
Figure 1. RIPK3- and MLKL-dependent necroptosis mediated by various stimuli.
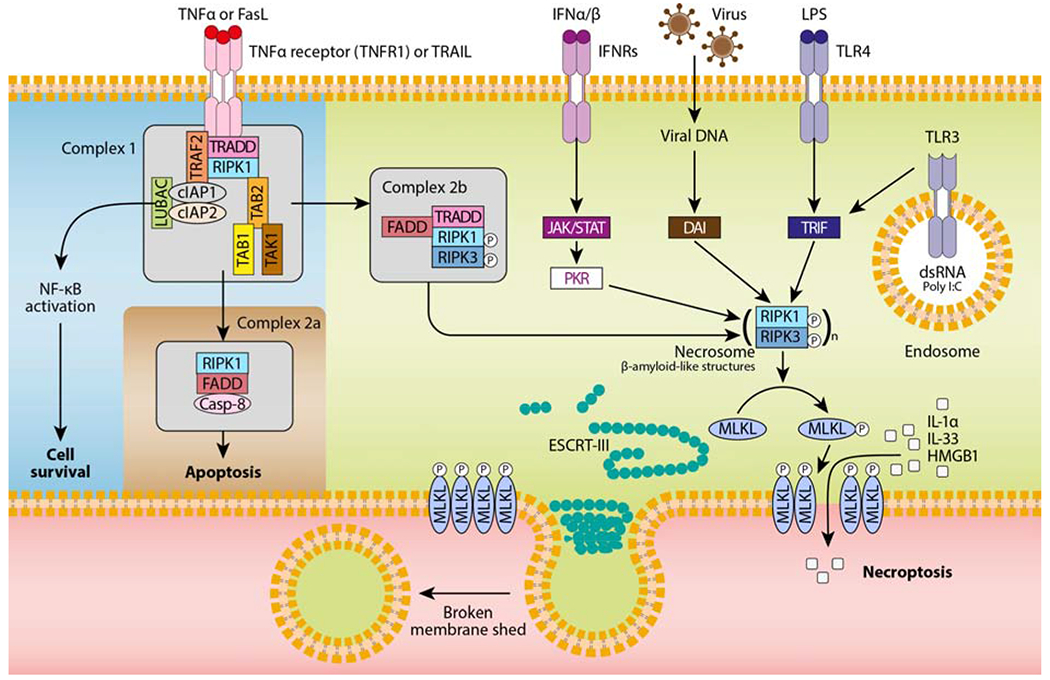
TNF-α or FasL binding to their receptor induces the formation of a receptor proximal complex (complex 1), which consists of adaptor proteins (e.g., TRADD, tumor necrosis factor receptor type 1-associated death domain; FADD, fas-associated protein with death domain; TAB, transforming growth factor beta-binding protein), ubiquitin ligases (e.g., TRAF2, TNF receptor associated factor 2; LUBAC, linear ubiquitin chain assembly complex; cIAPs, cellular inhibitors of apoptosis), and kinases (e.g., TAK, transforming growth factor beta-activated kinase 1; RIPK1, receptor interacting protein kinase 1). The ubiquitination of RIPK1 provides docking sites for the recruitment of key proteins leading to cell survival by nuclear factor κB (NF-κB)-dependent upregulation of pro-survival genes. Then, complex 1 internalization leads to the formation of complex 2a, which ultimately results in caspase-8-dependent apoptosis. Finally, when caspases or cIAPs are inhibited, RIPK1 associates with RIPK3 to form the necrosome (complex 2b), which in turn recruits the pseudokinase MLKL. RIPK3-mediated phosphorylation of MLKL results in MLKL translocation to the plasma membrane and pore formation. Other necroptotic stimuli such as IFNα/β, virus, pathogen-associated molecular patterns (e.g., LPS, poly I:C) are also depicted. Although the nature of these signalling complexes is not completely characterized, the main mediators activating the necrosome are shown. To antagonize necroptosis, ESCRT-III components are recruited to localized sites of MLKL-directed membrane damage to shed broken membrane into blebs.