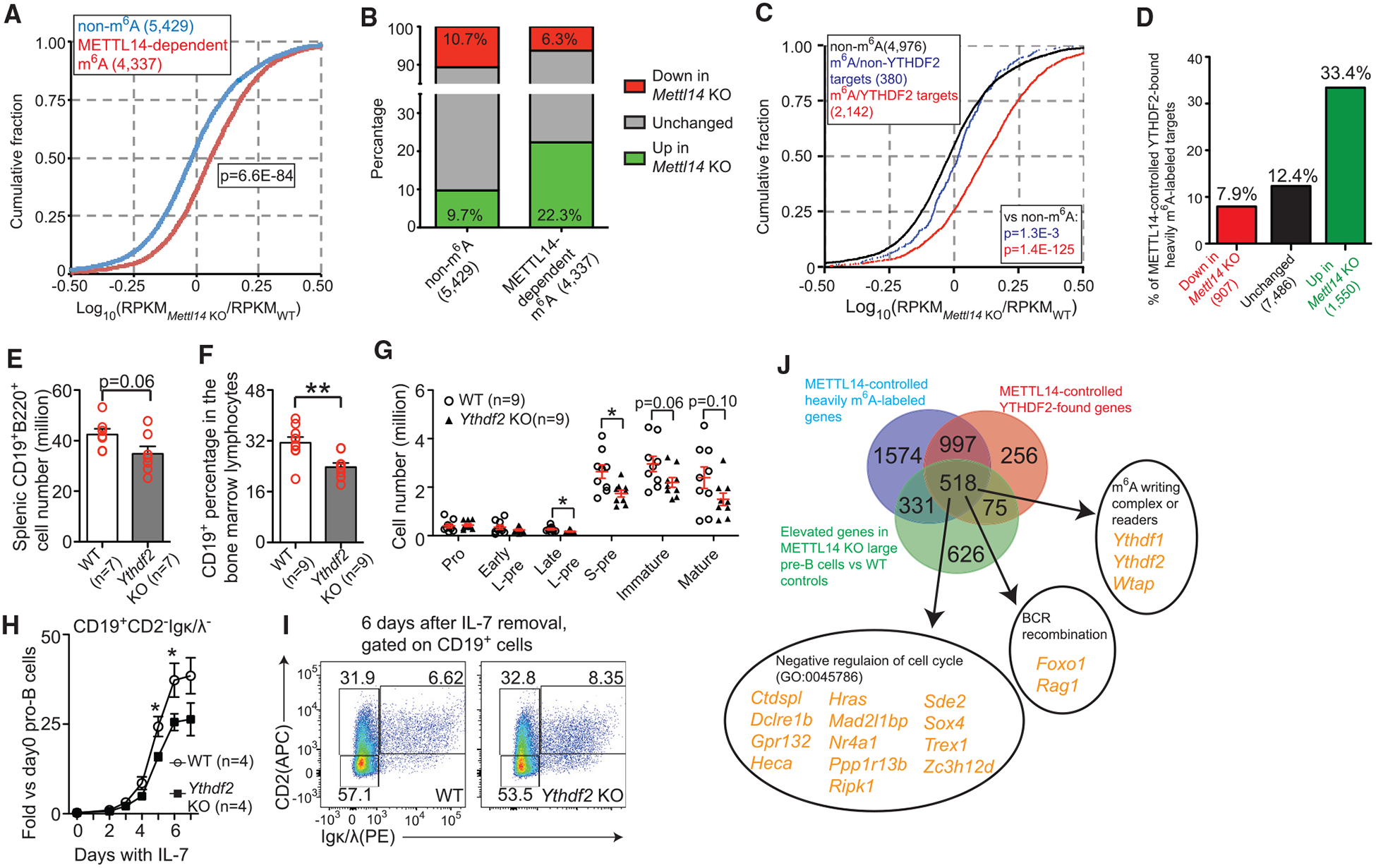
(A) Cumulated distribution of log10 fold changes between Metl14 KO cells and WT cells for non-m6A-labeled genes (blue) and METTL14-dependent m6A-labeled genes (red). (B) Distribution of DEGs identified by RNA-seq from the Mettl14 KO large pre-B cells in non-m6A-labeled genes and METTL14-dependent m6A-labeled genes. (C) Cumulated distribution of log10 fold changes between Metl14 KO cells and WT cells for non-m6A-labeled genes, non-YTHDF2-bound, METTL14-dependent, m6A-labeled genes, and YTHDF2-bound, METTL14-dependent, m6A-labeled genes. (A and C) p value was calculated by Mann-Whitney-Wilcoxon test. (D) Percentage of YTHDF2-bound heavily m6A-methylated transcripts that lose both m6A methylation and YTHDF2 binding in the Mettl14 KO cells in indicated DEG groups. (A–D) The number of genes in each group is shown in the parentheses after the group name. (E) Quantification of splenic B cells in indicated mice. (F) The percentages of CD19+ cells in the bone marrow cells in indicated mice. (G) The numbers of indicated populations (as defined in Figure 1C) in indicated mice. The number of each bone marrow cell population from 2 femurs per mouse was calculated. (H) Fold changes of the CD19+CD2−Igκ/λ− cells at indicated times during IL-7-induced expansion are shown. (I) Flow cytometry plots of indicated markers in CD19+ cells of indicated groups 6 days after IL-7 removal. Data are representative of 3 mice per group. (J) Identification of the 518 YTHDF2-suppressed m6A-labeled genes in large pre-B cells (Table S7). SEM or ± SEM is shown; NS, not significant; *, p < 0.05; **, p < 0.01 and ***, p < 0.001.
YTHDF2-mediated decay is a well-known mechanism by which m6A decreases mRNA levels (Wang et al., 2014b). To investigate whether the elevated expression of m6A-labeled genes in the KO cells was linked to YTHDF2, we identified transcripts bound by YTHDF2 in developing B cells by RNA immunoprecipitation and sequencing (RIP-seq). About 27% of transcripts of the whole transcriptome were identified as YTHDF2 targets in the WT large pre-B cells (Table S6). YTHDF2 clearly showed a preference to recognize m6A-containing transcripts (Figures S7C and S7D), and loss of METTL14 significantly reduced the binding of YTHDF2 to the majority of its targets (Figures S7E and S7F). Importantly, among the heavily m6A-labeled transcripts, only YTHDF2-bound transcripts were more likely to be elevated in the Mettl14 KO cells (Figure 6C). Approximately one-third of the elevated DEGs in the Mettl14 KO large pre-B cells (Table S2) were YTHDF2-recognized heavily m6A-methylated genes in the WT cells that lost both m6A methylation and YTHDF2 binding in the Mettl14 KO cells; this percentage was nearly 3-fold higher than those in reduced DEGs or unchanged genes (Figure 6D). YTHDF2 RIP-seq in WT pro-B cell showed that YTHDF2 also preferentially binds to m6A-labeled transcripts and YTHDF2-bound m6A-labeled transcripts tend to be elevated in Mettl14 KO pro-B cells (data not shown). These data strongly suggested that m6A directly decreases a group of transcripts via YTHDF2 in developing B cells.
Next, we generated mb1cre/+Ythdf2fl/fl (Ythdf2 KO) mice to evaluate the role of YTHDF2 in B cell development. We found that loss of YTHDF2 caused a 20% reduction of B cells in the spleen (Figure 6E). A significant block between the pro-B stage and the late large pre-B stage was detected in the bone marrow of the Ythdf2 KO mice (Figures 6F and 6G), but this block was milder than the one observed in the Mettl14 KO mice (Figures 1C and 1D). In vitro, Ythdf2 KO B-lineage cells exhibited a significant defect in the IL-7-induced expansion compared to WT controls (Figure 6H). However, loss of YTHDF2 did not cause any detectable block during the large-pre-B-to-small-pre-B transition in vitro (Figure 6I). These data indicated that the mRNA m6A mediates the IL-7-induced pro-B cell proliferation via YTHDF2 but modulates the large-pre-B-to-small-pre-B transition in an YTHDF2-independent way.
By integrative analysis of three sequencing datasets, we identified 518 genes as m6A-labeled, YTHDF2-recognized genes whose expressions were suppressed by m6A in the large pre-B cells (Figure 6J; Table S7). These genes contained a group of genes known to negatively regulate cell cycle (Figure 6J), which is consistent with the decreased proliferation observed in Mettl14 KO large pre-B cells (Figure 2). Both Foxo1 and Rag1 are among the list, suggesting that the YTHDF2-mediated RNA decay might also be critical to tuning down BCR recombination machinery (Ochiai et al., 2012). Interestingly, Ythdf1 and Ythdf2, together with Wtap, an important component of the m6A writing complex (Liu et al., 2014; Ping et al., 2014), are also present in the list, suggesting that the gene-suppression effect of m6A could also impact its readers and writers. Myc, the well-known target that was suppressed by m6A in hematopoietic stem cells (Cheng et al., 2019; Lee et al., 2019; Weng et al., 2018), also showed up in our list. However, the increased Myc in the Mettl14 KO large pre-B cells is not likely responsible for the defect in proliferation, as elevated MYC usually causes increased proliferation and overexpression of MYC in B lineage cells causes a very minor block (Iritani and Eisenman, 1999) compared with the one in Mettl14 KO mice.
mRNA m6A Does Not Modulate Early B Cell Development via YTHDF1-Facilitated mRNA Translation
To investigate whether YTHDF1-facilitated mRNA translation (Wang et al., 2015) plays a role in B cell development, ribosome profiling experiments were performed and m6A-labeled transcripts did not show reduced translation efficiencies in the Mettl14 KO large pre-B cells (Figures S7G and S7H). Constitutive Ythdf1 knockout (Ythdf1 KO) mice did not display any defects in B cell development either (Figures S7I and S7J).
METTL14 Is Required for the Proper Transcriptional Activation of Key TFs during the Large-Pre-B-to-Small-Pre-B Transition
As our data suggest that RNA m6A does not likely modulate the large-pre-B-to-small-pre-B transition via post-transcriptional mechanisms, we investigated whether changes at the transcription level could play a role. We noticed that several key TFs that are essential for the large-pre-B-to-small-pre-B transition, including Ikzf3, Irf4, Spib, and Bcl6 (Clark et al., 2014; Mandal et al., 2009), were not properly upregulated in Mettl14 KO cells after IL-7 removal (Figure 7A), suggesting that, in the absence of METTL14, the switch of the transcriptional programs could not be completed. TF Ikzf3 (also known as Aiolos) has been shown to control the exit from cell cycle and initiation of Igk (Mandal et al., 2009). We confirmed that loss of METTL14 impaired the induction of IKZF3 protein in the large pre-B cells in vitro (Figure 7B) and in vivo (Figure 7C). Furthermore, the residual IKZF3+ cells in the Mettl14 KO mice did not express CD2−(Figure 7C), suggesting that the block during the large-pre-B-to-small-pre-B transition in Mettl14 KO mice is multifactorial and not likely to be rescued by expression of single TF.
Figure 7. Mettl14 KO Cells Are Unable to Properly Open Chromatin of Key TFs during the Large-Pre-B-to-Small-Pre-B Transition.
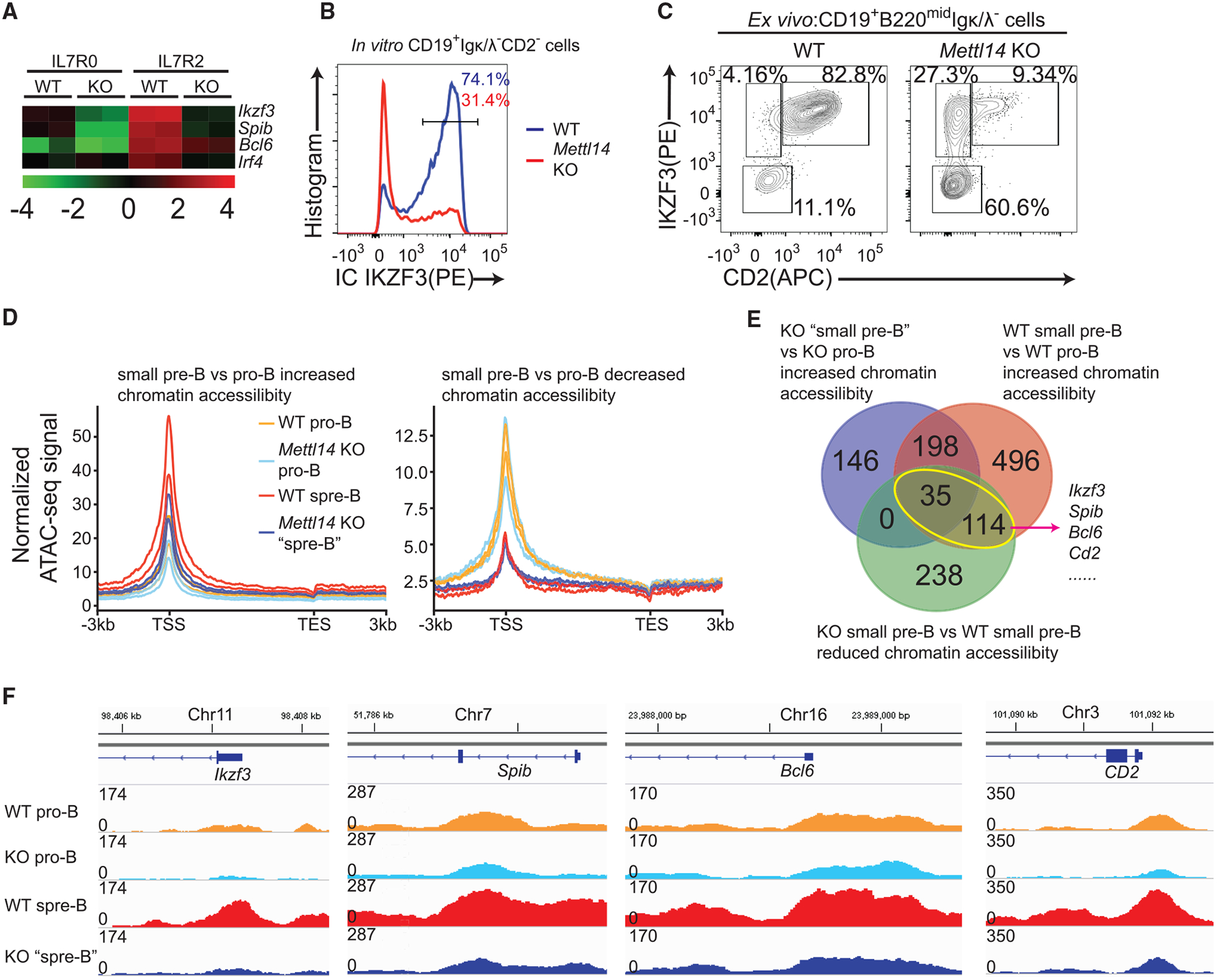
(A) Relative expression level of indicated genes in indicated samples. Data were from RNA-seq analysis (Tables S2 and S4). (B) Intracellular staining of IKZF3 in indicated in vitro expanded large pre-B cells at day 5. Data are representative of 3 mice per group. (C) Flow cytometry plots of CD2−by intracellular IKZF3 in the CD19+B220midIgκ/λ− cells sorted from indicated mice. Data are representative of 4 pairs of mice. (D) Normalized ATAC-seq signals of genes with elevated or reduced chromatin accessibility (WT small pre-B population versus WT pro-B population). Two biological replicates per group are shown. (E) Expressed genes in pro-B cells (Table S2) were grouped by comparing chromatin accessibility between four cell groups. Overlapping of indicated gene groups and representative genes are shown. (F) Track graphs showing the change of chromatin accessibilities of indicated genes from indicated samples. Graphs are representative of two biological replicates per group.
To understand how METTL14 regulates the transcription of these critical TFs and other genes, we conducted ATAC-seq (Buenrostro et al., 2015) on Mettl14 KO pro-B cells and the abnormal “small pre-B” cells and compared with respective WT controls. For the genes whose chromatin are closed in transition to small pre-B cells in WT mice, Mettl14 KO abnormal small pre-B cells did not show any global defect in closing chromatin of these genes (Figure 7D). However, for genes having elevated chromatin accessibility during the transition, Mettl14 KO cells appeared unable to properly open chromatin of these loci (Figure 7D). Only 28% of genes having increased accessibility (more open chromatin) during this transition in the WT group showed increased accessibility in the Mettl14 KO group (Figure 7E). We identified a total of 149 genes that show increased chromatin accessibility during the transition in the WT group but have lower chromatin accessibility in the Mettl14 KO group (Figure 7E, yellow circle). Several key TFs mediating this transition, including Ikzf3, Spib, Bcl6, as well as the small pre-B marker Cd2, belong to this list (Figures 7E and 7F). These data suggest that these key TFs cannot be upregulated properly at the transcription level in Mettl14 KO cells, resulting in the block from large pre-B cells to small pre-B cells.
DISCUSSION
Two separate blocks in B cell development due to METTL14 deficiency were clearly identified. They coincide with transitions of proliferation and recombination, strongly supporting the proposed role of m6A in coordinating gene expression switch during cell state transitions (Geula et al., 2015; Roundtree et al., 2017; Zhao et al., 2017a). The latter block during the large-pre-B-to-small-pre-B transition is the major defect in B cell development in Mettl14 KO mice. In contrast, the contribution of the defective IL-7-induced pro-B cell proliferation is relatively minor, as it only leads to reduced pro-B/early large pre-B cell proliferation in the Mettl14 KO mice. The significant defect in the IL-7-induced pro-B cell proliferation but very mild reduction in peripheral B cell numbers in the Ythdf2 KO mice also supports this conclusion. In two recent studies (Cheng et al., 2019; Lee et al., 2019), Mx1-Cre inducible Mettl3 conditional KO mice were generated to study the roles of m6A in hematopoietic stem cell identity and symmetric commitment. Both studies showed that there were no major defects in the bone marrow B cells after poly I:C treatment-induced Mettl3 deletion. Given the fast turnover of immature B cells and our data on early B cell developmental blocks, the unaffected immature B cell number in these mice might result from incomplete deletion of Mettl3 in the B lineage cells. However, it is also possible that RNA m6A methylation is not essential for the maintenance and survival of mature B cells once they have reached this stage.
mRNA m6A has been shown to inversely correlate with mRNA stability (Fu et al., 2014; Wang et al., 2014b). Loss of METTL3 or METTL14 causes elevations of m6A-containing transcripts in various cell types, and the subsequent increased expression of key m6A-labeled genes has been linked to abnormalities in these cells (Geula et al., 2015; Li et al., 2017a; Weng et al., 2018; Zhang et al., 2017). Our previous study identified the YTHDF2-mediated mRNA decay as a main regulatory pathway to post-transcriptionally reduce mRNA levels, which had been shown critical in processes such as embryo development (Zhao et al., 2017b) and hematopoietic stem cell expansion (Li et al., 2018b). Here, we found that, in developing B cells, loss of METTL14 also reduced the binding of YTHDF2 to its targets and specifically caused the elevations of a group of YTHDF2-bound transcripts. Both loss of METTL14 and loss of YTHDF2 caused significant block during the IL-7-induced pro-B cell proliferation, indicating that the YTHDF2-mediated mRNA decay is critical to the transition from the pro-B stage to the large pre-B stage. In contrast, although YTHDF1-facilitated, m6A-containing mRNA translation has been reported to play functional roles in several processes (Han et al., 2019; Shi et al., 2018), our data indicate that it is dispensable in B cell development.
Interestingly, loss of METTL14 causes block between the large pre-B stage and the small pre-B stage via a different mechanism. Loss of YTHDF1 or YTHDF2 alone does not affect this transition. It is possible that the m6A methylation affects multiple key TF transcripts through both YTHDF1 (increases translation of certain transcripts) and YTHDF2 (mediates decay of other transcripts), as well as processes independent of these two readers. This more synergistic effect could not be recapitulated with any single reader protein. Furthermore, METTL14 depletion also dramatically decays METTL3. These two proteins may play additional roles in the nucleus or affect stability of certain regulatory non-coding RNAs (Liu et al., 2020). We suspect it could be a combination of some of these potential pathways that warrants future investigations.
In summary, our data demonstrate important regulatory roles of the mRNA m6A methylation in early B cell development. The RNA m6A methylation modulates two cellular transitions by different mechanisms. The installation of m6A onto mRNA by the METTL3-METTL14 complex is important for the IL-7-induced pro-B cell proliferation through post-transcriptional reduction of a group of transcripts by YTHDF2. In contrast, the large-pre-B-to-small-pre-B transition is controlled by regulating gene expression at the transcriptional level.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Haochu Huang (huang.haochu@gene.com).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The accession numbers for the RNA-seq, m6A-seq, YTHDF2 RIP-seq, Ribosome profiling, and ATAC-seq data reported in this paper are Gene Expression Omnibus (GEO): GSE 112022, GSE 136419, and GSE 151071.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Mb1-cre(Hobeika et al., 2006), Mettl14-floxed(Yoon et al., 2017), Ythdf1-KO(Shi et al., 2018), Ythdf2-floxed(Li et al., 2018a) and SWHEL(Phan et al., 2003) mice have been described previously. Rag1−/− (Mombaerts et al., 1992) mice were purchased from the Jackson Laboratory. All mice were housed in a specific pathogen-free facility. Both males and females were used. Mice were used 6 24 weeks of age and all the experiments were approved by the University of Chicago Institutional Animal Care and Use Committee.
In vitro culture of B cell progenitors
B220+ cells were enriched from the bone marrow of mice by positively selecting B220+ cells with biotin-conjugated anti-mouse B220 antibody (Biolegend), the anti-Biotin MicroBeads (Miltenyi Biotec) and the LS column (Miltenyi Biotec). Enriched B220+ cells were co-cultured with OP9 cells in the culture medium (OPTI-MEM media supplemented with 10% FBS, 0.05 mM 2-mercaptoethanol, 100 units/ml penicillin, 100 μg/ml streptomycin, and 2 mM glutamine) with 15ng/mL IL-7 (Biolegend) for indicated times. For IL-7 removal, co-cultured B cell progenitors and OP9 cells were washed twice with pre-warmed medium without IL-7 and continued to be cultured in the same medium without IL-7 for indicated times.
METHOD DETAILS
Flow Cytometry
The antibodies for flow cytometry are summarized in the Key Resources Table. Cells were stained with fluorochrome-conjugated antibodies against surface antigens in DPBS (Thermo Fisher Scientific) containing 2% FBS (Thermo Fisher Scientific) prior to analysis, cell sorting or intracellular staining. To stain intracellular Igμ, Igk or VPREB, cells were fixed and permeabilized with a fixation/permeabilization solution (BD Biosciences) before being stained for intracellular proteins. To stain intracellular FOXO1, cyclin D3 or IKZF3, cells were fixed and permeabilized with the True-Nuclear transcription factor buffer set (BioLegend). For FOXO1 staining, after fixation and permeabilization, cells were stained with the Rabbit anti-FOXO1 antibody and washed three times, followed by being stained with a FITC-labeled anti-Rabbit IgG. For intracellular staining of p-STAT5, p-AKT or p-ERK, cells were fixed with BD Cytofix buffer and permeabilized with BD Phosflow Perm Buffer III as recommended by the manufacturer (BD Biosciences). Labeled cells were analyzed on a BD LSR II or a BD LSRFortessa, or sorted on a BD FACSAria, using Diva software (BD Biosciences). Flow cyto-metric data were analyzed using FlowJo software (BD Biosciences).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| APC-cy7 anti-mouse B220 antibody | Biolegend | RRID: AB_313007 |
| PE anti-mouse B220 antibody | Biolegend | RRID: AB_312993 |
| Biotin anti-mouse B220 antibody | Biolegend | RRID: AB_2794320 |
| Brilliant Violet 421 anti-mouse CD19 antibody | Biolegend | RRID: AB_10895761 |
| FITC anti-mouse CD19 antibody | Biolegend | RRID: AB_313641 |
| APC anti-mouse CD19 antibody | Biolegend | RRID: AB_313647 |
| PE Anti-mouse Igκ antibody | Biolegend | RRID: AB_2563581 |
| FITC Anti-mouse Igκ antibody | Biolegend | RRID: AB_2563585 |
| Biotin anti-mouse Igλ antibody | Biolegend | RRID: AB_345332 |
| PE anti-mouse Igλ antibody | Biolegend | RRID: AB_1027659 |
| PE-cy7 anti-mouse CD43 antibody | Biolegend | RRID: AB_2564349 |
| FITC anti-mouse CD2 antibody | Biolegend | RRID: AB_312652 |
| APC anti-mouse CD2 antibody | Biolegend | RRID: AB_2563090 |
| Brilliant Violet 711 anti-mouse c-Kit antibody | Biolegend | RRID: AB_2565956 |
| APC anti-mouse CD25 antibody | Biolegend | RRID: AB_312861 |
| FITC anti-mouse Ly-51(BP-1) antibody | Biolegend | RRID: AB_313362 |
| FITC anti-mouse CD4 antibody | Biolegend | RRID: AB_312691 |
| APC anti-mouse CD24 antibody | Biolegend | RRID: AB_2565651 |
| APC anti-mouse IL-7Rα antibody | Biolegend | RRID: AB_1937216 |
| Biotin anti-mouse Pre-B Cell Receptor antibody | BD Biosciences | RRID: AB_394277 |
| PE anti-mouse VpreB antibody | Biolegend | RRID: AB_11150774 |
| PE anti-Cyclin D3 antibody | Biolegend | RRID: AB_2686980 |
| PE anti-mouse Aiolos (IKZF3) antibody | Biolegend | RRID: AB_2561701 |
| Alexa Fluor 488 Donkey anti-rabbit IgG antibody | Biolegend | RRID: AB_2563203 |
| FITC anti-mouse IgM antibody | BD Biosciences | RRID: AB_394857 |
| Alexa Fluor 488 anti-ERK1/2 (pT202/pY204) antibody | BD Biosciences | RRID: AB_399875 |
| Alexa Fluor 488 anti-STAT5 (pY694) antibody | BD Biosciences | RRID: AB_11154039 |
| Alexa Fluor 488 anti-Akt (pS473) antibody | BD Biosciences | RRID: AB_1645342 |
| FITC anti-BrdU antibody | BD Biosciences | RRID: AB_396304 |
| Rabbit Anti-FoxO1 (C29H4) antibody | Cell Signaling Technology | RRID: AB_2106495 |
| Rabbit anti-METTL3 antibody | Cell Signaling Technology | RRID: AB_2800072 |
| Rabbit anti-METTL14 antibody | Cell Signaling Technology | RRID: AB_2799383 |
| HRP-conjugated Rabbit anti-GAPDH (D16H11) | Cell Signaling Technology | RRID: AB_11129865 |
| HRP-conjugated anti-rabbit IgG | Cell Signaling Technology | RRID: AB_2099233 |
| anti-m6A antibody | New England Biolabs | Catalog#:E1610s |
| anti-YTHDF2 antibody | Aviva Systems Biology | Catalog#:ARP67917_P050 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant Mouse IL-7 (carrier-free) | Biolegend | Cat#: 577802 |
| 5-Bromo-2’-deoxyuridine (BrdU) | Sigma-Aldrich | Cat#: B5002 |
| TRIzol Reagent | Thermo Fisher Scientific | Cat#: 15596026 |
| Critical Commercial Assays | ||
| FITC BrdU Flow Kit | BD Biosciences | Cat#:559619 |
| CellTrace CFSE Cell Proliferation Kit, for flow cytometry | Invitrogen | Cat#:C34554 |
| Dynabeads mRNA DIRECT Purification Kit | Invitrogen | Cat#:61012 |
| SMARTer Stranded RNA-Seq Kits | Takara | Cat#:634839 |
| EpiMark N6-Methyladenosine Enrichment Kit | New England Biolabs | Cat#: E1610S |
| RNA Clean & Concentractor-5 | Zymo Research | Cat#: R1013 |
| TruSeq Ribo Profile (Mammalian) Library Prep Kit | IIIumina | Cat#: RPYSC12116 |
| RiboMinus Eukaryote System v2 | Invitrogen | Cat#: A15026 |
| ZR small-RNA PAGE Recovery Kit | Zymo Research | Cat#: R1070 |
| NEBNext Small RNA Library Prep Set for Illumina | New England Biolabs | Cat#: E7330L |
| Deposited Data | ||
| Raw data: RNA-seq, ATAC-seq and ribosome profiling of large pre-B cells | This paper | GEO: GSE112022 |
| Raw data: m6A-seq and YTHDF2 RIP-seq of large pre-B cells | This paper | GEO: GSE136419 |
| Raw data: RNA-seq,m6A-seq and YTHDF2 RIP-seq of pro-B cells | This paper | GEO: GSE151071 |
| Experimental Models: Cell Lines | ||
| OP9 cells stromal cells | Gift from Dr Barbara Kee lab | N/A |
| Experimental Models: Organisms/Strains | ||
| Mettl14-floxed mouse | Yoon et al., 2017 | N/A |
| Mb1-cre mouse | Hobeika et al., 2006 | JAX:020505 |
| Ythdf1-KO mouse | Shi et al., 2018 | N/A |
| Ythdf2-floxed mouse | Li et al., 2018a | N/A |
| SWHEL mouse | Phan et al., 2003 | N/A |
| Rag1−/− mouse | Jackson Laboratory | JAX:002216 |
| Oligonucleotides | ||
| Mettl14 flox-Forward common primer: CTGCCAAGAAAATGGGAAAA | This paper | N/A |
| Mettl14 flox-WT&Flox reverse primer: TGCAGCCCCACAATTATAGC | This paper | N/A |
| Mettl14 flox- Deleted reverse primer: GGGACTGGGAACACTTGAAA | This paper | N/A |
| Primer degVk (Vκ1-Jκ1 primer a): GGCT GCAGSTTCAGTGGCAGTGGRTCGGRAC | Johnson et al., 2008 | N/A |
| Primer MAR (Vκ1-Jκ1 primer b): AACAC TGGATAAAGCAGTTTATGCCCTTTC | Johnson et al., 2008 | N/A |
| Primer k-meth-F (Vκ1-Jκ1 primer c): ATGACCCAGAGGATGAAAC | Johnson et al., 2008 | N/A |
| Forward primer for IgH recombination VH7183: CG GTACCAAGAASAMCCTGTWCCTGCAAATGASC | Liu et al., 2007 | N/A |
| Forward primer for IgH recombination VHGam3.8: CAAGGGACGGTTTGCCTTCTCTTTGGAA | Liu et al., 2007 | N/A |
| Forward primer for IgH recombination VHJ558: CGAGCTCTCCARCACAGCCTWCATGCARCTCARC | Liu et al., 2007 | N/A |
| Common reverse primer for IgH recombination: GTCTAGATTCTCACAAGAGTCCGATAGACCCTGG | Liu et al., 2007 | N/A |
| Hprt forward primer: TCCTCCTCAGACCGCTTTT | Inoue et al., 2015 | N/A |
| Hprt forward primer: CCTGGTTCATCATCGCTAATC | Inoue et al., 2015 | N/A |
| Software and Algorithms | ||
| FlowJo 10.0.8r.1 | BD Bioscinces | https://www.flowjo.com/solutions/flowjo/downloads/ |
| FACSDiva V8.0.1 | BD Biosciences | https://www.bdbiosciences.com/en-us/instruments/research-instruments/research-software/flow-cytometry-acquisition/facsdiva-software |
| Microsoft Exel | Microsoft | https://www.microsoft.com/en-us/microsoft-365 |
| GraphPad Prism | GraphPad Software Inc | https://www.graphpad.com/ |
| ABI Prism Sequence Detection v 1.9.1 | Thermo Fisher Scientific | https://www.thermofisher.com/us/en/home/technical-resources/software-downloads/ab-prism-7000-sequence-detection-system.html |
| Trim_galore v0.4.4 | Babraham Bioinformatics | https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ |
| Hisat2 v2.1.0 | Kim et al., 2015 | http://daehwankimlab.github.io/hisat2/ |
| Cufflinks v2.2.1 | Trapnell et al., 2013 | http://cole-trapnell-lab.github.io/cufflinks/releases/v2.2.1/ |
| Cluster 3.0 | Human Genome Center, University of Tokyo | http://bonsai.hgc.jp/~mdehoon/software/cluster/ |
| Java Treeview | Saldanha, 2004 | http://jtreeview.sourceforge.net/ |
| deeptools 2.0 tool suite | Ramírez et al., 2016 | https://deeptools.readthedocs.io/en/develop/ |
| featureCounts from R package Rsubread | Liao et al., 2019 | http://subread.sourceforge.net/ |
| RiboRex | Li et al., 2017b | https://github.com/smithlabcode/riborex |
| exomePeak | Meng et al., 2014 | https://bioconductor.riken.jp/packages/3.8/bioc/html/exomePeak.html |
| DESeq2 | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| HOMER | Heinz et al., 2010 | http://homer.ucsd.edu/homer/ |
| DAVID Bioinformatics Resources 6.8 | Huang et al., 2009 | https://david.ncifcrf.gov/ |
| JUMPp software | Wang et al., 2014a | https://github.com/hongwang198745/HGG_Source_Code |
| Other | ||
| DPBS, no calcium, no magnesium | Thermo Fisher Scientific | Cat#:14190250 |
| FBS | Thermo Fisher Scientific | Cat#:26140 |
| ACK cell lysis buffer | Thermo Fisher Scientific | Cat#: A1049201 |
| CountBright Absolute Counting Beads | Thermo Fisher Scientific | Cat#: C36950 |
| Opti-MEM I Reduced Serum Medium | Thermo Fisher Scientific | Cat#: 31985088 |
| L-Glutamine (200 mM) | Thermo Fisher Scientific | Cat#: 25030081 |
| Penicillin-Streptomycin (10,000 U/mL) | Thermo Fisher Scientific | Cat#: 15140122 |
| 2-Mercaptoethanol (50 mM) | Thermo Fisher Scientific | Cat#: 31350010 |
| Anti-Biotin MicroBeads | Miltenyi Biotec | Cat#:130–105–637 |
| MS Columns | Miltenyi Biotec | Cat#:130–042–201 |
| Fixation/Permeabilization Solution Kit | BD Biosciences | Cat#:554714 |
| True-Nuclear Transcription Factor Buffer Set | Biolegend | Cat#:424401 |
| BD Cytofix buffer | BD Biosciences | Cat#:554655 |
| Perm Buffer III | BD Biosciences | Cat#:558050 |
| Trypsin-EDTA (0.05%), phenol red | Thermo Fisher Scientific | Cat#: 25300120 |
| Nuclease P1 | FUJIFILM Wako Pure Chemical Corporation | Cat#: 145–08221 |
| FastAP Thermosensitive Alkaline Phosphatase | Thermo Fisher Scientific | Cat#: EF0651 |
| Dynabeads Protein G for Immunoprecipitation | Invitrogen | Cat#: 10004D |
| Micro Bio-Spin® Columns with Bio-Gel® P-30 | Bio-Rad | Cat#: 7326223 |
In vivo and in vitro proliferation assays
For in vivo BrdU labeling, one milligram of BrdU (Sigma) in 100 μL DPBS per mouse was injected intraperitoneally, before mice were sacrificed one hour later and BrdU cooperation in various B cell subpopulations was analyzed. For in vitro labeling, BrdU was added to the cell culture at a concentration of 10 μM for 45 mins at indicated time points. BrdU staining was performed with the BrdU flow cytometry kit following manufacturer instructions (BD Biosciences). For CFSE dilution assay, the CellTrace CFSE kit (Invitrogen) was used to label cells with CFSE.
Quantification of cells in vivo and in vitro
Spleens were mashed between 2 frosted microscope slides. Peritoneal cells were collected by injecting 5mL PBS to the peritoneal cavity of mouse to wash out cells. Bone marrow cells were collected by mashing bones with mortar and pestle. Splenocytes, peritoneal cavity cells, and bone marrow cells were counted using hemocytometer after red blood cells were lyzed with ACK RBC lysis buffer, and the cells were filtered. For bone marrow cells, the cell numbers represent either 2 (femur+ humerus+ tibia) per mouse or 2 femurs per mouse. For in vitro cultured cells, floating and attached cells were combined for counting. Attached cells were collected by incubating cells with 0.05% Trypsin/EDTA (Thermo Fisher Scientific) for 5 mins. The cultured cells were counted by flow cytometry with counting beads (Thermo Fisher Scientific).
PCR and quantitative PCR
The primers used for amplifying the Mettl14 flox and deleted alleles were: Forward common: CTGCCAAGAAAATGGGAAAA; Flox reverse: TGCAGCCCCACAATTATAGC; Deleted reverse: GGGACTGGGAACACTTGAAA. Primers for quantitative PCR of Vκ1-Jκ1 rearrangement with genomic DNA were as previously reported(Johnson et al., 2008). Primers “a” and “b” from the reference paper were used for the qPCR of the recombined Vκ1-Jκ1, while primers “b” and “c” were used for internal control reaction. Primers for quantitative PCR of recombination of various IgH families with genomic DNA were as previously reported(Liu et al., 2007) and Hprt was used as internal control.
RNA sequencing and data analysis
Total RNA from sorted cells was extracted with TRIzol (Thermo Fisher Scientific). Polyadenylated RNA was enriched with Dynabeads mRNA DIRECT Purification Kit (Invitrogen). The sequencing library was generated with SMARTer Stranded Total RNA-Seq Kit (Takara). All sequencing was performed by the Genomics Facility at the University of Chicago. Raw sequencing reads were trimmed by Trim_Galore software (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) to remove adaptor sequences and low-quality nucleotides. Clean reads were then mapped to the mm9 or mm10 reference genome using Hisat (Kim et al., 2015), and only uniquely mapped reads were kept for the following analyses. Cuffnorm(Trapnell et al., 2013) were used to calculate RPKM for all samples. Fold changes and FDRs were determined by Cuffdiff(Trapnell et al., 2013) with default parameters. Deferentially expressed genes between different groups were defined as genes with fold change > 1.5 and FDR < 0.05. GO term analysis was conducted with the Database for Annotation, Visualization and Integrated Discovery (DAVID)(Huang et al., 2009). Heatmaps were made with Cluster 3.0 (http://bonsai.hgc.jp/~mdehoon/software/cluster/) and Java Treeview (Saldanha, 2004) (http://jtreeview.sourceforge.net/).
Proteome profiling by 10-plex TMT-LC/LC-MS/MS
Sorted CD19+CD2−Igκ/λ− cells before and 2 days after IL-7 removal were subject to proteome profiling (biological duplicates for each group) as previously described(Bai et al., 2017; Du et al., 2018; Tan et al., 2017). Briefly, proteins in cell pellets were extracted, digested and labeled with TMT tags. The mixture of the TMT-labeled samples was desalted and separated on an XBridge C18 column (3.5 mm particle size, 2.1 mm × 10 cm, Waters) into about 40 fractions. Each fraction was then analyzed by LC-MS/MS. Mass spectrometer (Orbitrap HF, Thermo Fisher Scientific) was operated in data-dependent mode with a survey scan in Orbitrap (60,000 resolution, 1 × 106 AGC target and 50 ms maximal ion time) and 20 MS/MS high resolution scans (60,000 resolution, 1 × 105 AGC target, 105 ms maximal ion time, HCD, 35 normalized collision energy, 1.0 m/z isolation window, and 20 s dynamic exclusion). Data analysis was performed by the JUMP software(Wang et al., 2014a). Briefly, TMT reporter ion intensities of each PSM were extracted followed by removing the low intensity PSMs, correcting loading bias and adjusting intensities by isotopic distribution of each labeling reagent. The mean-centered intensities across samples were calculated and protein relative intensities were derived by averaging related PSMs. Finally, protein absolute intensities were determined by multiplying the relative intensities by the grand-mean of three most highly abundant PSMs(Niu et al., 2017). Protein differential expressions were evaluated by Student’s t test.
Quantification of m6A on mRNA by LC-MS/MS
Polyadenylated RNA from sorted cells was enriched with Dynabeads mRNA DIRECT Purification Kit (Invitrogen), digested with nuclease P1 (Wako) and dephosphorylated with FastAP Thermosensitive Alkaline Phosphatase (Thermo Fisher Scientifics). Then the samples were filtered and quantified by Agilent 6410 QQQ triple-quadrupole LC mass spectrometer.
m6A-seq and data analysis
Briefly, polyadenylated RNA isolated from indicated cells was fragmented to ~100nt with sonication. Four percent of the fragmented RNA was preserved as input. The rest was subject to m6A immunoprecipitation using the EpiMark N6-Methyladenosine Enrichment Kit (NEB). RNA was concentrated with RNA Clean & Concentractor-5 (Zymo Research) and the library was constructed with the SMARTerStranded Total RNA-Seq Kit (Takara). IP and Input sequencing data were sent to trim-galore to remove low quality reads and adaptor sequence contaminants under default parameters except for “−length 50.” Remaining reads were then aligned to the mouse transcriptome annotation based on mm9 assembly using hisat2(Kim et al., 2015) aligner (v2.1.0), with default parameters being used. Peaks enriched in the IP samples versus input or enriched in WT versus Mettl14 KO samples were identified using exomePeak (Meng et al., 2014) with default parameters. Profiles of m6A tag density were generated by plotProfile tool in deeptools 2.0 tool suite(Ramírez et al., 2016). For motif search, identified peaks were sorted according to the p values from lowest to highest, then the top 1000 peaks were chosen for the de novo motif analysis using HOMER Motif discovery tools(Heinz et al., 2010). Expressed genes (defined by RNA-seq) with at least one peak called with enrichment score > 2 (IP versus Input) in the WT group was defined as m6A-labeled genes. m6A-labeled genes with at least one peak significantly reduced by over two-fold in the Mettl14 KO group versus WT group were defined as METTL14-dependent m6A-labeled genes.
YTHDF2 RIP-seq and data analysis
Sorted cells were lysed with lysis buffer (50mM HEPES, 150mM KCl, 2mM EDTA, 0.5% NP40, 0.5mM DTT, protease inhibitor, RNase Inhibitor) on ice for 30 minutes. The lysate was spin at 21,000xg for 15 minutes. To pre-clear, the supernatant was incubated with Protein G beads (Invitrogen) at 4C for 1 hour. 5% of the lysate was kept as input. Antibody against YTHDF2 (Aviva Systems biology) was conjugated to Protein G beads. The protein-bead conjugate was added to the lysate and incubated for 4 hours. Beads were washed 6 times with NT2 buffer (50mM HEPES, 200mM NaCl, 2mM EDTA, 0.05% NP40, 0.5mM DTT, protease inhibitor, RNase inhibitor). RNA was eluted with TRIzol and library was constructed as described above. Reads from RIP-seq experiments were mapped to mm9 mouse genome sequence by hisat2(Kim et al., 2015), with the parameters setting as –rna-strandness R –mp 6,2 -k 5 –score-min L,0,−0.2. The FPKM value for each gene was calculated with Cufflinks(Trapnell et al., 2013). The number of the reads assigned to each gene was counted by the function of featureCounts from R package Rsubread(Liao et al., 2019). The target genes of YTHDF2 in WT and M14 KO samples were identified by the R package DESeq2(Love et al., 2014). The genes with p value less than 0.05 and log2 fold change bigger than 2 in RIP data contrast to input were defined as YTHDF2 target genes.
Ribosome profiling
Ribo-seq was performed using the Illumina TruSeq Ribo Profile kit (Illumina) following the manufacturer’s instructions with slight modifications (Ingolia et al., 2009). rRNA depletion was performed on input and RPF samples using RiboMinus Eukaryote System V2 (Invitrogen). 4.5 μL of TruSeq Ribo Profile PNK buffer was added to 12 μL of input RNA, and the mixture was heated at 94°C for 25 minutes to fragment the RNA. The buffer was replaced with Tris using Micro Bio-Spin P-30 Gel Columns (Bio-rad). End repair was performed on input and RPF samples using T4 PNK: (1) 3′ dephosphorylation: RNA was mixed with PNK buffer to 1X and 2 μL T4 PNK, and incubated at 37°C for 30 minutes; (2) 5′ phosphorylation: 1 μL 10 mM ATP and 1 μL extra T4 PNK were added to the mixture, which was incubated at 37°Cfor 30 minutes. The RNA was purified using RNA Clean and Concentrator 5 (Zymo Research). RPF samples were denatured in 2X denaturing gel loading dye at 95°C for 5 minutes and PAGE purified on a 10% TBU gel, and fragments between 28 nt and 30 nt were isolated and purified using ZR small-RNA PAGE Recovery Kit (Zymo Research). Library construction was performed using NEBNext Small RNA Library Prep Set (Illumina). Raw sequencing reads were trimmed by Trim_Galore to remove adaptor sequences and low-quality nucleotides. Clean reads were then mapped to mm9 reference genome using Hisat, and only uniquely mapped reads were kept for the following analyses. FeatureCounts (Liao et al., 2014) was used to count reads on all protein-coding genes, and differentially translated genes were identified by Riborex(Li et al., 2017b) with default parameters. Translation efficiency was defined as the ratio of ribosome-protected fragments to mRNA input, which reflected the relative occupancy of 80S ribosome per mRNA species.
ATAC-seq and data analysis
Pro-B (CD19+B220midIgκ/λ−CD2−CD43hi) cells and small pre-B (CD19+B220midIgκ/λ−CD43losmall) population were sorted from WT and Mettl14 KO mice and subject to ATAC-seq analysis (n = 2 per group). ATAC-seq was conducted as formerly reported (Buenrostro et al., 2015; Mandal et al., 2015). Raw sequencing reads were first trimmed by Trim_Galore to remove adaptor sequences and low-quality nucleotides, and then aligned to mm9 reference genome using bowtie2. PCR duplication and mitochondria-derived reads were discarded using Samtools, and normalized Bigwig files were generated by Deeptools. To quantify chromatin states’ change across different stages, all reads mapped to RefSeq TSS flanking 1 kb regions were counted by featureCounts and DESeq2 was used to search significantly changed promoters (p value < 0.01).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis
Unless otherwise noted in figure legend, differences between different comparisons were tested using two-tailed Student’s t test for data analysis, with NS, not significant; *, p < 0.05; **, p < 0.01 and ***, p < 0.001. For scatter figures with error bars and bar figures, each symbol represents an individual biological replicates, and mean with SEM or ± SEM is shown, unless otherwise noted in figure legend. Mann-Whitney-Wilcoxon test and K-S test are also used for some figures and are clearly indicated in figure legend. In RNA-seq analysis, the FDR of individual genes between samples were calculated by Cuffdiff. In m6A-seq or YTHDF2 RIP-seq analysis, the p values of individual gens between samples were calculated by exomePeak R/Bioconductor package or R package DESeq2, respectively.
Supplementary Material
Highlights.
RNA m6A methylation plays essential roles in early B cell development
Loss of RNA m6A-writer complex blocks two key transitions in B cell development
RNA m6A facilitates IL-7-induced pro-B cell proliferation via its reader YTHDF2
The large-pre-B-to-small-pre-B transition is independent of YTHDF1/2
ACKNOWLEDGMENTS
We would like to thank Drs. Demin Wang, Mei Yu, and Yuhong Chen for B cell signaling assays and discussions; Drs. Barbara Kee, Renee de Pooter, and Rosemary Morman for help in B cell progenitor culture; the flow cytometry core of the University of Chicago; Dr. Michael Reth for providing Mb1-cre mice; Dr. Robert Brink for providing SWHEL mice; Dr. Lou Dore and Dr. Bin Zhou for generating and providing Mettl14flox/flox, Ythdf1−/−, and Ythdf2flox/flox mice; and Drs. Thomas Lu, Hideki Terajima, Yawei Gao, Tong Wu, Dr. Hailing Shi, Zhike Lu, and Adam Rosa for help with experiments and discussions. This work was supported by the Howard Hughes Medical Institute (C.H.) and the National Institutes of Health (NIH) (AI128469 to C.H. and H.H.; HG008935 to C.H.; AI136318 and GM007281 to P.J.H.; AG053987 to J.P.; and GM08720 to L.Z.).
Footnotes
DECLARATION OF INTERESTS
C.H. is a scientific founder and a scientific advisory board member of Accent Therapeutic, Inc. H.H. is an employee of Genentech.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107819.
REFERENCES
- Bai B, Tan H, Pagala VR, High AA, Ichhaporia VP, Hendershot L, and Peng J (2017). Deep profiling of proteome and phosphoproteome by isobaric labeling, extensive liquid chromatography, and mass spectrometry. Methods Enzymol. 585, 377–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barneda-Zahonero B, Roman-Gonzalez L, Collazo O, Mahmoudi T, and Parra M (2012). Epigenetic regulation of B lymphocyte differentiation, trans-differentiation, and reprogramming. Comp. Funct. Genomics 2012, 564381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Chang HY, and Greenleaf WJ (2015). ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol 109, 21.29.1–21.29.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busslinger M (2004). Transcriptional control of early B cell development. Annu. Rev. Immunol 22, 55–79. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Luo H, Izzo F, Pickering BF, Nguyen D, Myers R, Schurer A, Gourkanti S, Brüning JC, Vu LP, et al. (2019). m6A RNA methylation maintains hematopoietic stem cell identity and symmetric commitment. Cell Rep. 28, 1703–1716 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark MR, Mandal M, Ochiai K, and Singh H (2014). Orchestrating B cell lymphopoiesis through interplay of IL-7 receptor and pre-B cell receptor signalling. Nat. Rev. Immunol 14, 69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffre M, Benhamou D, Rieß D, Blumenberg L, Snetkova V, Hines MJ, Chakraborty T, Bajwa S, Jensen K, Chong MMW, et al. (2016). miRNAs are essential for the regulation of the PI3K/AKT/FOXO pathway and receptor editing during B cell maturation. Cell Rep. 17, 2271–2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper AB, Sawai CM, Sicinska E, Powers SE, Sicinski P, Clark MR, and Aifantis I (2006). A unique function for cyclin D3 in early B cell development. Nat. Immunol 7, 489–497. [DOI] [PubMed] [Google Scholar]
- Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob-Hirsch J, Amariglio N, Kupiec M, et al. (2012). Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485, 201–206. [DOI] [PubMed] [Google Scholar]
- Du X, Wen J, Wang Y, Karmaus PWF, Khatamian A, Tan H, Li Y, Guy C, Nguyen TM, Dhungana Y, et al. (2018). Hippo/Mst signalling couples metabolic state and immune function of CD8a+ dendritic cells. Nature 558, 141–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Dominissini D, Rechavi G, and He C (2014). Gene expression regulation mediated through reversible m6A RNA methylation. Nat. Rev. Genet 15, 293–306. [DOI] [PubMed] [Google Scholar]
- Galloway A, Saveliev A, qukasiak S, Hodson DJ, Bolland D, Balmanno K, Ahlfors H, Monzón-Casanova E, Mannurita SC, Bell LS, et al. (2016). RNA-binding proteins ZFP36L1 and ZFP36L2 promote cell quiescence. Science 352, 453–459. [DOI] [PubMed] [Google Scholar]
- Geula S, Moshitch-Moshkovitz S, Dominissini D, Mansour AA, Kol N, Salmon-Divon M, Hershkovitz V, Peer E, Mor N, Manor YS, et al. (2015). Stem cells. m6A mRNA methylation facilitates resolution of naı¨ve pluripotency toward differentiation. Science 347, 1002–1006. [DOI] [PubMed] [Google Scholar]
- Hagman J, and Lukin K (2006). Transcription factors drive B cell development. Curr. Opin. Immunol 18, 127–134. [DOI] [PubMed] [Google Scholar]
- Han D, Liu J, Chen C, Dong L, Liu Y, Chang R, Huang X, Liu Y, Wang J, Dougherty U, et al. (2019). Anti-tumour immunity controlled through mRNA m6A methylation and YTHDF1 in dendritic cells. Nature 566, 270–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobeika E, Thiemann S, Storch B, Jumaa H, Nielsen PJ, Pelanda R, and Reth M (2006). Testing gene function early in the B cell lineage in mb1-cre mice. Proc. Natl. Acad. Sci. USA 103, 13789–13794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PJ, and He C (2018). Identifying the m6A methylome by affinity purification and sequencing. Methods Mol. Biol 1649, 49–57. [DOI] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, and Lempicki RA (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Ingolia NT, Ghaemmaghami S, Newman JR, and Weissman JS (2009). Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324, 218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue T, Morita M, Hijikata A, Fukuda-Yuzawa Y, Adachi S, Isono K, Ikawa T, Kawamoto H, Koseki H, Natsume T, et al. (2015). CNOT3 contributes to early B cell development by controlling Igh rearrangement and p53 mRNA stability. J. Exp. Med 212, 1465–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iritani BM, and Eisenman RN (1999). c-Myc enhances protein synthesis and cell size during B lymphocyte development. Proc. Natl. Acad. Sci. USA 96, 13180–13185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson K, Hashimshony T, Sawai CM, Pongubala JM, Skok JA, Aifantis I, and Singh H (2008). Regulation of immunoglobulin light-chain recombination by the transcription factor IRF-4 and the attenuation of interleukin-7 signaling. Immunity 28, 335–345. [DOI] [PubMed] [Google Scholar]
- Kikuchi K, Kasai H, Watanabe A, Lai AY, and Kondo M (2008). IL-7 specifies B cell fate at the common lymphoid progenitor to pre-proB transition stage by maintaining early B cell factor expression. J. Immunol 181, 383–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Langmead B, and Salzberg SL (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai M, Gonzalez-Martin A, Cooper AB, Oda H, Jin HY, Shepherd J, He L, Zhu J, Nemazee D, and Xiao C (2016). Regulation of B-cell development and tolerance by different members of the miR-17 92 family micro-RNAs. Nat. Commun 7, 12207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Bao S, Qian Y, Geula S, Leslie J, Zhang C, Hanna JH, and Ding L (2019). Stage-specific requirement for Mettl3-dependent m6A mRNA methylation during haematopoietic stem cell differentiation. Nat. Cell Biol 21, 700–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HB, Tong J, Zhu S, Batista PJ, Duffy EE, Zhao J, Bailis W, Cao G, Kroehling L, Chen Y, et al. (2017a). m6A mRNA methylation controls T cell homeostasis by targeting the IL-7/STAT5/SOCS pathways. Nature 548, 338–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Wang W, Uren PJ, Penalva LOF, and Smith AD (2017b). Riborex: fast and flexible identification of differential translation from Ribo-seq data. Bioinformatics 33, 1735–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Zhao X, Wang W, Shi H, Pan Q, Lu Z, Perez SP, Suganthan R, He C, Bjørås M, and Klungland A (2018a). Ythdf2-mediated m6A mRNA clearance modulates neural development in mice. Genome Biol. 19, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Qian P, Shao W, Shi H, He XC, Gogol M, Yu Z, Wang Y, Qi M, Zhu Y, et al. (2018b). Suppression of m6A reader Ythdf2 promotes hematopoietic stem cell expansion. Cell Res. 28, 904–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2019). The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 47, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Schmidt-Supprian M, Shi Y, Hobeika E, Barteneva N, Jumaa H, Pelanda R, Reth M, Skok J, Rajewsky K, and Shi Y (2007). Yin Yang 1 is a critical regulator of B-cell development. Genes Dev. 21, 1179–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, Jia G, Yu M, Lu Z, Deng X, et al. (2014). A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol 10, 93–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, You Y, Lu Z, Yang J, Li P, Liu L, Xu H, Niu Y, and Cao X (2019). N6-methyladenosine RNA modification-mediated cellular metabolism rewiring inhibits viral replication. Science 365, 1171–1176. [DOI] [PubMed] [Google Scholar]
- Liu J, Dou X, Chen C, Chen C, Liu C, Xu MM, Zhao S, Shen B, Gao Y, Han D, and He C (2020). N6-methyladenosine of chromosome-associated regulatory RNA regulates chromatin state and transcription. Science 367, 580–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malin S, McManus S, Cobaleda C, Novatchkova M, Delogu A, Bouillet P, Strasser A, and Busslinger M (2010). Role of STAT5 in controlling cell survival and immunoglobulin gene recombination during pro-B cell development. Nat. Immunol 11, 171–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal M, Powers SE, Ochiai K, Georgopoulos K, Kee BL, Singh H, and Clark MR (2009). Ras orchestrates exit from the cell cycle and light-chain recombination during early B cell development. Nat. Immunol 10, 1110–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal M, Hamel KM, Maienschein-Cline M, Tanaka A, Teng G, Tuteja JH, Bunker JJ, Bahroos N, Eppig JJ, Schatz DG, and Clark MR (2015). Histone reader BRWD1 targets and restricts recombination to the Igk locus. Nat. Immunol 16, 1094–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal M, Maienschein-Cline M, Maffucci P, Veselits M, Kennedy DE, McLean KC, Okoreeh MK, Karki S, Cunningham-Rundles C, and Clark MR (2018). BRWD1 orchestrates epigenetic landscape of late B lymphopoiesis. Nat. Commun 9, 3888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mårtensson IL, Almqvist N, Grimsholm O, and Bernardi AI (2010). The pre-B cell receptor checkpoint. FEBS Lett. 584, 2572–2579. [DOI] [PubMed] [Google Scholar]
- Mendel M, Chen K-M, Homolka D, Gos P, Pandey RR, McCarthy AA, and Pillai RS (2018). Methylation of structured RNA by the m6A writer METTL16 is essential for mouse embryonic development. Mol. Cell 71, 986–1000.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng J, Lu Z, Liu H, Zhang L, Zhang S, Chen Y, Rao MK, and Huang Y (2014). A protocol for RNA methylation differential analysis with MeRIP-seq data and exomePeak R/Bioconductor package. Methods 69, 274–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, and Papaioannou VE (1992). RAG-1-deficient mice have no mature B and T lymphocytes. Cell 68, 869–877. [DOI] [PubMed] [Google Scholar]
- Niu M, Cho JH, Kodali K, Pagala V, High AA, Wang H, Wu Z, Li Y, Bi W, Zhang H, et al. (2017). Extensive peptide fractionation and y1 ion-based interference detection method for enabling accurate quantification by isobaric labeling and mass spectrometry. Anal. Chem 89, 2956–2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochiai K, Maienschein-Cline M, Mandal M, Triggs JR, Bertolino E, Sciammas R, Dinner AR, Clark MR, and Singh H (2012). A self-reinforcing regulatory network triggered by limiting IL-7 activates pre-BCR signaling and differentiation. Nat. Immunol 13, 300–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan TG, Amesbury M, Gardam S, Crosbie J, Hasbold J, Hodgkin PD, Basten A, and Brink R (2003). B cell receptor-independent stimuli trigger immunoglobulin (Ig) class switch recombination and production of IgG autoantibodies by anergic self-reactive B cells. J. Exp. Med 197, 845–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ping XL, Sun BF, Wang L, Xiao W, Yang X, Wang WJ, Adhikari S, Shi Y, Lv Y, Chen YS, et al. (2014). Mammalian WTAP is a regulatory sub-unit of the RNA N6-methyladenosine methyltransferase. Cell Res. 24, 177–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44 (W1), W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez A, Vigorito E, Clare S, Warren MV, Couttet P, Soond DR, van Dongen S, Grocock RJ, Das PP, Miska EA, et al. (2007). Requirement of bic/microRNA-155 for normal immune function. Science 316, 608–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roundtree IA, Evans ME, Pan T, and He C (2017). Dynamic RNA modifications in gene expression regulation. Cell 169, 1187–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Zhang X, Weng YL, Lu Z, Liu Y, Lu Z, Li J, Hao P, Zhang Y, Zhang F, et al. (2018). m6A facilitates hippocampus-dependent learning and memory through YTHDF1. Nature 563, 249–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldanha AJ (2004). Java Treeview–extensible Visualization of Microarray Data. Bioinformatics 20, 3246–3248. [DOI] [PubMed] [Google Scholar]
- Shi H, Wei J, and He C (2019). Where, when, and how: context-dependent functions of RNA methylation writers, readers, and erasers. Mol. Cell 74, 640–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan H, Yang K, Li Y, Shaw TI, Wang Y, Blanco DB, Wang X, Cho JH, Wang H, Rankin S, et al. (2017). Integrative proteomics and phospho-proteomics profiling reveals dynamic signaling networks and bioenergetics pathways underlying T cell activation. Immunity 46, 488–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timblin GA, and Schlissel MS (2013). Ebf1 and c-Myb repress rag transcription downstream of Stat5 during early B cell development. J. Immunol 191, 4676–4687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, and Pachter L (2013). Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol 31, 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Li Y, Wu Z, Wang H, Tan H, and Peng J (2014a). JUMP: a tag-based database search tool for peptide identification with high sensitivity and accuracy. Mol. Cell. Proteomics 13, 3663–3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, Fu Y, Parisien M, Dai Q, Jia G, et al. (2014b). N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 505, 117–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, Weng X, Chen K, Shi H, and He C (2015). N(6)-methyladenosine modulates messenger RNA translation efficiency. Cell 161, 1388–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warda AS, Kretschmer J, Hackert P, Lenz C, Urlaub H, Höbartner C, Sloan KE, and Bohnsack MT (2017). Human METTL16 is a N6-methylade-nosine (m6A) methyltransferase that targets pre-mRNAs and various non-coding RNAs. EMBO Rep. 18, 2004–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng H, Huang H, Wu H, Qin X, Zhao BS, Dong L, Shi H, Skibbe J, Shen C, Hu C, et al. (2018). METTL14 inhibits hematopoietic stem/progenitor differentiation and promotes leukemogenesis via mRNA m6A modification. Cell Stem Cell 22, 191–205.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Deng Y, Feng Y, Long D, Ma K, Wang X, Zhao M, Lu L, and Lu Q (2018). Epigenetic regulation in B-cell maturation and its dysregulation in autoimmunity. Cell. Mol. Immunol 15, 676–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Z, Cui Y, Watford WT, Bream JH, Yamaoka K, Hissong BD, Li D, Durum SK, Jiang Q, Bhandoola A, et al. (2006). Stat5a/b are essential for normal lymphoid development and differentiation. Proc. Natl. Acad. Sci. USA 103, 1000–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon K-J, Ringeling FR, Vissers C, Jacob F, Pokrass M, Jimenez-Cyrus D, Su Y, Kim N-S, Zhu Y, Zheng L, et al. (2017). Temporal control of mammalian cortical neurogenesis by m6A methylation. Cell 171, 877–889.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue Y, Liu J, and He C (2015). RNA N6-methyladenosine methylation in post-transcriptional gene expression regulation. Genes Dev. 29, 1343–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Chen Y, Sun B, Wang L, Yang Y, Ma D, Lv J, Heng J, Ding Y, Xue Y, et al. (2017). m6A modulates haematopoietic stem and progenitor cell specification. Nature 549, 273–276. [DOI] [PubMed] [Google Scholar]
- Zhao BS, Roundtree IA, and He C (2017a). Post-transcriptional gene regulation by mRNA modifications. Nat. Rev. Mol. Cell Biol 18, 31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao BS, Wang X, Beadell AV, Lu Z, Shi H, Kuuspalu A, Ho RK, and He C (2017b). m6A-dependent maternal mRNA clearance facilitates zebrafish maternal-to-zygotic transition. Nature 542, 475–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Li YS, Bandi SR, Tang L, Shinton SA, Hayakawa K, and Hardy RR (2015). Lin28b promotes fetal B lymphopoiesis through the transcription factor Arid3a. J. Exp. Med 212, 569–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession numbers for the RNA-seq, m6A-seq, YTHDF2 RIP-seq, Ribosome profiling, and ATAC-seq data reported in this paper are Gene Expression Omnibus (GEO): GSE 112022, GSE 136419, and GSE 151071.