

Abstract
An adverse maternal in utero and lactation environment can program offspring for increased risk for metabolic disease. The aim of this study was to determine whether N-acetylcysteine (NAC), an anti-inflammatory antioxidant, attenuates programmed susceptibility to obesity and insulin resistance in offspring of mothers on a high-fat diet (HFD) during pregnancy. CD1 female mice were acutely fed a standard breeding chow or HFD. NAC was added to the drinking water (1 g/kg) of the treatment cohorts from embryonic day 0.5 until the end of lactation. NAC treatment normalized HFD-induced maternal weight gain and oxidative stress, improved the maternal lipidome, and prevented maternal leptin resistance. These favorable changes in the in utero environment normalized postnatal growth, decreased white adipose tissue (WAT) and hepatic fat, improved glucose and insulin tolerance and antioxidant capacity, reduced leptin and insulin, and increased adiponectin in HFD offspring. The lifelong metabolic improvements in the offspring were accompanied by reductions in proinflammatory gene expression in liver and WAT and increased thermogenic gene expression in brown adipose tissue. These results, for the first time, provide a mechanistic rationale for how NAC can prevent the onset of metabolic disease in the offspring of mothers who consume a typical Western HFD.
Introduction
A growing body of evidence suggests that obesity and type 2 diabetes may have their origins in early life. This process in which an adverse maternal in utero environment affects the offspring by increasing the risk of metabolic disease is known as “fetal programming” and results from perturbations in epigenetic phenomena (1). Reduced antioxidant capacity resulting from a high-fat diet (HFD) and/or maternal obesity produces unfavorable changes in the in utero environment, which has metabolic, physiological, behavioral, and molecular effects on the offspring.
N-acetylcysteine (NAC) displays both direct and indirect antioxidant properties (2). Once metabolized, NAC augments the intracellular pool of glutathione (GSH). NAC can also act as a direct scavenger of free radicals (3). In rodents, NAC has been used to ameliorate lipopolysaccharide-induced oxidative stress in pregnancy (4) and improve behavior in offspring of mothers on a HFD during pregnancy (5). NAC also reduces hepatic triglycerides and diacylglycerol (DAG) in rats fed HFD (6). Further, NAC attenuates increases in fat mass, inflammation, and metabolic derangements and upregulates thermogenic genes in high-fat-fed mice (7). In humans, NAC has been shown to be effective for a wide variety of human disorders, including nonalcoholic fatty liver disease and recurrent pregnancy loss (for review, see ref. 8).
Using a well-validated mouse model developed in our laboratory, we previously showed that a mother’s ability to utilize HFD as a substrate can program metabolic disease susceptibility and epigenetic changes in offspring exposed in utero (9–13). We further defined critical periods during development in which the offspring are more vulnerable to the insult of an HFD (14). We also recently reported that NAC prevents histopathologic changes in the placenta, suggesting that the placenta is a potential mediator of NAC’s favorable effects on offspring in our model of HFD-induced metabolic programming (15). The hypothesized mechanism whereby maternal consumption of an HFD results in unfavorable effects on offspring is by driving inflammation and oxidative stress, leading to increased production of reactive oxygen species in the mother, placenta, and offspring (16). Based on the published effects of NAC from our work and that of others, we have hypothesized that NAC would have salutatory effects on both dams and offspring exposed to an HFD. Here, we extend our hypothesis that NAC treatment of HFD-fed dams would impart improvements in both maternal and offspring metabolism and antioxidant capacity. To test our hypothesis, we fed mice normal diets or HFDs beginning 2 weeks prior to mating and continuing throughout pregnancy and lactation and investigated effects of a short treatment of NAC on maternal and offspring metabolic phenotypes.
Research Design and Methods
Animals and Experimental Design
Animal protocols were approved by the Institutional Animal Care and Use Committee at the Albert Einstein College of Medicine in accordance with Animal Welfare Act guidelines as previously described (11–15). Age- and body weight (BW)-matched 12- to 14-week-old wild-type (WT) female mice (CD1 background) were maintained on a control diet (C) (mouse diet no. 5058: 9% fat as soybean oil and fat, 3.59 kcal/g; PicoLab) or switched to HFD (product no. F3282: 35.5% fat as lard, 5.29 kcal/g; Bio-Serv) during the periconceptional period, 2 weeks prior to mating, and throughout pregnancy and lactation (11–15). Females were bred to nonlittermate GLUT4 heterozygous knockout males (Glut4+/−) as previously described (9,10). Pregnancy was determined by the presence of a copulatory plug and defined as embryonic day 0.5 (e0.5). All offspring were genotyped, and only WT (Glut4+/+) were included in the study. With the exception of analyses of offspring weights, each pregnant dam was considered a single experimental unit and care was taken to select a representative male and female from each litter that was neither the smallest nor the largest pup of the litter. Dams were given either plain drinking water (vehicle) or 1 g/kg NAC in the drinking water from e0.5 until e17.5 and restarted on 1 g/kg NAC at postnatal day (PD)3 and continued until weaning (PD21) as previously described (15,17). Genotyping of offspring was performed as previously described (18). Offspring were weaned onto PicoLab mouse diet no. 5053 (low fat: 4.5% fat as soybean oil and animal fat, 54.8% carbohydrate, 3.4 kcal/g) at PD21. Blood was drawn at e18.5 for serum lipidomic analysis and leptin measurement.
Four experimental groups were studied, described as maternal diet+treatment as follows: 1) C+vehicle, 2) C+NAC, 3) HFD+vehicle, and 4) HFD+NAC (Fig. 1A). For elimination of confounding effects of genetic factors related to insulin resistance previously reported in Glut4+/− mice, WT mice were the focus of this study (19,20). Both male and female offspring were studied because of the well-documented sexual dimorphism in metabolic disease sensitivity in rodents (21). Although no definite sexual dimorphism was observed in any outcome measures, some antioxidant effects were slightly greater in males than in females; the results observed in males are presented here in the main text, while the results of the females are presented in Supplementary Material.
Figure 1.

NAC treatment normalized HFD-induced pregnancy weight gain patterns, preserved maternal antioxidant capacity, and improved the HFD maternal lipidome. A: Study design illustrating maternal/offspring diet and treatment, resulting in four groups: C+vehicle (n = 9), C+NAC (n = 13), HFD+vehicle (n = 9), and HFD+NAC (n = 16). B: NAC prevented the excess in weight gain in the first half of pregnancy and the deficiency in weight gain in the second half of pregnancy seen in HFD+vehicle dams. C: NAC normalized the reduction in litter size seen in HFD+vehicle mice. D: NAC prevented the increase in serum leptin found in HFD+vehicle dams at e18.5. E: NAC normalized serum and hepatic GR and GPx activities and hepatic GSH observed in HFD+vehicle dams. F: NAC reduced hepatic diacylglycerol (DG) and TG in HFD+vehicle dams at e18.5. G: NAC reduced serum C12:0, C14:0, and C16:1 fatty acids in HFD+vehicle dams at e18.5. H: NAC increased serum C16:0, C20:0, C22:0, C24:1, and C24:0 ceramides in HFD+vehicle dams. I: NAC increased hepatic C24:0 and decreased C18:0 ceramide in HFD+NAC dams at e18.5. J: NAC reduced mRNA of hepatic ceramide–synthesizing genes measured in HFD+vehicle dams. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; #P < 0.05 by ANOVA (B–E) or Student t test (F–J). In E, significance is denoted by different letters. In E–J, n = 6/group. FFA, free fatty acid; veh, vehicle; wt, weight.
Phenotypic Evaluation
Pregnant mice were placed in individual cages on e0.5; food intake and BW were assessed daily from e0.5 until e18.5. Phenotypic and genotypic evaluation of the offspring was performed as previously described (18). Food intake was determined in 8–10 randomly selected mice from five to seven litters per group housed individually for 3 days before recording of BW and daily food consumption (kilocalories per body weight per day) for a period of 7–10 days. Core body temperature was determined using a digital rectal thermometer.
Indirect Calorimetry
Animals were individually housed in metabolic chambers on a 12 h light, 12 h dark cycle, with lights on at 0700 h. Metabolic measurements (including respiratory exchange ratio to assess substrate utilization and energy expenditure to determine the daily energy demands) were obtained continuously using a Columbus Instruments Comprehensive Lab Animal Monitoring System (CLAMS) (Columbus Instruments, Columbus, OH) open-circuit indirect calorimetry system. Calorimetry data were normalized to individual lean body mass. Results contain data collected over 5 days after at least 2–3 days of adaptation to the metabolic chambers (22).
Intraperitoneal Insulin and Glucose Tolerance Tests and Oral Lipid Challenge Test
Insulin tolerance tests, glucose tolerance tests, and lipid challenge test were done as previously described (9,23).
Lipidomic Analysis
Global lipidomics was performed with an Agilent 1200 Series high-performance liquid chromatography system online with an Agilent 6220 ESI-TOF (Agilent Technologies, Wilmington, DE) (24). For negative mode, a Gemini (Phenomenex, Torrance, CA) or Inspire (Dikma Technologies, Lake Forest, CA) C18 column (5 µm, 4.6 mm × 50 mm) was used with a guard column (C18, 2 µm frit, 2 mm × 20 mm). For positive mode, a Luna (Phenomenex) C5 or Bio-Bond (Dikma Technologies) C4 column (5 µm, 4.6 mm × 50 mm) was used with a guard column (C4, 2 µm frit, 2 mm × 20 mm). Data were collected in both profile and centroid modes using a mass range of 100–1,500 Da. For untargeted analysis, raw data were converted to mzXML format and analyzed by XCMS (20). XCMS output files were filtered by statistical significance (P ≤ 0.05), fold change (≥3), and reproducibility across four independent data sets, and the remaining ions were further verified by manual integration using the MassHunter Qualitative Analysis Program (Agilent, Santa Clara, CA). Lipid classes were assigned using the accurate mass of the ion, the retention times, and the ion intensity. For targeted analysis, samples were prepared as previously described for liquid chromatography/mass spectroscopy analysis (24).
Plasma Serum Analysis
Fed-state blood samples were obtained between 11:00 p.m. and 1:00 a.m. Serum concentrations of insulin (Linco Research, St. Charles, MO), glycerol and triglycerides (Sigma-Aldrich, St. Louis, MO), nonesterified fatty acids (Wako Chemicals, Neuss, Germany), adiponectin (R&D Systems, Minneapolis, MN), and leptin (EMD Millipore, Darmstadt, Germany) were determined by commercially available kits. Glucose levels were determined by use of a glucometer (Precision Q.I.D., a gift from Abbott Laboratories, Chicago, IL). HOMA of insulin resistance (HOMA-IR) was calculated by a formula adapted in by van Dijk et al. (25).
Liver and Fecal Lipid Content
Liver and fecal lipid was extracted using a modification of the Folch method (26,27).
Antioxidant Assays
Reduced GSH was determined by a reaction with DTNB (Ellman’s reagent: 5,5′-dithio-bis-[2-nitrobenzoic acid]). Glutathione reductase (GR) activity was determined in tissue homogenates by the decrease in absorbance at 340 nm as NADPH is oxidized to NADP+. Glutathione peroxidase (GPx) activity was determined in either tissue homogenates or serum by a coupled reaction with GR (28).
Histology
Liver, white adipose tissue (WAT), and brown adipose tissue (BAT) were formalin fixed and paraffin embedded, sectioned at 5 μm, stained with hematoxylin-eosin (H-E), and visualized using a Zeiss Axioskop 2 light microscope.
Quantitative Real-time PCR Analysis
Total RNA was extracted from liver, WAT, and BAT using TRIzol Reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. Quantitative real-time PCR was performed using the Roche LightCycler 480 Real-Time PCR System with SYBR Green Master Mix (Roche, Indianapolis, IN). Primer sets were designed using Universal Probe Library Assay Design Center. For quantitative analysis, all samples were normalized to cyclophilin B and relative mRNA expression levels were determined by the ΔΔ−CT method and expressed as fold change compared with C+vehicle mice. Samples were measured in triplicate for each gene to assess technical variability (n = 6–8/group from six litters). Primers are listed in Supplementary Table 1.
Statistical Analysis
Data represent the mean ± SEM. Statistical analyses were performed using GraphPad Prism software, version 6.0, for Macintosh (www.graphpad.com) (GraphPad Software, San Diego, CA). Comparisons between groups were performed using unpaired Student t test and one-way ANOVA. Post hoc analyses of ANOVA were conducted using Tukey post hoc tests. Acceptable study power was agreed a priori to be ≥80% (type I error of ≤0.20). P < 0.05 was considered statistically significant.
Data and Resource Availability
All data generated or analyzed during this study are included here in the published article (and Supplementary Material). No applicable resources were generated or analyzed during the current study.
Results
NAC Treatment Normalized HFD-Induced Pregnancy Weight Gain Patterns and Preserved Maternal Antioxidant Capacity
To test our hypothesis that NAC treatment would prevent metabolic dysfunction in dams consuming an HFD, we first tested NAC’s effect on maternal weight gain throughout gestation. NAC treatment prevented the excessive weight gain associated with consumption of an HFD in the first half of pregnancy and normalized the deficiency in weight gain observed in HFD+vehicle mice in the second half of pregnancy (Fig. 1B). Changes in maternal BW were not due to changes in food intake when NAC treatment was added to the HFD (20.95 ± 1.45 vs. 20.89 ± 1.70 kcal/day [not significant]). Unlike its effects in HFD dams, NAC treatment had no effect on the weight gain of C dams during pregnancy (Fig. 1B). Total weight gain during pregnancy was not different among the groups (Fig. 1B). NAC treatment normalized the reduction in litter size seen in HFD+vehicle offspring (Fig. 1C). Finally, as anticipated, serum leptin increased in HFD dams; however, remarkably, NAC significantly reduced serum leptin in both C and HFD dams (Fig. 1D).
Having shown that NAC prevents changes in weight gain patterns in HFD dams, we tested its effect on lipotoxicity-driven oxidative stress. HFD consumption was associated with reduced antioxidant capacity as demonstrated by reduced serum and hepatic GR activity and reduced hepatic GPx activity in HFD+vehicle dams compared with C+vehicle dams (Fig. 1E). NAC treatment restored serum GR to normal levels and partially restored hepatic GR and GPx levels (Fig. 1E). Similarly, HFD consumption was associated with reduced levels of hepatic GSH, which was restored to control levels by NAC treatment (Fig. 1E). NAC treatment did not alter serum or hepatic antioxidant parameters of C dams (Fig. 1E).
NAC Treatment Improved the HFD Maternal Lipidome
We also tested whether NAC’s favorable effects on maternal weight gain patterns and oxidative stress in HFD dams were accompanied by improvements in the maternal lipidome. NAC treatment significantly reduced hepatic DAG and triacylglycerol (TG) levels (Fig. 1F) and serum levels of lauric, myristic, and palmitoleic acid in HFD dams (Fig. 1G). Surprisingly, HFD+NAC dams had increased serum levels of several long- and very-long-chain ceramides, including C16:0, C20:0, C22:0, C24:1, and C24:0 (Fig. 1H). Conversely, the hepatic concentrations of pathogenic long-chain C18:0 ceramides were significantly decreased in the livers of NAC-treated animals, while the levels of benign very-long-chain C24:0 ceramides were increased (Fig. 1I). NAC treatment also significantly reduced hepatic expression of numerous ceramide-synthesizing genes, including Sptcl1, Sptcl2, and Cers1–Cers6 (Fig. 1J). NAC had similar, but milder, effects in C dams, with reductions in hepatic TG and several plasma fatty acids (C14:0, C16:0, C17:0, C18:0, C20:4, and C22:6) but was without effect on plasma and hepatic ceramides (Supplementary Fig. 1).
NAC Treatment Normalized Abnormal Prenatal and Postnatal Growth Patterns in HFD Offspring
Having established NAC’s significant effects on HFD-induced lipotoxicity in the dams, we proceeded to test NAC’s effects on the HFD offspring phenotype. NAC treatment normalized birth weight of HFD offspring (Fig. 2A). NAC treatment prevented HFD-induced early-life catch-up growth (CUG) in offspring (Fig. 2B and Supplementary Fig. 2A and B). Interestingly, normalization of the CUG of HFD+NAC offspring was associated with a reduction in food intake compared with the hyperphagia observed in HFD+vehicle in male offspring (Fig. 2C), while in female offspring, HFD+NAC actually consumed more food than HFD+vehicle (Supplementary Fig. 2C). NAC’s effect on postnatal growth was maintained over a long period, as BWs of HFD+NAC offspring were ∼15–20 g lower than those of offspring from untreated HFD dams at 36 weeks of age (Fig. 2D and Supplementary Fig. 2D). Despite lowered birth weight (Fig. 2A), NAC treatment of C dams did not affect postnatal growth (Fig. 2B and Supplementary Fig. 2A and B) or food intake (Fig. 2C and Supplementary Fig. 2C) of their offspring. Neither maternal NAC treatment nor maternal HFD exposure was associated with altered fecal lipid content in offspring, indicating that phenotypic differences seen are not accounted for by altered lipid absorption (Supplementary Fig. 3).
Figure 2.
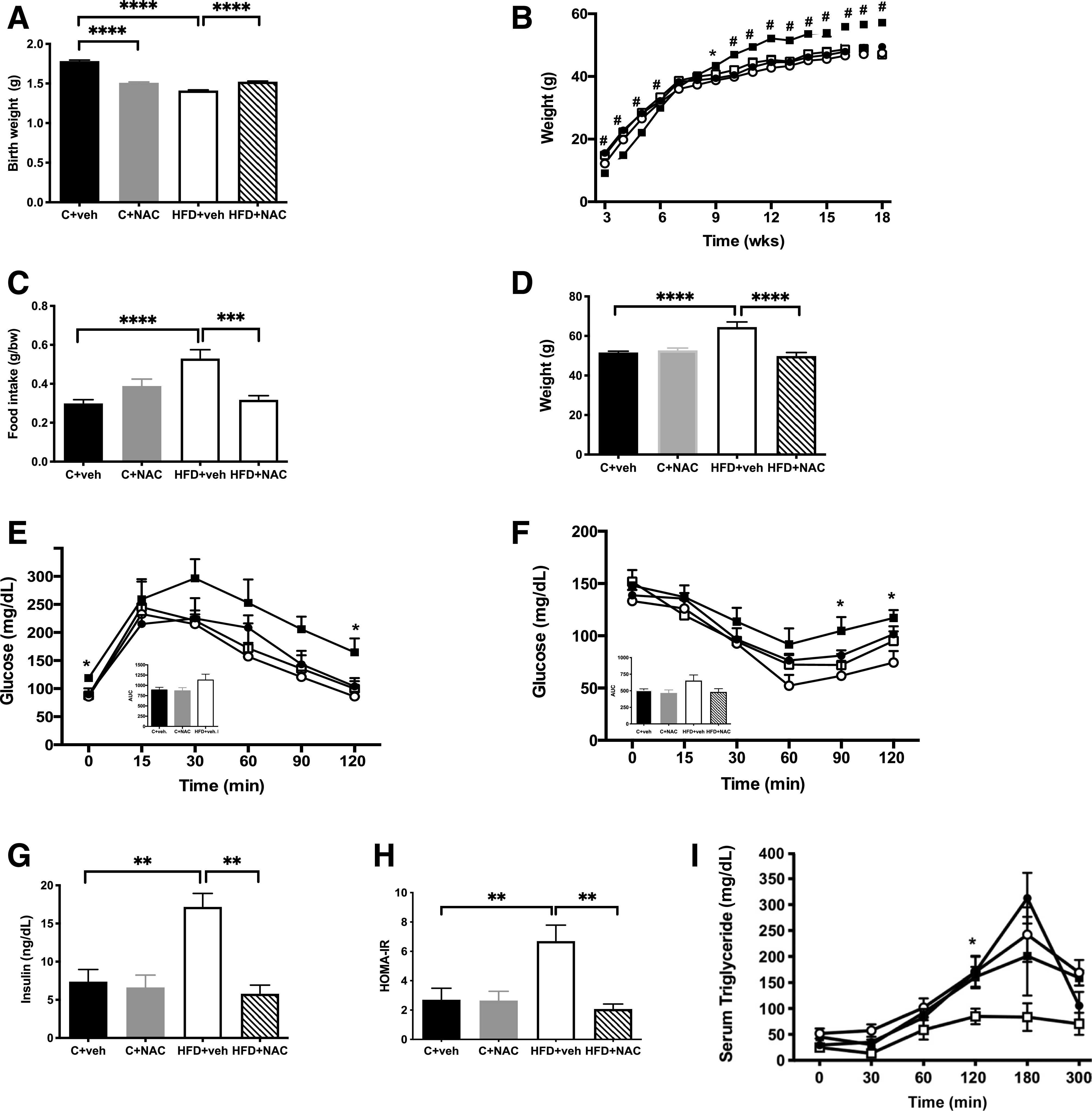
NAC treatment improved metabolic function in HFD offspring. A: Exposure to NAC normalized the birth weights seen in HFD+vehicle offspring (n = 25–40/group). B: NAC prevented the early-life CUG seen in HFD+vehicle offspring (n = 25–40/group). C: NAC normalized food intake in HFD+vehicle offspring (n = 8–10/group). D: NAC normalized the increase in terminal BW observed at 36 weeks in HFD+vehicle offspring (n = 25–40/group). E: NAC improved glucose tolerance compared with HFD+vehicle (▪) at 7 weeks of age (n = 6/group). Inset represents area under the curve (AUC). F: HFD+NAC resulted in improved insulin sensitivity in offspring at 8 weeks of age compared with HFD+vehicle offspring (n = 6/group). Inset represents AUC. G: Fed insulin (n = 5–8). H: HOMA-IR (n = 5–8/group). I: NAC improved the ability to clear lipids in response to an oral lipid load assessed at 28 weeks (n = 5/group). ●, C+vehicle; ○, C+NAC; ▪, HFD+vehicle; □, HFD+NAC. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; #P < 0.0001 by ANOVA. veh, vehicle; wks, weeks.
NAC Treatment Improved Metabolic Function in HFD Offspring
NAC’s favorable effects on growth patterns in HFD offspring were accompanied by favorable effects on metabolic function. NAC treatment normalized elevated fasting and postprandial glucose levels at 30 min (Fig. 2E and Supplementary Fig. 2E), improved insulin sensitivity as determined by insulin tolerance tests (Fig. 2F and Supplementary Fig. 2F) and insulin resistance as determined by reduced fed serum insulin levels (Fig. 2G and Supplementary Fig. 2G) and HOMA-IR (Fig. 2H and Supplementary Fig. 2H), and improved the ability to clear lipids in response to an oral lipid load in HFD+NAC offspring (Fig. 2I). NAC treatment had no effect on the metabolic profile of offspring born to C dams (Fig. 2D–I and Supplementary Fig. 2C–H).
NAC Treatment Normalized Adiposity and Prevented WAT Inflammation in HFD Offspring
To further investigate NAC’s effect on the HFD offspring phenotype, we examined changes in gonadal WAT across the different groups. NAC treatment normalized the increase in WAT pad size (Fig. 3A) and weight (Fig. 3B and Supplementary Fig. 4A) in HFD+NAC offspring and the HFD-induced interstitial histiocytic WAT inflammation observed only in males (Fig. 3C and Supplementary Fig. 4C). Consistent with the overall reduction in WAT mass (Supplementary Fig. 4B), NAC treatment was associated with a microvesicular pattern, more pronounced in females than in males (Fig. 3C and Supplementary Fig. 4C). NAC also decreased mRNA expression of proinflammatory cytokines, Mcp1, Tnfα, Il-1β, and Il-6, and inflammatory markers, F4/80 and Cd11c (Fig. 3D). In addition, NAC treatment prevented HFD-induced interstitial histiocytic WAT inflammation in males (Fig. 3C [indicated with arrow in HFD+vehicle panel]). NAC treatment significantly decreased serum leptin (Fig. 3E and Supplementary Fig. 4D), increased serum adiponectin in males but not females (Fig. 3F and Supplementary Fig. 4E), and increased WAT adiponectin mRNA compared with HFD+vehicle (Fig. 3D). NAC treatment of C dams had no effect on adipocyte size (Fig. 3C and Supplementary Fig. 4C); mRNA expression of F4/80, Cd11c, Mcp1, Tnfα, Il-6, adiponectin, or leptin (Fig. 3D); or serum leptin (Fig. 3E and Supplementary Fig. 4D) or adiponectin (Fig. 3F).
Figure 3.
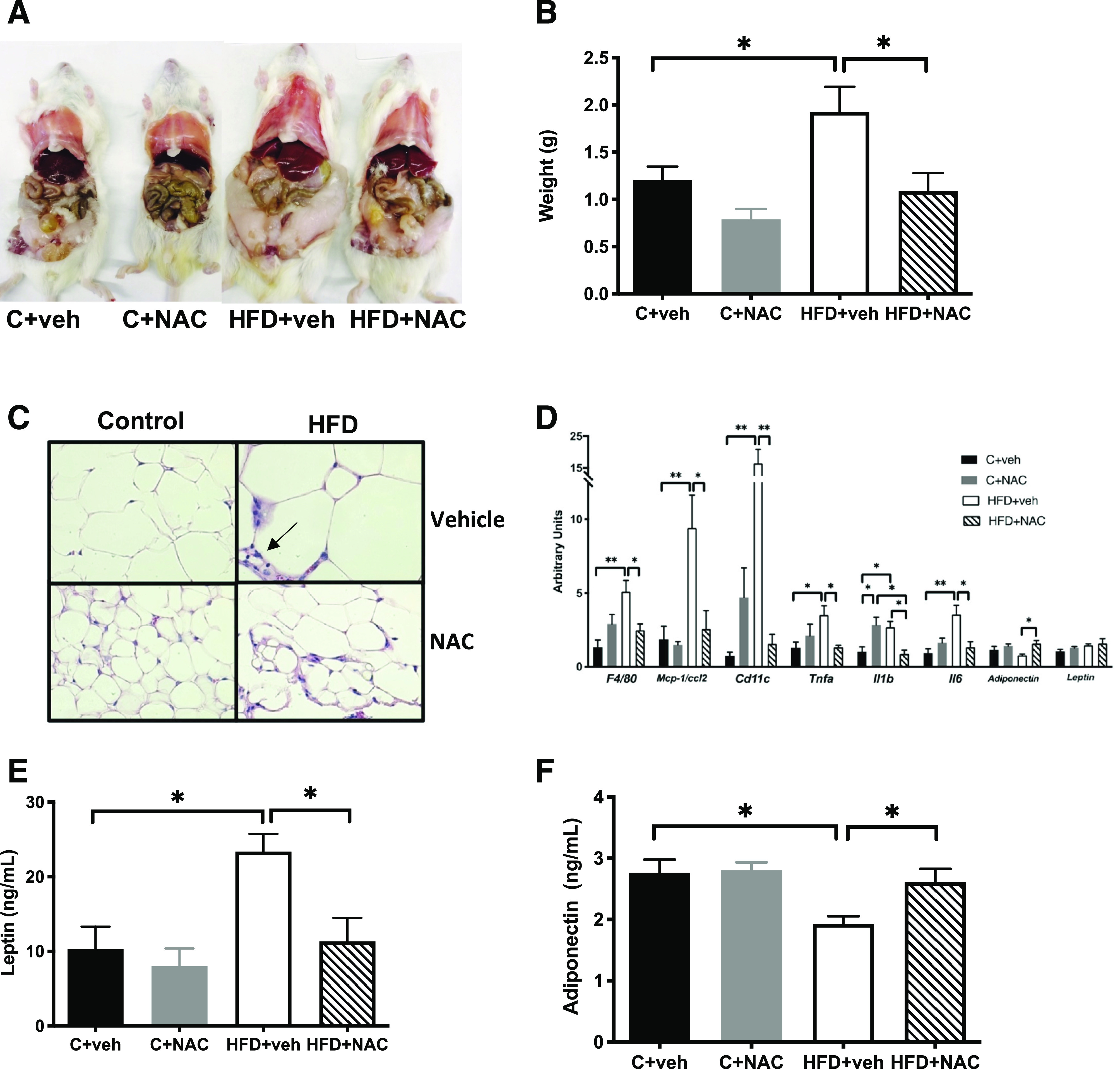
NAC treatment normalized adiposity and prevented WAT inflammation in HFD offspring. A and B: NAC normalized gonadal fat pad size and weight in HFD+vehicle. C: NAC normalized adipocyte hypertrophy and prevented inflammation seen with H-E staining in HFD+vehicle and produced a microvesicular pattern characteristic of “beiging” of WAT (original magnification ×600). D: NAC decreased mRNA expression of proinflammatory cytokine and inflammatory marker genes. NAC normalized serum leptin (E) and adiponectin (F) (n = 5–6/group except n = 3/group in C). *P < 0.05, **P < 0.01 by ANOVA. veh, vehicle.
NAC Treatment Prevented BAT Hypertrophy and Increased Expression of Thermogenic Genes and Core Body Temperature in HFD Offspring
We also tested whether NAC’s favorable effects on WAT would extend to the more metabolically active BAT. NAC treatment normalized BAT weight in HFD offspring (Fig. 4A and Supplementary Fig. 5A), in part by decreasing lipid droplet size (Fig. 4B and Supplementary Fig. 5B). Similarly, NAC exposure significantly increased mRNA expression of the thermogenic genes (Ucp1, Ucp3, and Prdm16) (Fig. 4C). In line with these changes, NAC treatment had a thermogenic effect on male offspring such that NAC-treated HFD offspring displayed core body temperatures that were 0.5°C higher than HFD offspring from untreated dams (Fig. 4D and Supplementary Fig. 5C). Results of indirect calorimetry revealed alterations in substrate utilization as seen as higher respiratory exchange ratio in HFD+NAC offspring compared with HFD+vehicle offspring (Fig. 4E). NAC treatment of C dams had no effect on BAT weight (Fig. 4A and Supplementary Fig. 5A), lipid droplet size (Fig. 4B and Supplementary Fig. 5B), or mRNA expression of Ucp1, Ucp3, and Prdm16 (Fig. 4C).
Figure 4.
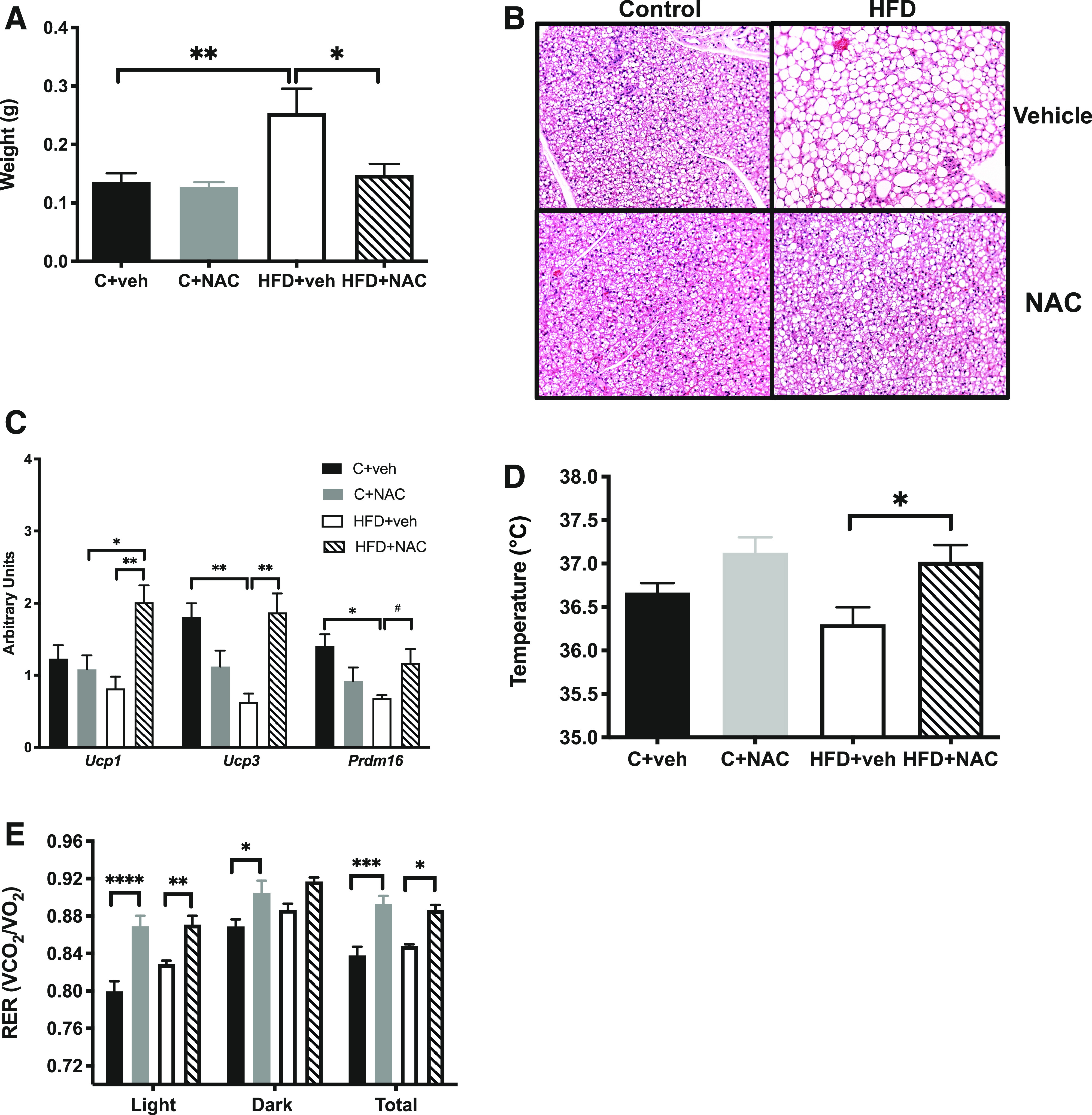
NAC treatment prevented BAT hypertrophy and increased expression of thermogenic genes and core body temperature in HFD offspring. A: NAC prevented the increase in BAT weight compared with HFD+vehicle (n = 5–6/group). B: NAC decreased the lipid droplet size seen with H-E staining in HFD+vehicle (original magnification ×200) (n = 3/group). C: NAC increased mRNA expression of thermogenic genes (n = 5–6/group). D: NAC increased core body temperature of HFD offspring (n = 5–6/group). E: NAC increased respiratory exchange ratio (RER) in the light and dark cycle of C and HFD offspring (n = 4/group). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; #P < 0.05 by ANOVA. veh, vehicle.
NAC Treatment Prevented Hepatic Steatosis and Inflammation in HFD Offspring
NAC treatment normalized the increased liver weight (Fig. 5A and Supplementary Fig. 6A) and TG content (Fig. 5B and Supplementary Fig. 6B) observed in HFD+vehicle. NAC also prevented the development of hepatic steatosis characterized by centrilobular vacuolation seen in these HFD+vehicle (Fig. 5C and Supplementary Fig. 6C) and normalized the HFD-induced increased mRNA expression of genes involved in modulation of cellular lipid content, including Scd1, Srebp1c, Pparα, and Cidec (Fig. 5D). NAC treatment of HFD dams also normalized mRNA expression of the proinflammatory genes Nf-κb and Il-1β in HFD+NAC offspring liver (Fig. 5D). NAC treatment of C dams had no effect on liver weight, histology (Fig. 5A and C and Supplementary Fig. 6A and C), or mRNA expression in offspring (Fig. 5D).
Figure 5.
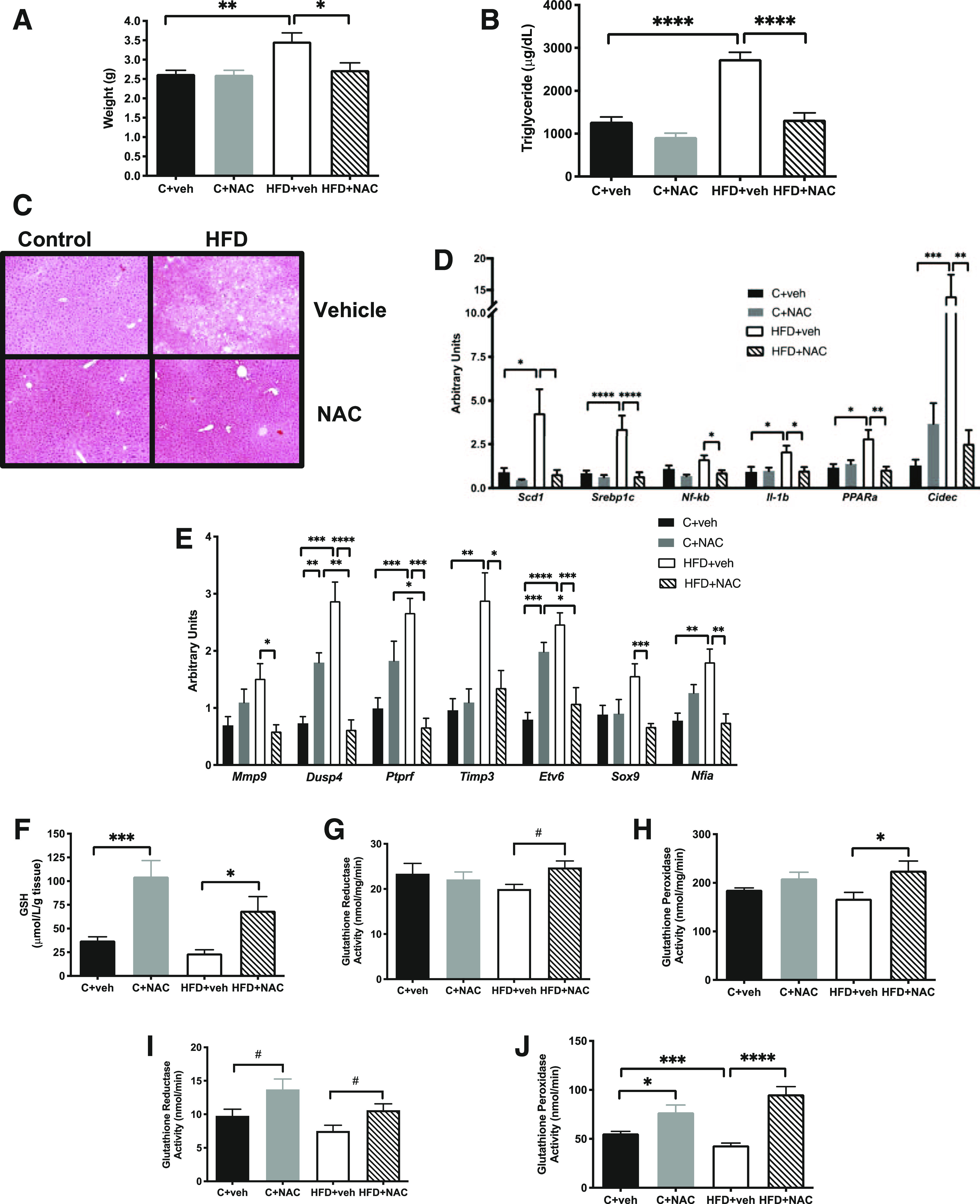
NAC treatment prevented hepatic steatosis and inflammation and preserved antioxidant capacity in HFD offspring. A: NAC normalized liver weight. B and C: The rise in hepatic triglycerides (B) and the increase in hepatic lipid vacuolization (C) (original magnification ×200) observed by H-E staining in HFD+vehicle offspring. NAC also normalized mRNA expression of proinflammatory and lipid-associated genes (D) and genes previously identified as differentially expressed and differentially methylated in HFD+vehicle offspring (E). NAC exposure increased hepatic GSH (F), GR (G), and GPx (H) activity and serum GR (I) and GPx (J) activity. n = 5–6/group except n = 3/group in C. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; #P < 0.05 by ANOVA. veh, vehicle.
NAC Treatment Prevented Lifelong Changes in Hepatic Gene Expression in Differentially Methylated Genes in HFD Offspring
In line with our previous findings detailing the DNA methylation signatures linked to lifelong alterations in gene expression associated with exposure to maternal HFD (11), additional differentially expressed hepatic genes were identified in HFD offspring, including Mmp9, Dusp4, Ptprf, and Timp3 and transcription factors Etv6, Sox9, and Nfia (Fig. 5E). Expression levels of all of these genes in offspring were normalized by NAC treatment of the HFD dams (Fig. 5E). NAC treatment of C dams also resulted in increased Dusp4 and Etv6 mRNA expression in offspring (Fig. 5E).
NAC Treatment Preserved Antioxidant Capacity in HFD Offspring
NAC’s ability to attenuate oxidative stress in HFD dams extended to HFD offspring. NAC treatment increased offspring hepatic GSH in both C and HFD offspring (Fig. 5F and Supplementary Fig. 6D). HFD offspring hepatic GR and GPx activities were also increased with NAC treatment in male offspring (Fig. 5G and H, respectively), while in females NAC treatment only increased GPx activity in HFD offspring (Supplementary Fig. 6E and F). NAC treatment increased serum GR activity in all offspring (Fig. 5I and Supplementary Fig. 6G). Finally, decreased serum GPx activity was seen in HFD+vehicle compared with C+vehicle offspring, while NAC treatment increased serum GPx activity in C male and both male and female HFD offspring (Fig. 5J and Supplementary Fig. 6H).
Discussion
Using a well-validated animal model of fetal programming previously characterized by our laboratory (9,11,12,15), we show here, for the first time, that NAC treatment of HFD-fed dams has profound effects on maternal antioxidant capacity and lipidome with favorable changes in lipid species and maternal leptin levels that are associated with lifelong improvements in the offspring metabolic phenotype. NAC normalized an abnormal pattern of early pregnancy weight gain (9). This is an important finding, as abnormal pregnancy weight gain has been associated with childhood obesity (29), as well as reduced insulin sensitivity and glucose tolerance in the mother, which exposes the fetus to elevated maternal glucose levels (29). In addition, NAC treatment resulted in increased antioxidant capacity as well as decreased maternal leptin levels. Disruption of the signaling capacity of leptin associated with gestational obesity may lead to pregnancy complications as a result of aberrant fuel partitioning in utero (30), as well as lead to inflammation (31). Leptin regulates proinflammatory cytokines that are able to cross the placenta and the blood-brain barrier, and exposing the offspring to high levels of leptin may result in neurodevelopmental dysfunction (32). Consistent with NAC’s effects reported here, recent studies show that elevated maternal leptin levels in humans interfere with normal decidual vessel remodeling, leading to preeclampsia (33). In addition, elevated leptin levels in offspring due to an HFD in utero reduced hypothalamic phosphorylated STAT3 (34), which may explain the hyperphagia and obesity observed in HFD offspring as well as the normalization of the food intake of the HFD+NAC offspring in this study.
NAC treatment shifts the maternal lipidome to a more metabolically favorable profile and decreases serum lauric, myristic, palmitoleic, and linolenic acid. Both lauric and myristic acid are major contributors to the increase in cholesterol caused by dietary saturated FAs (35). NAC increased hepatic levels of C24:0 ceramide, believed to be protective against development of glucose intolerance and hepatic insulin resistance (36), while it decreased C18:0, which has previously been linked to obesity and insulin resistance (37). Surprisingly, HFD+NAC dams had increased serum levels of several long- and very-long-chain pathogenic ceramides. As the liver contributes only 50% of serum ceramides, this increase in serum ceramides may reflect compensatory increases from other sources, consistent with adipose-liver cross talk (38 and S.A.S., personal communication). Interestingly, NAC decreased expression of all genes encoding the CerS isoforms in liver of HFD+NAC dams. CerS1 is involved in synthesis of C18:0 and is increased in HFD mice, along with alterations in glucose tolerance and ceramide levels (39). CerS6 is positively correlated with adiposity and hyperglycemia (40). Furthermore, mRNA expression of CerS6 and levels of C16:0 ceramide are increased in WAT of obese humans and these changes correlate with insulin resistance (40). Both CerS5 and CerS6 maintain levels of C16:0 ceramide in mice (40). As expected, the gene expression levels of the ceramide palmitoyltransferases Sptlc1 and Sptlc2 were decreased in HFD+NAC dams in comparison with HFD+vehicle; haploinsufficiency of Sptlc2 has been shown to improve insulin sensitivity after HFD feeding (39). Lastly, NAC treatment reduced the elevations in hepatic DAG and TG in HFD+vehicle typically associated with insulin resistance. In future studies, it will be important to determine whether the effects on offspring of the altered maternal lipidome, leptin sensitivity, and redox status vary depending on the stage of fetal development. Thus, NAC produces profound changes in the maternal lipidome, which may represent part of the mechanism by which NAC produces a spectrum of salutary changes in the offspring phenotype.
In the offspring, NAC treatment resulted in normalization of birth weight and also prevented the CUG seen in HFD+vehicle offspring. Being born small for gestational age (SGA) is associated with both a hyperlipidemic in utero environment and metabolic disease in adulthood (41); additionally, accelerated postnatal growth during early life is an independent risk factor for metabolic disease (42) as seen in HFD+vehicle offspring born small for gestational age in this study. NAC supplementation prevented insulin resistance, fasting hyperglycemia, and glucose intolerance in HFD offspring.
The hyperphagia that we observed in HFD offspring may account for their rapid CUG in early life and may suggest alterations in the regulation of appetite-controlling pathways. Both leptin and insulin are important in long-term feeding regulation, and both are dysregulated in HFD+vehicle offspring (43). NAC resulted in a reduction in both serum leptin and insulin, which is accompanied by changes in body composition, including a reduction in BW and fat mass.
NAC treatment also resulted in changes in WAT and BAT. An increase in multilocular lipid droplets in WAT, and an increase in thermogenesis, evidenced by increased core body temperature and suggested by upregulation of Ucp1, Ucp3, and Prdm16 gene expression in BAT, was observed in the HFD offspring, consistent with NAC’s ability to increase expression of thermogenic genes (7). Future studies will target changes in subcutaneous fat and changes in levels of proteins encoded by the upregulated BAT genes to determine whether there is a thermogenic mechanism by which early developmental NAC exposure in an HFD milieu programs compensatory changes persisting through 36 weeks of age.
NAC treatment prevented multiple programmed changes in the liver linked with exposure to a maternal HFD (44,45). Importantly, NAC normalized differential expression of several hepatic genes regulating metabolism and cellular signaling/development (Dusp4, Ptprf, Timp3, and Etv6), transcription regulators (Sox9 and Nfia), and a human obesity epigenetic marker (Mmp9) (46), all previously shown to have differential DNA methylation and mRNA expression following exposure to maternal HFD (11). Curiously, Dusp4, which acts as an important regulator of cell growth within the mitogen-activated protein kinase (MAPK) pathway, and Etv6, which regulates the development and growth of diverse cell types, were increased in C+NAC compared with C+vehicle offspring, which could potentially be of negative impact on the aged offspring. Future studies should be performed to determine the optimal dose of NAC needed to obtain the beneficial effects to the C+NAC dams, placenta, and offspring while minimizing any adverse effects. NAC also prevented increased hepatic TG, increased liver weight, and hepatic steatosis—changes found to be caused by HFD (44,45). Finally, NAC increased mRNA expression of Scd1, Srebp1c, and Cidec, which play important roles in TG storage in liver and WAT (47). NAC exposure resulted in decreased mRNA expression of these genes and prevented hepatic steatosis.
In summary, we show here, for the first time, a molecular basis for improvements in fetal programming of metabolic disease produced by an exposure to an HFD during development. Our data indicate that NAC treatment of HFD dams reduces oxidative stress, which leads to improved metabolic function, an improved lipidome, and decreased leptin resistance. These changes result in a healthier in utero environment, which, as we show here, not only alters the offspring phenotype but, importantly, also prevents differential expression of offspring hepatic genes previously shown to be differentially methylated in response to HFD-programmed metabolic disease. Hence, NAC, an antioxidant which is known to prevent fetal epigenetic programming (48), produces favorable effects on the in utero environment that provide offspring with lifelong protection from metabolic disease. A pictorial representation of our mechanistic model of NAC’s role in decreasing HFD offspring susceptibility to metabolic dysfunction is provided in Fig. 6.
Figure 6.

Unified mechanism for NAC effects on HFD dams that program the metabolic health of offspring. ROS, reactive oxygen species.
Article Information
Acknowledgments. The authors are very grateful to Dr. Ryan Pekson, Albert Einstein College of Medicine, for his valuable assistance with the preparation of some of the figures. This article is dedicated to the authors’ beloved co-author, colleague, and friend, Dr. Ellen B. Katz, who bravely worked on this manuscript in spite of her illness until the end. The authors regret that she was not able to see this work published.
Funding. This work was supported by National Institutes of Health (NIH) grant R21 DK081194 (to M.J.C.), American Diabetes Association grant 1-13-CE-06 (to M.J.C.), and an NIH Ruth Kirschstein predoctoral fellowship (F31 DK093332 [to L.W.]).
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. M.J.C. conceived the idea of the project, supervised all of the experiments, revised the figures, edited the manuscript, and provided funding. L.W. performed the experiments and produced the first draft of the manuscript. Y.S., X.Q.D., B.C., A.S., S.A.S., and E.B.K. provided intellectual input into the design of the project and assisted with some experiments. P.M.V. provided intellectual input into the design of the project and edited the manuscript. S.E.R. evaluated the data, interpreted the histopathology, and revised the manuscript. M.J.C. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
This article contains supplementary material online at https://doi.org/10.2337/figshare.12340631.
References
- 1.Williams L, Seki Y, Vuguin PM, Charron MJ. Animal models of in utero exposure to a high fat diet: a review. Biochim Biophys Acta 2014;1842:507–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Radomska-Lesniewska Dm, Skopinski P. N-acetylcysteine as an anti-oxidant and anti-inflammatory drug and its some clinical applications. Cent Eur J Immunol 2012;37:57–66 [Google Scholar]
- 3.Sadowska AM, Manuel-Y-Keenoy B, De Backer WA. Antioxidant and anti-inflammatory efficacy of NAC in the treatment of COPD: discordant in vitro and in vivo dose-effects: a review. Pulm Pharmacol Ther 2007;20:9–22 [DOI] [PubMed] [Google Scholar]
- 4.Awad N, Khatib N, Ginsberg Y, et al. . N-acetyl-cysteine (NAC) attenuates LPS-induced maternal and amniotic fluid oxidative stress and inflammatory responses in the preterm gestation. Am J Obstet Gynecol 2011;204:450.e15–450.e20 [DOI] [PubMed] [Google Scholar]
- 5.Berry A, Bellisario V, Panetta P, et al. . Administration of the antioxidant N-acetyl-cysteine in pregnant mice has long-term positive effects on metabolic and behavioral endpoints of male and female offspring prenatally exposed to a high-fat diet. Front Behav Neurosci 2018;12:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang R, Le G, Li A, Zheng J, Shi Y. Effect of antioxidant capacity on blood lipid metabolism and lipoprotein lipase activity of rats fed a high-fat diet. Nutrition 2006;22:1185–1191 [DOI] [PubMed] [Google Scholar]
- 7.Ma Y, Gao M, Liu D. N-acetylcysteine protects mice from high fat diet-induced metabolic disorders. Pharm Res 2016;33:2033–2042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mokhtari V, Afsharian P, Shahhoseini M, Kalantar SM, Moini A. A review on various uses of N-acetyl cysteine. Cell J 2017;19:11–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hartil K, Vuguin PM, Kruse M, et al. . Maternal substrate utilization programs the development of the metabolic syndrome in male mice exposed to high fat in utero. Pediatr Res 2009;66:368–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vuguin PM, Hartil K, Kruse M, et al. . Shared effects of genetic and intrauterine and perinatal environment on the development of metabolic syndrome. PLoS One 2013;8:e63021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seki Y, Suzuki M, Guo X, et al. . In utero exposure to a high-fat diet programs hepatic hypermethylation and gene dysregulation and development of metabolic syndrome in male mice. Endocrinology 2017;158:2860–2872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Suter MA, Ma J, Vuguin PM, et al. . In utero exposure to a maternal high-fat diet alters the epigenetic histone code in a murine model. Am J Obstet Gynecol 2014;210:463.e1–463.e11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kruse M, Seki Y, Vuguin PM, et al. . High-fat intake during pregnancy and lactation exacerbates high-fat diet-induced complications in male offspring in mice. Endocrinology 2013;154:3565–3576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Plata MdelM, Williams L, Seki Y, et al. . Critical periods of increased fetal vulnerability to a maternal high fat diet. Reprod Biol Endocrinol 2014;12:80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Williams L, Burgos ES, Vuguin PM, et al. . N-acetylcysteine resolves placental inflammatory-vasculopathic changes in mice consuming a high-fat diet. Am J Pathol 2019;189:2246–2257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McMurray F, MacFarlane M, Kim K, et al. . Maternal diet-induced obesity alters muscle mitochondrial function in offspring without changing insulin sensitivity. FASEB J 2019;33:13515–13526 [DOI] [PubMed] [Google Scholar]
- 17.Williams L, Charron MJ, Sellers RS. High post-natal mortality associated with defects in lung maturation and reduced adiposity in mice with gestational exposure to high fat and N-acetylcysteine. Res Vet Sci 2017;114:262–265 [DOI] [PubMed] [Google Scholar]
- 18.Katz EB, Stenbit AE, Hatton K, DePinho R, Charron MJ. Cardiac and adipose tissue abnormalities but not diabetes in mice deficient in GLUT4. Nature 1995;377:151–155 [DOI] [PubMed] [Google Scholar]
- 19.Stenbit AE, Tsao TS, Li J, et al. . GLUT4 heterozygous knockout mice develop muscle insulin resistance and diabetes. Nat Med 1997;3:1096–1101 [DOI] [PubMed] [Google Scholar]
- 20.Stenbit AE, Burcelin R, Katz EB, et al. . Diverse effects of Glut 4 ablation on glucose uptake and glycogen synthesis in red and white skeletal muscle. J Clin Invest 1996;98:629–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mauvais-Jarvis F, Arnold AP, Reue K. A guide for the design of pre-clinical studies on sex differences in metabolism. Cell Metab 2017;25:1216–1230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van de Wall E, Leshan R, Xu AW, et al. . Collective and individual functions of leptin receptor modulated neurons controlling metabolism and ingestion. Endocrinology 2008;149:1773–1785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ranalletta M, Jiang H, Li J, et al. . Altered hepatic and muscle substrate utilization provoked by GLUT4 ablation. Diabetes 2005;54:935–943 [DOI] [PubMed] [Google Scholar]
- 24.Holland WL, Bikman BT, Wang LP, et al. . Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid-induced ceramide biosynthesis in mice. J Clin Invest 2011;121:1858–1870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van Dijk TH, Laskewitz AJ, Grefhorst A, et al. . A novel approach to monitor glucose metabolism using stable isotopically labelled glucose in longitudinal studies in mice. Lab Anim 2013;47:79–88 [DOI] [PubMed] [Google Scholar]
- 26.Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem 1957;226:497–509 [PubMed] [Google Scholar]
- 27.Kraus D, Yang Q, Kong D, et al. . Nicotinamide N-methyltransferase knockdown protects against diet-induced obesity. Nature 2014;508:258–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weydert CJ, Cullen JJ. Measurement of superoxide dismutase, catalase and glutathione peroxidase in cultured cells and tissue. Nat Protoc 2010;5:51–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lau EY, Liu J, Archer E, McDonald SM, Liu J. Maternal weight gain in pregnancy and risk of obesity among offspring: a systematic review. J Obes 2014;2014:524939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Briffa JF, McAinch AJ, Romano T, Wlodek ME, Hryciw DH. Leptin in pregnancy and development: a contributor to adulthood disease? Am J Physiol Endocrinol Metab 2015;308:E335–E350 [DOI] [PubMed] [Google Scholar]
- 31.Jung UJ, Choi MS. Obesity and its metabolic complications: the role of adipokines and the relationship between obesity, inflammation, insulin resistance, dyslipidemia and nonalcoholic fatty liver disease. Int J Mol Sci 2014;15:6184–6223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Valleau JC, Sullivan EL. The impact of leptin on perinatal development and psychopathology. J Chem Neuroanat 2014;61-62:221–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Molvarec A, Szarka A, Walentin S, et al. . Serum leptin levels in relation to circulating cytokines, chemokines, adhesion molecules and angiogenic factors in normal pregnancy and preeclampsia. Reprod Biol Endocrinol 2011;9:124–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun B, Purcell RH, Terrillion CE, Yan J, Moran TH, Tamashiro KL. Maternal high-fat diet during gestation or suckling differentially affects offspring leptin sensitivity and obesity. Diabetes 2012;61:2833–2841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zock PL, de Vries JH, Katan MB. Impact of myristic acid versus palmitic acid on serum lipid and lipoprotein levels in healthy women and men. Arterioscler Thromb 1994;14:567–575 [DOI] [PubMed] [Google Scholar]
- 36.Montgomery MK, Brown SH, Lim XY, et al. . Regulation of glucose homeostasis and insulin action by ceramide acyl-chain length: a beneficial role for very long-chain sphingolipid species. Biochim Biophys Acta 2016;1861:1828–1839 [DOI] [PubMed] [Google Scholar]
- 37.Bergman BC, Brozinick JT, Strauss A, et al. . Muscle sphingolipids during rest and exercise: a C18:0 signature for insulin resistance in humans. Diabetologia 2016;59:785–798 [DOI] [PubMed] [Google Scholar]
- 38.Cai J, Pires KM, Ferhat M, et al. . Autophagy ablation in adipocytes induces insulin resistance and reveals roles for lipid peroxide and Nrf2 signaling in adipose-liver crosstalk. Cell Rep 2018;25:1708–1717.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chavez JA, Summers SA. A ceramide-centric view of insulin resistance. Cell Metab 2012;15:585–594 [DOI] [PubMed] [Google Scholar]
- 40.Turpin SM, Nicholls HT, Willmes DM, et al. . Obesity-induced CerS6-dependent C16:0 ceramide production promotes weight gain and glucose intolerance. Cell Metab 2014;20:678–686 [DOI] [PubMed] [Google Scholar]
- 41.Barker DJ, Hales CN, Fall CH, Osmond C, Phipps K, Clark PM. Type 2 (non-insulin-dependent) diabetes mellitus, hypertension and hyperlipidaemia (syndrome X): relation to reduced fetal growth. Diabetologia 1993;36:62–67 [DOI] [PubMed] [Google Scholar]
- 42.Hermann GM, Miller RL, Erkonen GE, et al. . Neonatal catch up growth increases diabetes susceptibility but improves behavioral and cardiovascular outcomes of low birth weight male mice. Pediatr Res 2009;66:53–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Alfaradhi MZ, Ozanne SE. Developmental programming in response to maternal overnutrition. Front Genet 2011;2:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McCurdy CE, Bishop JM, Williams SM, et al. . Maternal high-fat diet triggers lipotoxicity in the fetal livers of nonhuman primates. J Clin Invest 2009;119:323–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oben JA, Mouralidarane A, Samuelsson AM, et al. . Maternal obesity during pregnancy and lactation programs the development of offspring non-alcoholic fatty liver disease in mice. J Hepatol 2010;52:913–920 [DOI] [PubMed] [Google Scholar]
- 46.Feinberg AP, Irizarry RA, Fradin D, et al. . Personalized epigenomic signatures that are stable over time and covary with body mass index. Sci Transl Med 2010;2:49ra67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Reynolds TH IV, Banerjee S, Sharma VM, et al. . Effects of a high fat diet and voluntary wheel running exercise on cidea and cidec expression in liver and adipose tissue of mice. PLoS One 2015;10:e0130259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Herrera EA, Cifuentes-Zúñiga F, Figueroa E, et al. . N-acetylcysteine, a glutathione precursor, reverts vascular dysfunction and endothelial epigenetic programming in intrauterine growth restricted guinea pigs. J Physiol 2017;595:1077–1092 [DOI] [PMC free article] [PubMed] [Google Scholar]