

Abstract
Aims
Chronic heart failure (CHF) can be caused by autoantibodies stimulating the heart via binding to first and/or second extracellular loops of cardiac β1‐adrenoceptors. Allosteric receptor activation depends on conformational features of the autoantibody binding site. Elucidating these features will pave the way for the development of specific diagnostics and therapeutics. Our aim was (i) to fine‐map the conformational epitope within the second extracellular loop of the human β1‐adrenoceptor (β1ECII) that is targeted by stimulating β1‐receptor (auto)antibodies and (ii) to generate competitive cyclopeptide inhibitors of allosteric receptor activation, which faithfully conserve the conformational auto‐epitope.
Methods and results
Non‐conserved amino acids within the β1ECII loop (compared with the amino acids constituting the ECII loop of the β2‐adrenoceptor) were one by one replaced with alanine; potential intra‐loop disulfide bridges were probed by cysteine–serine exchanges. Effects on antibody binding and allosteric receptor activation were assessed (i) by (auto)antibody neutralization using cyclopeptides mimicking β1ECII ± the above replacements, and (ii) by (auto)antibody stimulation of human β1‐adrenoceptors bearing corresponding point mutations. With the use of stimulating β1‐receptor (auto)antibodies raised in mice, rats, or rabbits and isolated from exemplary dilated cardiomyopathy patients, our series of experiments unmasked two features of the β1ECII loop essential for (auto)antibody binding and allosteric receptor activation: (i) the NDPK211–214 motif and (ii) the intra‐loop disulfide bond C209↔C215. Of note, aberrant intra‐loop disulfide bond C209↔C216 almost fully disrupted the functional auto‐epitope in cyclopeptides.
Conclusions
The conformational auto‐epitope targeted by cardio‐pathogenic β1‐receptor autoantibodies is faithfully conserved in cyclopeptide homologues of the β1ECII loop bearing the NDPK211–214 motif and the C209↔C215 bridge while lacking cysteine C216. Such molecules provide promising tools for novel diagnostic and therapeutic approaches in β1‐autoantibody‐positive CHF.
Keywords: Antibody/autoantibody, β1‐adrenoceptor/β1‐adrenergic receptor, Chronic heart failure, Conformational auto‐epitope, Cyclic peptides/cyclopeptides, Cyclopeptide therapy
Introduction
Dilated cardiomyopathy (DCM)—defined as progressive cardiac dilatation and dysfunction without coronary heart disease—mostly affects younger adults. It frequently entails severe heart failure and transplantation. Prevalence and annual incidence amount to 0.5% and 0.1%, respectively. 1 According to the 2008 ESC cardiomyopathy classification—within the non‐familial forms—autoimmune DCM has been recognized as a proper pathogenetic entity. 2 Autoimmunity arising from acute myocardial inflammation induced by microbial or viral agents is considered one key factor in the pathogenesis of autoimmune DCM. Progress to chronic immune‐cardiomyopathy and chronic heart failure (CHF) is common in patients unable to clear the infecting agent, 3 showing persistent inflammation, 4 and/or having a familial pre‐disposition to autoimmune reactions. 5 DCM autoimmunity encompasses autoantibodies directed against myocardial antigens 6 , 7 , 8 and cardiovascular G‐protein‐coupled receptors (for review, see Boivin‐Jahns and Jahns 9 ), most notably the β1‐AR. 10 , 11 , 12 , 13 The latter (termed anti‐β1‐aabs) are suspected to play a pivotal pathogenic role because anti‐β1‐aab‐positive compared with aab‐negative DCM patients have a more severely depressed cardiac function 13 and a threefold increased risk for cardiovascular death. 10 The incidence of anti‐β1‐aabs can precede DCM symptoms by many years, 14 and selective removal of anti‐β1‐aabs can decelerate disease progression and improve outcome in β1‐aab‐positive DCM patients. 15 , 16 Most DCM‐relevant anti‐β1‐aabs trigger a similar conformational switch of the β1‐AR molecule as the true agonists 17 and elicit detrimental cardiac effects similar to chronic catecholamine challenge. The pathogenic relevance of stimulating anti‐β1‐aabs in DCM is corroborated by DCM equivalents induced in rodents by β1‐AR immunization, isogenic antibody transfer, and corresponding rescue experiments. 11 , 18 , 19 , 20 At bottom line, chronic cAMP stimulation by anti‐β1‐aabs appears to play a major role in DCM pathogenesis. Because blockade of sympathetic hormone receptors by beta‐receptor antagonists has proven beneficial in CHF, a first logical step was to explore whether these drugs could also (pharmacologically) neutralize the cellular effects of stimulating anti‐β1‐aabs. 21 Although β1‐selective (e.g. bisoprolol and metoprolol) and non‐selective beta‐blockers (e.g. alprenolol and carvedilol) were able to significantly reduce anti‐β1‐aab‐stimulated cAMP production, in the end, all tested substances—even at saturating concentrations—blocked anti‐β1‐aab‐mediated effects only partially [resulting in a 50% (alprenolol) up to 70% (carvedilol) reduction of the antibody‐induced fluorescence resonance energy transfer (FRET) signals]. Consequently, clinical disease management would greatly profit from diagnostic determination and direct targeted intervention against stimulating anti‐β1‐aabs, but the prerequisite knowledge of the structural features of the auto‐epitope crucial for cAMP stimulation is yet too limited for that.
DCM‐relevant anti‐β1‐aabs target the first (β1‐ECI) and/or second extracellular loops (β1‐ECII) of the β1‐AR. 22 , 23 IgG binding to immobilized linear peptides representing these domains has been used to determine the prevalence of anti‐β1‐aabs in various diseases. However, these assays exhibit low sensitivity/specificity and, in general, fail to discriminate patients from healthy subjects in a non‐random fashion, because the disease‐relevant anti‐β1‐aabs target specific conformations of β1‐ECI and/or β1‐ECII that are not properly represented by immobile linear peptides. To date, DCM‐relevant anti‐β1‐aabs can only be detected by native assays or functional readouts. 19 , 24 , 25 However, the many patients potentially to be screened for anti‐β1‐aabs (i) require clinical diagnostics based on conventional procedures [e.g. enzyme‐linked immunosorbent assay (ELISA)] for measuring IgG binding to synthetic representations of the very auto‐epitope conformation specifically targeted in the course of allosteric β1‐AR activation. 17 , 24 Interestingly, circular peptide representations of β1‐ECII (alone or as an add‐on to cardioselective β1‐blockers) could stop progression and even revert CHF in rats subjected to β1ECII immunization(s), whereas cardioselective β1‐blockers alone were only able to stabilize the dilated cardiomyopathic phenotype. 19 , 20 Therefore, cyclopeptides might provide an adequate synthetic mimic of the DCM‐relevant conformational auto‐epitope. Following up on this idea, we unmask here the structural features within β1‐ECII that are essential and specific for allosteric receptor activation by anti‐β1‐aabs and required in cyclopeptides for a neutralization of that effect.
Methods
Anti‐β1ECII antibodies
Anti‐β1ECII antibodies were raised in male Lewis rats 11 or rabbits (Dianova, Hamburg, Germany) or mice (mouse Mab 23‐6‐7, BioGenes, Berlin, Germany 26 ) by immunization with β1‐ECII/glutathione S‐transferase (GST) fusion proteins. In addition, a monoclonal rat antibody was generated by fusing spleen cells from anti‐β1ECII‐positive immunized rats with a multiple myeloma Sp2/0‐Ag14 cell‐line (rat Mab 13F6 26 ). Hybridoma cells expressing mouse Mab 23‐6‐7 and rat Mab 13F6 were submitted to DSMZ‐Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH (Braunschweig). Depositing was carried out according to the rules of the Budapest Treaty (accession numbers DSM ACC3121 and ACC3174).
Human anti‐β1‐aabs were gained from representative CHF patients. 21 Because of the relatively large amounts of serum necessary to isolate human IgG for the serial functional assays, as a proof of concept in the present work, IgG was isolated from three DCM patients only {two men [56a, New York Heart Association (NYHA) IV, left ventricular ejection fraction (LVEF) 29%, and 53a, NYHA III, LVEF 31%] and one woman (29a, NYHA IV, LVEF 26%)}. In all three patients, LVEF was determined by ventriculography, and coronary heart disease was excluded by coronary angiography. At the time of invasive diagnostics, all three patients were stable under standard CHF medication including angiotensin‐converting enzyme inhibitor/AT1 receptor blockers, beta‐blockers, and aldosterone antagonists. In none of the patients, exposure to cardio‐toxic substances, myocarditis, or other systemic heart diseases was evident from clinical history. IgG was freshly isolated from the respective (human or animal) sera and dialyzed against the appropriate assay buffers. Immuno‐reactivity of rodent IgG against a linear 25 amino acid (AA) β1ECII peptide (residues 199–123) was determined by ELISA. 12 , 13
Cyclopeptides
Cyclopeptides corresponding to AA residues 200–220 of the human β1‐AR and constituting the entire β1‐ECII 27 were cyclized between the C‐terminal glutamate and the free N‐terminal amino group (Peptide Speciality Laboratories, Heidelberg, Germany). Non‐conserved residues differing between the β1‐AR and β2‐AR were sequentially substituted by alanine. Cysteine216 (C216) was replaced with α‐aminobutyric acid (B) to prevent aberrant disulfide bridging to C209. Corresponding 22‐mer cyclopeptides of the second extracellular loop of the human β2‐adrenergic receptor (β2‐ECII) served as negative controls. In 18‐mer cyclopeptides representing a minimal ECII structure encompassing residues 204–219 of the human β1‐AR, C215 or C216 was replaced with serine to selectively disrupt potential intra‐loop disulfide bridges (for further details, see Figure 2 and Table S1). Cyclopeptides were freeze‐dried, reconstituted in water, and stored at −20°C. For the functional assays, anti‐β1‐abs/β1‐aabs were pre‐incubated with 40 mol of cyclopeptides/mol IgG, assuming a 1:1 stoichiometry between the cyclic 22AA‐ECII peptide variants (~3.0 kDa) and one IgG molecule (~150 kDa). In fact, the molar excess was only 20‐fold, assuming two variable (Fab) chains with two antigen‐binding regions per IgG molecule. Antigen‐binding regions are further subdivided into hypervariable (HV) and framework (FR) regions; HV regions comprise about five to eight AAs directly contacting a portion of the antigen's surface. 28 IgG was freshly isolated from the respective (human or animal) sera by caprylic acid precipitation, dialyzed against the appropriate assay buffers, and allowed to interact with the Cyclopeptides (CPs) for 16 h at 4°C (cold room) in an Eppendorf cup on a gently rotating incubator. After short spin, the supernatant was used for the assays.
Figure 2.
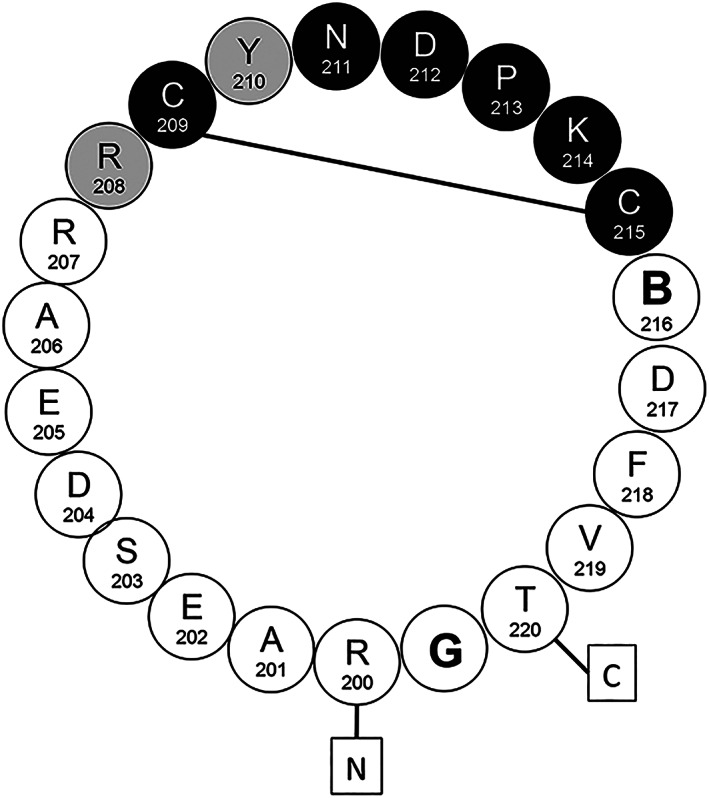
Schematic representation of the 22‐mer C/C/B‐β1 cyclopeptide mimicking the human β1ECII bearing the essential motif of the clinically relevant auto‐epitope: circles with capitals specify amino acids (single letter code). Numbers specify their position in the sequence of the human β1‐AR. ‘B' stands for the artificial amino acid α‐aminobutyrate substituting C216 in order to prevent a non‐physiological S‐S‐bond C209↔C216. Boxed letters N and C indicate the termini of the predicted β1ECII loop. The full line between C209 and C215 indicates the essential intra‐loop disulfide bridge; black circles depict amino acid residues essential for the neutralization of stimulating rodent anti‐β1ECII‐abs. Grey circles represent additional amino acid residues supposed to be relevant for the neutralization of stimulating human anti‐β1ECII‐aabs. Amino acid residues R207, E205, S203, and T220 were not found essential by directed mutational analysis.
cAMP measurements by a fluorescence resonance energy transfer sensor
cAMP measurements by a FRET sensor using HEK‐β1E1 cells stably co‐expressing the human β1‐AR and a FRET sensor for intracellular cAMP followed published procedures. 21 Cells were grown on poly‐d‐lysin‐coated 96‐well plates (Ibidi, Martinsried, Germany). Endogenous β2‐AR was blocked with ICI‐118551 (0.1 μM). Cells were imaged with a fluorescence microscope optimized for the cyan fluorescent protein (CFP)/yellow fluorescent protein (YFP) donor/acceptor pair of the cAMP sensor (iMIC 2000, TILLPhotonics, Martinsried, Germany); CFP/YFP emissions were continuously recorded for 32 min. FRET efficiency reflecting intracellular cAMP levels was calculated every 6 s using TillvisION v4.5.59 (TILLPhotonics) and OriginPro 8G (OriginLab, Northampton, USA). Antibody effects normalized to agonist maximum were derived from successive determinations of baseline (10 min) and effect of the antibody tested (20 min) and of (−)‐isoproterenol (1 μM, 2 min) performed on sets of ≈500 cells.
β1‐AR mutants
cDNA constructs of the human β1‐AR bearing point mutations E202A, R207A, D212N, P213A, or V219A were generated by site‐directed mutagenesis (Table S2), confirmed by sequencing, transiently expressed in HEK293 cells, and characterized by Western blotting and radioligand displacement. 21 To ensure reproducibility (and sufficient supply) of the test agent ‘anti‐β1ECII antibody' for our experiments with human β1‐AR mutants, we used monoclonal anti‐β1ECII‐abs raised in mice (Mab 23‐6‐7 26 ) or rats (Mab 13F6 26 ), thereby reducing the variability due to potentially different HV regions of polyclonal rabbit, rat, or human IgGs used in the experiments before. 28 HEK293 cells expressing human β1‐AR with the indicated point mutations were exposed (60 min, 37°C) to anti‐β1ECII‐Mabs (0.2 μg/μl) or (−)‐isoproterenol (1 μM) in the presence of 3‐isobutyl‐1‐methylxanthine (100 μM) and ICI‐118551 (37.5 pM). Cellular cAMP was measured by RIA (Coulter‐Immunotech, Krefeld, Germany).
Statistics
After testing for normal distribution (comparable variances on the means of each group), data were analysed by one‐way analysis of variance (ANOVA) with subsequent Dunnett's post‐hoc test for multiple comparisons (comparing all means against the control/reference mean) using GraphPad PRISM 8 (version 8.3.1, GraphPad Software Inc., USA).
Ethics
Animal experiments were approved by the animal ethics committee of the Government of Lower Franconia (Vote No. 55.2‐2531.01‐52/08, Experimental Animal Use and Care Committee, Government of Lower Franconia, Bavaria, Germany). The study of patient's biomaterials complies with the Declaration of Helsinki and was approved by the Medical Ethics Committee of the Medical Faculty of the University of Würzburg (Vote No. 186/07). Informed consent of the donors was obtained.
Results
Role of intra‐loop disulfide bridges in auto‐epitope conformation
The β1‐ECII contains three cysteines significantly determining loop conformation: C209 and C215 form an intra‐loop disulfide bridge, while C216 forms a bridge to β1‐ECI. 29 In cyclopeptides, a possible aberrant bridge C209↔C216 can alter the physiological conformation. To address whether this plays a role for binding and allosteric receptor activation by anti‐β1ECII‐abs, we synthesized two minimal (18‐mer) cyclopeptides representing AA residues 204–219 of the β1‐ECII, which had either C215 (termed 18 C/C/S) or C216 (termed 18 C/S/C) replaced by serine, thereby allowing either the physiological bridge C209↔C215 or the aberrant bridge C209↔C216 (Table S1). The reactivity of these cyclopeptides with stimulating anti‐β1ECII‐abs was assessed using sera of 99 rats having developed high titres of anti‐β1ECII‐abs and a cardiac DCM phenotype after repeated immunizations with β1ECII/GST fusion proteins. 19 , 20 First, we investigated pre‐adsorption of these rat anti‐β1ECII‐abs to either 18‐mer cyclopeptide (at 40‐fold molar excess) prior to ELISA with linear 25‐mer β1ECII peptides. Binding of the large majority of sera (83/99 = 84%) was neutralized by 18 C/C/S (bearing the physiological bond C209↔C215), but not by 18 C/S/C (bearing the aberrant bond C209↔C216). Only four sera were neutralized by 18 C/S/C, and 12 rat sera could not be fully blocked with either cyclopeptide (Table 1).
Table 1.
Effect of intra‐loop disulfide bonds on the binding of anti‐β1‐ECII‐abs a
| Neutralizing cyclopeptides (40 mol/mol IgG) | Effective pre‐adsorption of anti‐β1ECII rat sera (n) (≥90% reduction in ELISA reactivity with β1‐ECII peptides) |
|---|---|
| 18‐mer C/C/S (C209↔C215) | 83 (84%) |
| 18‐mer C/S/C (C209↔C216) | 4 (4%) |
| Not fully blocked | 12 (12%) |
Because β1‐AR‐mediated cAMP stimulation is the readout delineating the clinically relevant cardio‐pathogenic potential of anti‐β1ECII‐abs, 10 we next analysed neutralization of the cAMP stimulatory effect of rabbit anti‐β1ECII‐abs using HEK‐β1E1 cells co‐expressing human β1‐AR with a cAMP FRET reporter. 21 In these cells, intracellular cAMP levels can be continuously monitored via the CFP/YFP emission ratio of the FRET sensor, and cAMP stimulation by anti‐β1ECII‐abs can be normalized to the maximal stimulation obtained with 1 μM (−)‐isoproterenol, measured in the same batch of cells. Rabbit anti‐β1ECII‐abs increased cAMP levels by about 18.8 ± 1.2% of the maximum achieved with the full agonist (−)‐isoproterenol. Pre‐absorption of rabbit anti‐β1ECII‐abs to 18 C/C/S (the 18‐mer cyclopeptide with the physiological bond C209↔C215; 40‐fold molar excess) reduced the stimulatory effect of the antibodies by more than 50% (9.4 ± 2.2%; P < 0.01). By contrast, pre‐absorption of rabbit anti‐β1ECII‐abs to 18 C/S/C (the cyclopeptide with the aberrant bond C209↔C216; 40‐fold molar excess) failed to block the antibody‐induced increases in intracellular cAMP (18.1 ± 2.0%, n.s.; Figure 1). In conclusion, inhibition of anti‐β1ECII‐induced β1‐AR activation by β1ECII‐mimicking cyclopeptides requires the natural intra‐loop disulfide bridge C209↔C215 and is disrupted by the non‐physiological C209↔C216 bond.
Figure 1.
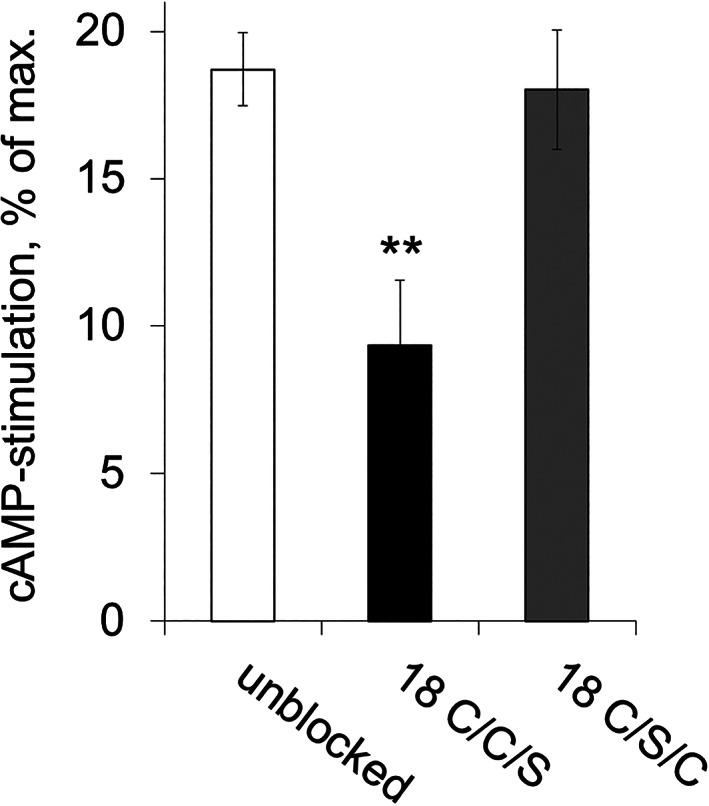
Neutralization of the stimulatory effects of rabbit anti‐β1ECII‐abs by pre‐incubation with cyclopeptides containing intra‐loop bonds C209↔C215 (18 C/C/S) or C209↔C216 (18 C/S/C): Stimulatory effects of polyclonal rabbit anti‐β1ECII‐abs on the human β1‐AR coupled to a CFP/YFP‐FRET sensor for intracellular cAMP, normalized to maximal stimulatory effects obtained by 1 μM (−)‐isoproterenol. Rabbit anti‐β1ECII‐abs were pre‐absorbed with the indicated cyclopeptides (40 mol CP/mol IgG). Unblocked: effect of stimulating rabbit anti‐β1ECII‐abs alone. Data are given as mean ± SEM (n ≥ 5 per experiment; differences between the conditions were analysed by one‐way ANOVA with subsequent Dunnett's post‐hoc test for multiple comparisons; **P < 0.01).
β1ECII loop residues essential for proper conformation of the auto‐epitope
Even the 18‐mer cyclopeptide with the appropriate disulfide bridge yielded only incomplete inhibition, indicating that an optimal representation of the auto‐epitope conformation might require longer constructs with a less bulky C216 substitution. Accordingly, a 22‐mer cyclopeptide was generated, representing the entire predicted β1ECII loop and having cysteine C216 replaced by α‐aminobutyric acid (B), an artificial cysteine homologue not engaging in disulfide bridges. This optimized construct (22 C/C/B‐β1, depicted in Figure 2) inhibited anti‐β1ECII antibody‐induced stimulation by ≥80% (Figure 3, first black column on the left) and served as a template to map further residues within β1ECII essential for the proper conformation of the auto‐epitope. Nine variants of 22 C/C/B‐β1 were synthesized, each having a different non‐conserved AA (compared with the AA constituting the ECII loop of the β2‐AR) sequentially replaced by alanine (E202A, S203A, D204A, R207A, R208A, N211A, D212A, P213A, and K214A). Conserved AA residues essential for adrenoceptor function per se were omitted from the scan (for details, see Table S1).
Figure 3.
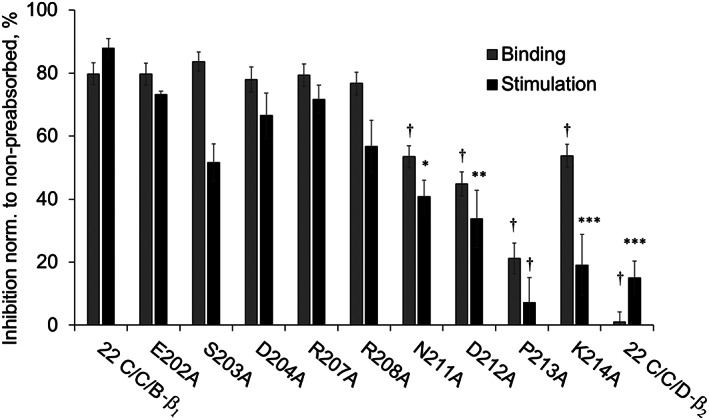
Neutralization of the functional effects of polyclonal rat anti‐β1ECII‐IgG by 22‐mer cyclopeptides mimicking the human β1‐ECII with non‐conserved amino acids (compared with the amino acids constituting the ECII loop of the β2‐AR) sequentially replaced by alanine: sera of 20 cardiomyopathic Lewis rats (immunized with β1ECII/GST fusion proteins) were pre‐absorbed with the indicated cyclopeptide mutants (40 mol CP/mol IgG, 4°C, 16 h). Grey bars: IgG binding to the linear 25 AA (199–223) β1ECII peptide as determined by ELISA (triplicates). Black bars: β1‐AR‐mediated cAMP stimulation in HEK293 cells expressing native β1‐AR functionally coupled to a cAMP FRET sensor. Decreases in ELISA reactivity (grey bars) or receptor activation/cAMP stimulation (black bars) following pre‐absorption with the different cyclopeptide mutants are shown, normalized to the values obtained without blocking cyclopeptides. Columns represent mean values ± SEM of n = 20 rat sera. Differences between the non‐mutated 22 C/C/B‐β1 cyclopeptide and CP mutations were analysed by one‐way ANOVA with subsequent Dunnett's post‐hoc test for multiple comparisons; *P < 0.05; **P < 0.01; ***P < 0.001; †P < 0.0001. Internal negative control: non‐mutated 22C/C/D‐β2 (sequence and alignment, see Table S1).
The reactivity of the 22‐mer C/C/B‐β1 variants with clinically relevant stimulating anti‐β1ECII‐abs was determined by (i) pre‐absorption of autoantibody binding to the linear 25 AA β1ECII peptide (determined by ELISA) and by (ii) pre‐absorption of the receptor‐stimulating capacities of anti‐β1ECII‐abs (determined with cAMP FRET reporter cells). Sera of β1ECII/GST‐immunized cardiomyopathic rats served as a source for DCM‐relevant autoantibodies. Mean results obtained with sera from 20 individual rats are summarized in Figure 3, demonstrating that receptor binding (grey columns) and receptor stimulation (black columns) were efficiently pre‐absorbed (>80%) by all cyclopeptide mutants tested, except for those having disrupted an NDPK motif encompassing AA residues 211–214. The most disruptive mutation was P213A, which almost completely abolished the scavenger effect of the respective cyclopeptide (blocking capacity 22 C/C/B‐β1 vs. P213A: 87.9 ± 3 vs. 7.2 ± 8%, P < 0.0001). The same was observed with a stimulating monoclonal mouse anti‐β1ECII antibody (Figure S1).
To confirm the clinical relevance of these results, comparable neutralization experiments were performed with stimulating anti‐β1‐aabs isolated from selected DCM patients. Figure 4A shows a representative recording of cAMP reporter signals obtained with patient‐derived anti‐β1‐aabs, which induced an increase in β1‐AR‐mediated cAMP production of ≈30% of the maximal signal obtained with saturating concentrations (1 μM) of the full agonist (−)‐isoproterenol. That stimulatory effect was almost fully abolished by pre‐incubation with the (non‐mutated) cyclopeptide 22 C/C/B‐β1, whereas the mutant P213A (earlier found ineffective after pre‐absorption of rodent anti‐β1ECII‐abs; Figure 3 and Figure S1) also failed to block the stimulatory effect of human anti‐β1‐aabs (Figure 4A). The blocking profile of the entire set of mutated cyclopeptides on cAMP stimulation by human anti‐β1‐aabs isolated from selected antibody‐positive DCM patients (male/female) is shown in Figure 4B.
Figure 4.
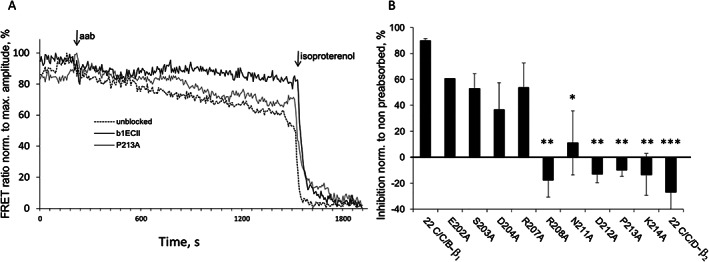
Neutralization of human anti‐β1‐aabs from DCM patients by 22‐mer cyclopeptides mimicking β1ECII with non‐conserved amino acids (compared with the amino acids constituting the ECII loop of the β2‐AR) sequentially replaced by alanine: human anti‐β1‐aabs (IgG fractions) were pre‐absorbed with the indicated cyclopeptide mutants (40 mol/mol IgG, 4°C, 16 h). Anti‐β1‐aab‐induced cAMP production was measured in HEK293 cells expressing the native β1‐AR functionally coupled to a cAMP FRET sensor. (A) Representative recordings of FRET ratios obtained upon addition of anti‐β1‐aabs prepared from a male DCM patient followed by the maximal signal achieved with 1.0 μM of (−)‐isoproterenol (Iso). (B) Prevention of cAMP stimulation after pre‐absorption of patient anti‐β1‐aabs. Results are normalized to the values without pre‐absorption. Columns represent mean ± SEM from at least three to four independent experiments with IgG prepared from different exemplary DCM patients (two men and one woman). Differences between the non‐mutated 22 C/C/B‐β1 cyclopeptide and CP mutations were analysed by one‐way ANOVA with subsequent Dunnett's post‐hoc test for multiple comparisons; *P < 0.05; **P < 0.01; ***P < 0.001. Internal negative control: non‐mutated 22C/C/D‐β2 (sequence and alignment, see Table S1).
In summary, our data suggest that the NDPK211–214 motif within β1‐ECII is of paramount relevance for β1‐AR‐induced cAMP stimulation by potentially cardio‐pathogenic anti‐β1‐aabs. The stimulatory effect of human DCM‐associated autoantibodies appeared even more sensitive to disruptions of the NDPK211–214 motif than the effect of anti‐β1ECII‐abs raised in rodents. Moreover, cAMP stimulation by human anti‐β1‐aabs was also affected by the adjacent mutation R208A (Figure 4B vs. Figure 3, black columns), suggesting that the auto‐epitope targeted by human anti‐β1‐aabs is slightly larger, encompassing the motif R (CY)NDPK208–214. This finding is not unexpected because our rodents through repeated immunizations have undergone a maturation process in their adapted immune system, entailing increases in antibody avidity and epitope narrowing, 30 which human DCM patients most probably have not undergone (explaining the generally lower levels and lower avidity of human anti‐β1‐aabs 25 ).
The black labels in Figure 2 summarize the results obtained in our experiments aiming at a further refinement of cyclopeptides intended as synthetic targets or therapeutic scavengers of stimulating anti‐β1‐aabs. Minimal requirements for such agents encompass a core domain of eight essential AA ranging from positions 208 to 215 of the human β1‐AR sequence, which harbours the essential intra‐loop bridge C209↔C215. Aberrant disulfide bridging to C216 must be precluded. The core domain should be embedded in a protein circle encompassing the entire β1ECII loop to be fully efficient.
Directed mutational analysis of the presumed auto‐epitope in intact human β1‐ARs
Our above results regarding the auto‐epitope of stimulatory anti‐β1ECII‐abs led to the assumption that the generated neutralizing (scavenger) cyclopeptides represent competitive inhibitors faithfully mimicking the three‐dimensional structure of the auto‐epitope as presented on native β1‐ARs. To confirm this assumption, we then investigated whether the very same AA residues essential for antibody neutralization by cyclopeptides would also be relevant for the interaction of stimulating anti‐β1ECII‐abs with full‐length human β1‐ARs. For this purpose, we generated β1‐AR constructs bearing point mutations of AA residues either presumed to be essential components of the core auto‐epitope (D212N and P213A) or flanking the core auto‐epitope while not essentially contributing to its formation (R207A and V219A). The mutant β1‐ARs were expressed in HEK293 cells. Expression and functionality of the β1‐AR mutants were ascertained (i) by ligand binding and (ii) by cAMP responses compared with the wild‐type β1‐AR expressed under same conditions (Table S3).
To ensure reproducibility of the test agent anti‐β1ECII antibody, we then measured the cAMP responses induced via the mutant β1‐ARs by two different monoclonal anti‐β1ECII‐abs (Mab 23‐6‐7 or Mab 13F6, raised in mice or rats, respectively 26 ). Whereas the cardio‐noxious potential of anti‐β1ECII‐Mabs generated in mice (as Mab 23‐6‐7) has been clearly shown independently by many research groups, 30 , 31 , 32 the clinical relevance of rat anti‐β1ECII‐Mabs remained to be demonstrated. Thus, according to our previously described strategy 11 —after purification from the cell supernatant—we (intravenously) transferred Mab 13F6 into healthy rats every 4 weeks. Compared with animals receiving a rat IgG isotype control, rats receiving Mab 13F6 within (6 to) 9 months of Mab transfer developed a dilated cardiomyopathic phenotype (Figure S2), demonstrating its disease‐inducing potential.
To rule out effects of the mutations on β1‐AR function per se, increases in cAMP were again normalized to the maximal stimulation achieved for each β1‐AR mutant by the full agonist (−)‐isoproterenol (1 μM). As summarized in Figure 5, β1‐ARs bearing mutations within the presumed auto‐epitope (D212N and P213A) were significantly (P < 0.0001) less sensitive to stimulation by rat or mouse anti‐β1ECII‐Mabs than the wild‐type β1‐AR. In contrast, cAMP stimulation by rat or mouse anti‐β1ECII‐Mabs was not significantly (P ≥ 0.01) affected by mutations in the flanking regions (R207A and V219A), previously found to be not involved in the scavenger effect of cyclopeptides (Figures 2 and 3B). In conclusion, the same AA residues essential for antibody neutralization by cyclopeptides (Figures 3 and 4) appear also essential for the anti‐β1ECII‐induced activation of native human β1‐AR (Figure 5). Consequently, cyclopeptides that faithfully mimic the auto‐epitope conformation relevant for allosteric β1‐AR activation should also efficiently block the effects of stimulating anti‐β1ECII‐abs.
Figure 5.
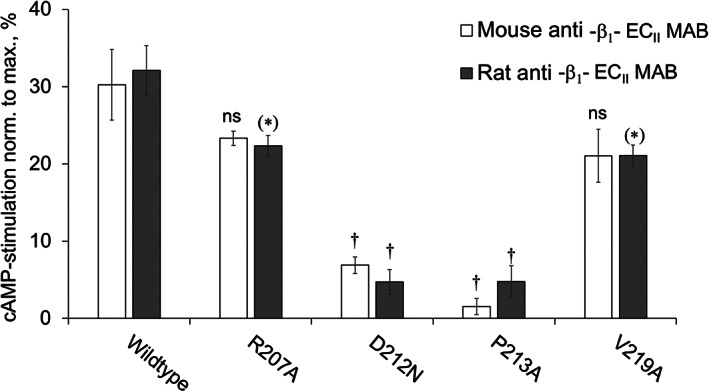
Stimulation of mutant β1‐ARs bearing point mutations within or flanking the presumed auto‐epitope of functional anti‐β1ECII antibodies: to ensure reproducibility of the test agent anti‐β1ECII antibody, HEK293 cells expressing human β1‐AR with the indicated point mutations were exposed to monoclonal anti‐β1ECII‐abs raised in mice (mouse Mab 23‐6‐7, 26 white columns) or rats (rat Mab 13F6, 26 grey columns). cAMP levels were determined in the cell lysates by radio‐immunoassay. For each experiment, increases in cAMP following antibody exposure were normalized to the maximal responses achieved with 1.0 μM of the full agonist (−)‐isoproterenol. Data are given as mean ± SEM of at least six independent experiments per column. Differences between the wild‐type β1‐AR and the indicated mutants were analysed by one‐way ANOVA with subsequent Dunnett's post‐hoc test for multiple comparisons; **P < 0.01; †P < 0.0001.
Discussion
We here provide a set of direct and indirect evidence delineating the minimal structure and conformation of the β1‐AR epitope targeted by anti‐β1‐aabs supposed to play an important role in various cardiovascular disorders. 9 Allosteric β1‐AR activation by these autoantibodies is considered a severe risk factor if not a cause of CHF. 10 , 11 It is long known that such stimulating aabs mostly target the β1‐ECII. 23 However, attempts at further narrowing the target epitope have yielded ambiguous results: autoantibodies involved in Chagas cardiomyopathy are thought to target residues 201–205 33 ; in post‐partum cardiomyopathy, the autoantibody target has been placed at residues 200–210; and for DCM, a whole series of overlapping sequences have been published (residues 183–208, 197–202, 206–212, and 213–218 22 , 23 , 33 ), encompassing almost the entire β1‐ECII and even portions of the adjacent transmembrane domain VI. Almost all these data have been obtained with linear peptides as probes. However, the surface of a protein is essentially a continuum of potential epitopes, but the fact that functional epitopes are often found to be discontinuous shows that mapping by linear peptide scanning may not be generally useful for comprehensive mapping or analysis of functional (energetic) epitopes. 34 By now, it seems quite clear that disease‐relevant anti‐β1‐aabs are only those that stimulate the β1‐AR 21 and that such aabs bind to a conformational epitope, thereby stabilizing an active conformation and prolonging the active state of the β1‐AR. 17 Consequently, stimulating anti‐β1‐aabs poorly cross‐react with linear peptides, 12 , 13 , 24 which probably explains why epitope mapping with linear peptides has not yielded a clearer picture of the relevant β1‐AR auto‐epitope(s). However, such a clearer picture is desperately needed for the development of specific diagnostics and therapeutics addressing this disease mechanism.
One crucial and potentially clinically relevant finding of our study is the identification of a minimal peptide structure that efficiently hinders receptor stimulation by DCM‐associated human anti‐β1‐aabs in an experimental approach using cells expressing functionally coupled human β1‐ARs. We have previously demonstrated that cyclopeptides mimicking that structure can stop progression of CHF and even partially revert the CHF phenotype in a human‐analogous rat model of autoimmune DCM. 19 , 20 This therapeutic effect is conveyed not only by scavenging (and thus neutralizing) cardio‐pathogenic anti‐β1ECII‐abs but possibly also by induction of B‐cell tolerance. 35 In support of this hypothesis, we observed in the above therapy model not only a reduction of autoantibody titres due to in vivo scavenging of circulating anti‐β1ECII‐abs but also a reduction of anti‐β1ECII‐secreting B cells. 19 While long‐lived plasma cells express very little or no immunoglobulins on the cell surface, 36 B cells do and could thus also serve as targets of β1ECII‐CPs. To detect the few antigen‐specific memory B cells within splenocytes of CP‐treated vs. untreated immunized rats, in our previous study, we differentiated memory B cells into short‐lived plasma blasts by boosting the rats with β1ECII/GST‐FPs 3 days prior to the analysis of the spleens. Whereas long‐lived plasma cells were not targeted by β1ECII‐CPs, preventive as well as therapeutic application of the same CPs resulted in a ~ 80% reduction of the frequencies of splenocytes secreting anti‐β1ECII‐abs. 19 This finding indicates that in immunized anti‐β1ECII‐positive rats, repeated injections of β1ECII‐CPs may lead to impaired B‐cell receptor (BCR)‐mediated β1ECII‐specific memory B‐cell expansion (and differentiation into anti‐β1ECII‐producing plasma cells). Thus, cyclopeptides based on the criteria outlined in the present study may complement therapeutic strategies that exclusively aim at (non‐specific) autoantibody scavenging, such as extracorporeal IgG absorption 37 or the systemic application of aptamers. 38
Furthermore, the very same peptide core conveying to cyclopeptides the potency to prevent allosteric β1‐AR activation by anti‐β1ECII‐aabs is obviously also a crucial prerequisite of the allosteric stimulation mechanism itself, which implies that the cyclopeptide actually mimics the 3D structure of the auto‐epitope that provides the allosteric trigger. The proposed antibody‐binding region as outlined in Figure 2 comprises the essential motif NDPK211–214 framed by the equally essential disulfide bridge between cysteines C209↔C215. In the intact receptor, this structure is constrained by another adjacent disulfide bridge linking C216 to the transmembrane region next to ECI. Both pairs of disulfide bridges are relevant for receptor function. 39 , 40 According to the 3D structure of the turkey β1‐AR, 41 the assumed auto‐epitope is located at the C‐terminal end of the backward‐oriented α‐helix constituting β1‐ECII. Several features pre‐dispose the NDPK211–214 motif as an autoantigen and allosteric trigger: N211, D212, and K214 are prone to bond with the binding surface of the antibody, while P213 is needed to interfere with the H‐bond formation to neighbouring helix residues, thereby bending the local α‐helix 42 and creating a unique structure for antibody recognition. The allosteric activation trigger is most probably provided by D212, the exchange of which to asparagine is known to cause paradoxical receptor activation by antagonistic ligands. 43
Limitations
The fact that human anti‐β1‐aabs are polyclonal and that the concentration of specific anti‐β1‐aabs in the circulation of a patient is probably much lower than in specifically immunized anti‐β1ECII‐positive rodents represents a major challenge for a therapeutic use of the here described cyclopeptides in the future. Immunoglobulin G after maturation by its (antigen‐binding) HV regions assumingly recognize stretches of five (to eight) AAs as a target epitope. 28 , 34 The ECII loop of the β1‐AR is thought to be composed of 30 AAs (AA 195–225 of the receptor protein), and most functionally active human anti‐β1‐aabs were shown to be directed against this segment of the β1‐AR membrane protein, representing a readily accessible target on the cell surface. Thus, it seems conceivable that polyclonal human anti‐β1‐aabs may target different epitopes (stretches of five to eight AAs) even within the 30 AAs that constitute β1ECII, which might result in diverging functional effects and/or downstream effects. 12 Moreover, not only the specific target epitope (at the surface of a same target protein) but also the target protein itself (which might be sarcolemmal proteins, such as myosin or troponin, or myocyte membrane proteins such as β1‐AR, β2‐AR, β3‐AR, M2‐muscarinergic, and/or angiotensin II AT1 receptors), or cross‐reactions (or molecular mimicry 44 ) between the target protein and other functionally relevant proteins may contribute to the diversity of functional effects and/or downstream effects of human aabs. In case of cross‐activation of β1‐AR by human anti‐myosin aabs, 45 β1‐derived cyclopeptides would probably fail to hinder anti‐myosin‐aab‐induced stimulation of cardiac β1‐AR. Assessment of the effect of human aabs on downstream signalling might be even further complicated by the co‐existence of aabs against different target proteins, which could then enhance (additive effect) or inhibit (opposing effect) downstream signalling and would require a panel of different epitope‐mimicking CPs to be therapeutically used on a case‐to‐case basis (e.g. human anti‐β1‐aabs were shown to increase cAMP production, whereas human anti‐M2‐aabs inhibit cAMP production, 46 or myocardial damage induced by anti‐β1‐aabs in heart failure was found to be alleviated by human anti‐β2‐aabs 47 ).
Next steps and clinical perspective
As a consequence (and because of the pilot character of our study, testing only a few exemplary DCM patients), it will be necessary to fine‐map the target epitopes of anti‐β1ECII‐aabs of a larger number of CHF patients (with different aetiologies, e.g. DCM vs. ICM vs. hypertensive heart disease) to confirm our findings. Because human anti‐β1‐aabs are polyclonal, their target epitope(s) might encompass not only the here unmasked structural motif in β1‐ECII, but also structural motifs in other extracellular domains of the human β1‐AR, for example, in β1‐ECI, 10 , 22 which were not addressed in the present study. Moreover, in future (prospective) CHF studies, the patients should be systematically screened also for the presence of autoantibodies against other cardiac membrane proteins. The functional effects and the specific target epitopes of each of these aabs should then be analysed more in detail (e.g. for functional tests by adapting the here described FRET approach and for epitope fine‐mapping by adapting the here described Ala‐scan approach). In the end, this could lead to the identification of functionally relevant (targetable) key epitopes also in other G‐protein‐coupled membrane receptors. Fine‐mapping of functionally relevant key epitopes would allow to tailor epitope‐specific cyclopeptides that (i) act as autoantibody scavengers in the circulation and—in the light of the data obtained by β1ECII‐CP treatment of anti‐β1ECII antibody‐positive rats 19 —might equally (ii) impair BCR‐mediated autoantibody‐specific memory B‐cell expansion. Because the general risk of non‐specific immune (e.g. T‐)cell activation increases with the size of a given (cyclo‐)peptide from >25 AAs on, 48 the exact knowledge of a key epitope required to neutralize (or scavenge) functional human aabs would allow to design much shorter (per se less immunogenic) cyclopeptides, as we started in the present study by testing 22‐mer instead of 25‐mer β1ECII‐CPs (used in our previous therapy study in anti‐β1ECII antibody‐positive rats 19 ). Even shorter CPs could be imagined, comprising 18, 16, or even less AAs.
With the availability of a wide panel of autoantibody‐specific (preferably short) scavenger CPs, a future therapeutic strategy could—in the end—comprise a kind of personalized approach in autoantibody‐associated CHF, that is, the injection of an individualized mixture of tailored cyclopeptides, depending on the autoantibody profile and the downstream ‘net effect' of cardio‐noxious vs. cardio‐protective autoantibodies in an individual CHF patient.
Conflict of interest
The University of Würzburg has filed for patent protection of the HEKβ1E1 cells (DE 102009 019578.5.2009) and for the methods and substances described in the present manuscript (EP 05 00 7056.4, WO 2006/103101 A2, EP 07 01 6637.6, WO 2009/027063 A2, WO 2010/61/2006.091, EP 11 01 2535.6). R.J. and M.J.L. are stockholders of the biotech company AdvanceCor GmbH (formerly CorImmun GmbH), München‐Martinsried, Germany. The remaining authors have no conflicts of interest to declare.
Funding
The present study was funded by the German Federal Ministry of Research and Education (Bundesministerium für Bildung, Wissenschaft, Forschung und Technologie and Bundesministerium für Bildung und Forschung), Grant GoBio No. 0315363 and Grant MolDiag No. 01ES0901.
Supporting information
Table S1 ‐ Amino‐acid sequences and denomination of cyclopeptides.
Table S2 ‐ Primer for site directed mutagenesis by nested PCR
Table S3 ‐ Characteristics of β1AR‐mutants expressed in HEK293‐cells
Figure S1 ‐ Neutralization of a monoclonal mouse anti‐β1ECII‐antibody by 22mer‐cyclopeptides corresponding to the human β1‐ECII, each having a different non‐conserved amino‐acid (compared to the amino‐acids constituting the ECII‐loop of the β2‐AR) sequentially replaced with alanine.
Figure S2 ‐ Time course of left ventricular diameters from rats immunized against β1ECII or receiving a monoclonal rat anti‐β1ECII and corresponding control animals, determined by echocardiography.
Wölfel, A. , Sättele, M. , Zechmeister, C. , Nikolaev, V. O. , Lohse, M. J. , Boege, F. , Jahns, R. , and Boivin‐Jahns, V. (2020) Unmasking features of the auto‐epitope essential for β1‐adrenoceptor activation by autoantibodies in chronic heart failure. ESC Heart Failure, 7: 1830–1841. 10.1002/ehf2.12747.
Angela Wölfel and Mathias Sättele contributed equally to this work.
References
- 1. Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, de Ferranti S, Despres JP, Fullerton HJ, Howard VJ, Huffman MD, Isasi CR, Jimenez MC, Judd SE, Kissela BM, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Magid DJ, McGuire DK, Mohler ER 3rd, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Rosamond W, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Woo D, Yeh RW, Turner MB. Executive summary: heart disease and stroke statistics—2016 update: a report from the American Heart Association. Circulation 2016. Jan 26; 133: 447–454. [DOI] [PubMed] [Google Scholar]
- 2. Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, Charron P, Dubourg O, Kuhl U, Maisch B, McKenna WJ, Monserrat L, Pankuweit S, Rapezzi C, Seferovic P, Tavazzi L, Keren A. Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J 2008; 29: 270–276. [DOI] [PubMed] [Google Scholar]
- 3. Kühl U, Pauschinger M, Seeberg B, Lassner D, Noutsias M, Poller W, Schultheiss HP. Viral persistence in the myocardium is associated with progressive cardiac dysfunction. Circulation 2005; 112: 1965–1970. [DOI] [PubMed] [Google Scholar]
- 4. Kindermann I, Kindermann M, Kandolf R, Klingel K, Bultmann B, Muller T, Lindinger A, Bohm M. Predictors of outcome in patients with suspected myocarditis. Circulation 2008; 118: 639–648. [DOI] [PubMed] [Google Scholar]
- 5. Caforio AL, Vinci A, Iliceto S. Anti‐heart autoantibodies in familial dilated cardiomyopathy. Autoimmunity 2008; 41: 462–469. [DOI] [PubMed] [Google Scholar]
- 6. Göser S, Andrassy M, Buss SJ, Leuschner F, Volz CH, Öttl R, Zittrich S, Blaudeck N, Hardt SE, Pfitzer G, Rose NR, Katus HA, Kaya Z. Cardiac troponin I but not cardiac troponin T induces severe autoimmune inflammation in the myocardium. Circulation 2006; 114: 1693–1702. [DOI] [PubMed] [Google Scholar]
- 7. Caforio AL, Mahon NJ, Tona F, McKenna WJ. Circulating cardiac autoantibodies in dilated cardiomyopathy and myocarditis: pathogenetic and clinical significance. Eur J Heart Fail 2002; 4: 411–417. [DOI] [PubMed] [Google Scholar]
- 8. Jahns R. Autoantibodies against cardiac troponin I: friend or foe? Eur J Heart Fail 2010; 12: 645–648. [DOI] [PubMed] [Google Scholar]
- 9. Boivin‐Jahns V, Jahns R. GPCR‐autoantibodies in chronic heart failure. Front Biosci (Landmark Ed) 2018. Jun 1; 23: 2065–2081. [DOI] [PubMed] [Google Scholar]
- 10. Störk S, Boivin V, Horf R, Hein L, Lohse MJ, Angermann CE, Jahns R. Stimulating autoantibodies directed against the cardiac beta1‐adrenergic receptor predict increased mortality in idiopathic cardiomyopathy. Am Heart J 2006; 152: 697–704. [DOI] [PubMed] [Google Scholar]
- 11. Jahns R, Boivin V, Hein L, Triebel S, Angermann CE, Ertl G, Lohse MJ. Direct evidence for a beta 1‐adrenergic receptor‐directed autoimmune attack as a cause of idiopathic dilated cardiomyopathy. J Clin Invest 2004; 113: 1419–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jahns R, Boivin V, Krapf T, Wallukat G, Boege F, Lohse MJ. Modulation of beta1‐adrenoceptor activity by domain‐specific antibodies and heart failure‐associated autoantibodies. J Am Coll Cardiol 2000; 36: 1280–1287. [DOI] [PubMed] [Google Scholar]
- 13. Jahns R, Boivin V, Siegmund C, Inselmann G, Lohse MJ, Boege F. Autoantibodies activating human beta1‐adrenergic receptors are associated with reduced cardiac function in chronic heart failure. Circulation 1999; 99: 649–654. [DOI] [PubMed] [Google Scholar]
- 14. Yu X, Patterson E, Stavrakis S, Huang S, De Aos I, Hamlett S, Cunningham MW, Lazarra R, Kem DC. Development of cardiomyopathy and atrial tachyarrhythmias associated with activating autoantibodies to beta‐adrenergic and muscarinic receptors. J Am Soc Hypertens 2009. ‐Apr; 3: 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dandel M, Wallukat G, Englert A, Lehmkuhl HB, Knosalla C, Hetzer R. Long‐term benefits of immunoadsorption in beta(1)‐adrenoceptor autoantibody‐positive transplant candidates with dilated cardiomyopathy. Eur J Heart Fail 2012; 14: 1374–1388. [DOI] [PubMed] [Google Scholar]
- 16. Schimke I, Muller J, Dandel M, Gremmels HD, Bayer W, Wallukat B, Wallukat G, Hetzer R. Reduced oxidative stress in parallel to improved cardiac performance one year after selective removal of anti‐beta 1‐adrenoreceptor autoantibodies in patients with idiopathic dilated cardiomyopathy: data of a preliminary study. J Clin Apher 2005; 20: 137–142. [DOI] [PubMed] [Google Scholar]
- 17. Bornholz B, Weidtkamp‐Peters S, Schmitmeier S, Seidel CA, Herda LR, Felix SB, Lemoine H, Hescheler J, Nguemo F, Schafer C, Christensen MO, Mielke C, Boege F. Impact of human autoantibodies on beta1‐adrenergic receptor conformation, activity, and internalization. Cardiovasc Res 2013; 97: 472–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fukuda Y, Miyoshi S, Tanimoto K, Oota K, Fujikura K, Iwata M, Baba A, Hagiwara Y, Yoshikawa T, Mitamura H, Ogawa S. Autoimmunity against the second extracellular loop of beta(1)‐adrenergic receptors induces early after depolarization and decreases in K‐channel density in rabbits. J Am Coll Cardiol 2004; 43: 1090–1100. [DOI] [PubMed] [Google Scholar]
- 19. Boivin V, Beyersdorf N, Palm D, Nikolaev VO, Schlipp A, Muller J, Schmidt D, Kocoski V, Kerkau T, Hunig T, Ertl G, Lohse MJ, Jahns R. Novel receptor‐derived cyclopeptides to treat heart failure caused by anti‐beta1‐adrenoceptor antibodies in a human‐analogous rat model. PLoS ONE 2015; 10: e0117589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Boivin‐Jahns V, Jahns R, Boege F. Relevant effects of beta1‐adrenoceptor autoantibodies in chronic heart failure. Front Biosci (Landmark Ed) 2018. Jun 1; 23: 2146–2156. [DOI] [PubMed] [Google Scholar]
- 21. Nikolaev VO, Boivin V, Stork S, Angermann CE, Ertl G, Lohse MJ, Jahns R. A novel fluorescence method for the rapid detection of functional beta1‐adrenergic receptor autoantibodies in heart failure. J Am Coll Cardiol 2007; 50: 423–431. [DOI] [PubMed] [Google Scholar]
- 22. Wallukat G, Wollenberger A, Morwinski R, Pitschner HF. Anti‐beta 1‐adrenoceptor autoantibodies with chronotropic activity from the serum of patients with dilated cardiomyopathy: mapping of epitopes in the first and second extracellular loops. J Mol Cell Cardiol 1995; 27: 397–406. [DOI] [PubMed] [Google Scholar]
- 23. Magnusson Y, Marullo S, Hoyer S, Waagstein F, Andersson B, Vahlne A, Guillet JG, Strosberg AD, Hjalmarson A, Hoebeke J. Mapping of a functional autoimmune epitope on the beta 1‐adrenergic receptor in patients with idiopathic dilated cardiomyopathy. J Clin Invest 1990; 86: 1658–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bornholz B, Hanzen B, Reinke Y, Felix SB, Jahns R, Schimke I, Wallukat G, Boege F. Detection of DCM‐associated beta1‐adrenergic receptor autoantibodies requires functional readouts or native human beta1‐receptors as targets. Int J Cardiol 2016; 202: 728–730. [DOI] [PubMed] [Google Scholar]
- 25. Wenzel K, Schulze‐Rothe S, Muller J, Wallukat G, Haberland A. Difference between beta1‐adrenoceptor autoantibodies of human and animal origin‐Limitations detecting beta1‐adrenoceptor autoantibodies using peptide based ELISA technology. PLoS ONE 2018; 13: e0192615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Holthoff HP, Zeibig S, Jahns‐Boivin V, Bauer J, Lohse MJ, Kaab S, Clauss S, Jahns R, Schlipp A, Munch G, Ungerer M. Detection of anti‐beta1‐AR autoantibodies in heart failure by a cell‐based competition ELISA. Circ Res 2012; 111: 675–684. [DOI] [PubMed] [Google Scholar]
- 27. Frielle T, Collins S, Daniel KW, Caron MG, Lefkowitz RJ, Kobilka BK. Cloning of the cDNA for the human beta1‐adrenergic receptor. Proc Natl Acad Sci U S A 1987; 84: 7920–7924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Starks MAA. Immunology and Animal Biotechnology. EDTECH; 2019. [Google Scholar]
- 29. Rasmussen SG, Devree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, Mathiesen JM, Shah ST, Lyons JA, Caffrey M, Gellman SH, Steyaert J, Skiniotis G, Weis WI, Sunahara RK, Kobilka BK. Crystal structure of the beta(2) adrenergic receptor‐Gs protein complex. Nature 2011; 477: 549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hutchings CJ, Cseke G, Osborne G, Woolard J, Zhukov A, Koglin M, Jazayeri A, Pandya‐Pathak J, Langmead CJ, Hill SJ, Weir M, Marshall FH. Monoclonal anti‐beta 1‐adrenergic receptor antibodies activate G protein signaling in the absence of beta‐arrestin recruitment. MAbs 2014. Jan 1; 6: 246–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Du Y, Zhang S, Yu H, Wu Y, Cao N, Wang W, Xu W, Li Y, Liu H. Autoantibodies against beta1‐adrenoceptor exaggerated ventricular remodeling by inhibiting CTRP9 expression. J Am Heart Assoc 2019; 8: e010475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lv T, Du Y, Cao N, Zhang S, Gong Y, Bai Y, Wang W, Liu H. Proliferation in cardiac fibroblasts induced by beta1‐adrenoceptor autoantibody and the underlying mechanisms. Sci Rep 2016; 6: 32430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wallukat G, Schimke I. Agonistic autoantibodies directed against G‐protein‐coupled receptors and their relationship to cardiovascular diseases. Semin Immunopathol 2014; 36: 351–363. [DOI] [PubMed] [Google Scholar]
- 34. Benjamin DC, Perdue SS. Site‐directed mutagenesis in epitope mapping. Methods 1996; 9: 508–515. [DOI] [PubMed] [Google Scholar]
- 35. Ungerer M, Fabbender J, Holthoff HP. Antigen‐specific therapy of Graves disease and orbitopathy by induction of tolerance. Front Biosci (Landmark Ed) 2018. Jun 1; 23: 2044–2052. [DOI] [PubMed] [Google Scholar]
- 36. Manz RA, Lohning M, Cassese G, Thiel A, Radbruch A. Survival of long‐lived plasma cells is independent of antigen. Int Immunol 1998; 10: 1703–1711. [DOI] [PubMed] [Google Scholar]
- 37. Patel PA, Hernandez AF. Targeting anti‐beta‐1‐adrenergic receptor antibodies for dilated cardiomyopathy. Eur J Heart Fail 2013; 15: 724–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Haberland A, Holtzhauer M, Schlichtiger A, Bartel S, Schimke I, Muller J, Dandel M, Luppa PB, Wallukat G. Aptamer BC 007—a broad spectrum neutralizer of pathogenic autoantibodies against G‐protein‐coupled receptors. Eur J Pharmacol 2016; 789: 37–45. [DOI] [PubMed] [Google Scholar]
- 39. Peeters MC, van Westen GJ, Li Q, Ijzerman AP. Importance of the extracellular loops in G protein‐coupled receptors for ligand recognition and receptor activation. Trends Pharmacol Sci 2010; 32: 35–42. [DOI] [PubMed] [Google Scholar]
- 40. Klco JM, Wiegand CB, Narzinski K, Baranski TJ. Essential role for the second extracellular loop in C5a receptor activation. Nat Struct Mol Biol 2005; 12: 320–326. [DOI] [PubMed] [Google Scholar]
- 41. Warne T, Serrano‐Vega MJ, Baker JG, Moukhametzianov R, Edwards PC, Henderson R, Leslie AG, Tate CG, Schertler GF. Structure of a beta1‐adrenergic G‐protein‐coupled receptor. Nature 2008; 454: 486–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Branden C, Tooze J. Introduction to Protein Structure, 2nd ed. New York: Garland Publishing; 1998. [Google Scholar]
- 43. Behr B, Hoffmann C, Ottolina G, Klotz KN. Novel mutants of the human beta1‐adrenergic receptor reveal amino acids relevant for receptor activation. J Biol Chem 2006; 281: 18120–18125. [DOI] [PubMed] [Google Scholar]
- 44. Rojas M, Restrepo‐Jimenez P, Monsalve DM, Pacheco Y, Acosta‐Ampudia Y, Ramirez‐Santana C, Leung PSC, Ansari AA, Gershwin ME, Anaya JM. Molecular mimicry and autoimmunity. J Autoimmun 2018; 95: 100–123. [DOI] [PubMed] [Google Scholar]
- 45. Mascaro‐Blanco A, Alvarez K, Yu X, Lindenfeld J, Olansky L, Lyons T, Duvall D, Heuser JS, Gosmanova A, Rubenstein CJ, Cooper LT, Kem DC, Cunningham MW. Consequences of unlocking the cardiac myosin molecule in human myocarditis and cardiomyopathies. Autoimmunity 2008; 41: 442–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chiale PA, Ferrari I, Mahler E, Vallazza MA, Elizari MV, Rosenbaum MB, Levin MJ. Differential profile and biochemical effects of antiautonomic membrane receptor antibodies in ventricular arrhythmias and sinus node dysfunction. Circulation 2001; 103: 1765–1771. [DOI] [PubMed] [Google Scholar]
- 47. Cao N, Chen H, Bai Y, Yang X, Xu W, Hao W, Zhou Y, Chai J, Wu Y, Wang Z, Yin X, Wang L, Wang W, Liu H, Fu MLX. Beta2‐adrenergic receptor autoantibodies alleviated myocardial damage induced by beta1‐adrenergic receptor autoantibodies in heart failure. Cardiovasc Res 2018; 114: 1487–1498. [DOI] [PubMed] [Google Scholar]
- 48. Paul S, Kolla RV, Sidney J, Weiskopf D, Fleri W, Kim Y, Peters B, Sette A. Evaluating the immunogenicity of protein drugs by applying in vitro MHC binding data and the immune epitope database and analysis resource. Clin Dev Immunol 2013; 2013: 467852. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 ‐ Amino‐acid sequences and denomination of cyclopeptides.
Table S2 ‐ Primer for site directed mutagenesis by nested PCR
Table S3 ‐ Characteristics of β1AR‐mutants expressed in HEK293‐cells
Figure S1 ‐ Neutralization of a monoclonal mouse anti‐β1ECII‐antibody by 22mer‐cyclopeptides corresponding to the human β1‐ECII, each having a different non‐conserved amino‐acid (compared to the amino‐acids constituting the ECII‐loop of the β2‐AR) sequentially replaced with alanine.
Figure S2 ‐ Time course of left ventricular diameters from rats immunized against β1ECII or receiving a monoclonal rat anti‐β1ECII and corresponding control animals, determined by echocardiography.