

Abstract
Effective therapies are lacking to treat gastrointestinal infections caused by the genus Cryptosporidium, which can be fatal in the immunocompromised. One target of interest is Cryptosporidium hominis (C. hominis) thymidylate synthase-dihydrofolate reductase (ChTS-DHFR), a bifunctional enzyme necessary for DNA biosynthesis. Targeting the TS-TS dimer interface is a novel strategy previously used to identify inhibitors against the related bifunctional enzyme in Toxoplasma gondii. In the present study, we target the ChTS dimer interface through homology modeling and high-throughput virtual screening to identifying allosteric, ChTS-specific inhibitors. Our work led to the discovery of methylenedioxyphenyl-aminophenoxypropanol analogues which inhibit ChTS activity in a manner that is both dose-dependent and influenced by the conformation of the enzyme. Preliminary results presented here include an analysis of structure activity relationships and a ChTS-apo crystal structure of ChTS-DHFR supporting the continued development of inhibitors that stabilize a novel pocket formed in the open conformation of ChTSTS.
Keywords: Cryptosporidium hominis, Cryptosporidiosis, Thymidylate synthase, TS-DHFR, allosteric inhibitor, TS/TS interface, Virtual Screen, GLIDE, X-ray Crystallography
Cryptosporidium hominis (C. hominis) is an apicomplexan parasite found on every continent except Antarctica which causes moderate-to-severe gastrointestinal illness (cryptosporidiosis) in humans. The 2012 Global Enteric Multicenter Study in resource-poor[1] countries identified cryptosporidiosis as the second-leading cause of diarrheal disease and deaths in children <2 years old.[2–5] Additionally, outbreaks of cryptosporidiosis have occurred in industrialized countries, and is responsible for 58% of recreational water-associated disease attributed to infectious organisms in the United States from 2000–2014.[6–8] C. parvum, which is genetically similar to C. hominis, is capable of surviving without a host for many months and at a wide range of temperatures.[9–12] Infection occurs when an individual ingests the parasite in contaminated food or water. In immunocompetent individuals, it is often a transient, self-limiting illness. However, in immunocompromised individuals, children, and the elderly, infections from C. hominis can cause serious complications, including muscle wasting and death, due to cellular ablation and malabsorption in the gastrointestinal tract.[13–16] Currently, nitazoxanide is the only FDA-approved drug for the treatment of cryptosporidiosis, but due to its limited efficacy there is a pressing need for improved therapies and therapeutic targets.[2, 17, 18]
One protein which has been extensively studied and validated as a therapeutic target in C. hominis is thymidylate synthase-dihydrofolate reductase (ChTS-DHFR), a bifunctional enzyme in parasitic protozoa that is necessary for DNA biosynthesis and survival.[19, 20] TS catalyzes the transfer of a methylene group from 5,10-methylene tetrahydrofolate (CH2H4F) to deoxyuridine monophosphate (dUMP) to produce deoxythymidine monophosphate (dTMP) necessary for DNA replication with dihydrofolate (H2F) as a byproduct. DHFR utilizes NADPH in order to reduce H2F to tetrahydrofolate (H4F). While most drug discovery efforts against TS-DHFR from protozoan parasites have focused on inhibiting catalytic activity of DHFR or TS,[21–23] targeting non-active site regions in the bifunctional enzyme of parasites has become increasingly recognized as a desirable strategy for improving drug selectivity and overcoming active site resistance mutations.[7, 24–26]
As with human TS, proper orientation of the TS domain in ChTS-DHFR and arrangement of active site residues is required for catalysis to occur.[27–31] Specifically, catalysis requires that TS dimerize such that conserved arginine residues from one monomer transverse the dimer interface and form contacts with incoming dUMP in the adjacent monomer.[31, 32] Since TS exists in equilibrium between its active and inactive conformations,[30, 33, 34] dUMP binding helps stabilize TS in its active conformation, while the apo-structure without dUMP favors the inactive conformation.[30, 34]
Previous studies have shown that peptides can be designed to target the dimer interface of human TS, stabilize the inactive conformation, and, therefore, disrupt catalytic activity.[35, 36] Two such peptides were crystallized with human TS in its inactive conformation, revealing a new pocket formed by an opening at the base of the TS/TS interface (PDB IDs: 3N5E, 4FGT). The TS/TS interface pocket is generally absent in both dUMP-apo and dUMP-liganded human TS structures;[35, 37] therefore, formation of the new pocket in 3N5E and 4FGT appears to be dependent on peptide binding. The two crystallized peptides form a disulfide bond with Cys-180 in the new pocket, suggesting that targeting specific residues at the TS/TS interface may be a promising strategy for the discovery of novel TS allosteric inhibitors.
The case for inhibiting TS activity allosterically is further supported by a peptidomimetic study from our group focused on a related bifunctional protozoal enzyme from Toxoplasma gondii.[24] This effort revealed that short peptides can be designed to non-competitively inhibit T. gondii TS activity. Moreover, since the sequence for the parasitic enzymes is distinct in the TS/TS interface region compared with the human TS enzyme, inhibition was selective for the T. gondii TS activity.[24] This early work provided the first evidence that the TS/TS interface can be targeted in bifunctional enzymes, not just monofunctional ones like human TS. A subsequent virtual screen campaign by our group resulted in the discovery of small molecules with non-competitive inhibition against TS activity in T. gondii TS.[38] In the virtual screen study, homology modeling was used to generate a model of T. gondii TS with a TS/TS interface pocket like the one found in the peptide-bound human TS structure.[38]
In principle, a similar strategy can be used to search for allosteric inhibitors of TS from C. hominis. In the present study, we set out to target the ChTS dimer interface by homology modeling and high-throughput virtual screening with the goal of identifying allosteric, ChTS-specific inhibitors. Our work led to the discovery of analogues originating from the Maybridge Diversity Library which inhibit ChTS activity in a manner that is both dose-dependent and influenced by the conformation of the enzyme. Our experimental approach, as well as a discussion of the results and implications from our work, are described below.
As illustrated in Figure 1, we used the Structure Prediction Wizard from Schrödinger to generate a homology model of ChTS containing the TS/TS interface pocket by matching the amino acid sequence of ChTS to the peptide-bound human TS structure (PDB ID: 3N5E). As with the human TS structure, the interface region at the base of the dimer complex in the C. hominis homology model was now more open, revealing a novel pocket not previously observed in ChTS crystallographically. The SiteMap tool from Schrödinger was used to evaluate the ligandability of the new pocket and produced a SiteScore > 1.0, suggesting high similarity in size, volume, and hydrophilic character with known ligand-binding pockets. Notably, the pocket consists of a buried hydrophobic cavity at its center and extends to a solvent-exposed surface with hydrophilic residue contributions from both monomers (right of Figure 1).
Figure 1.
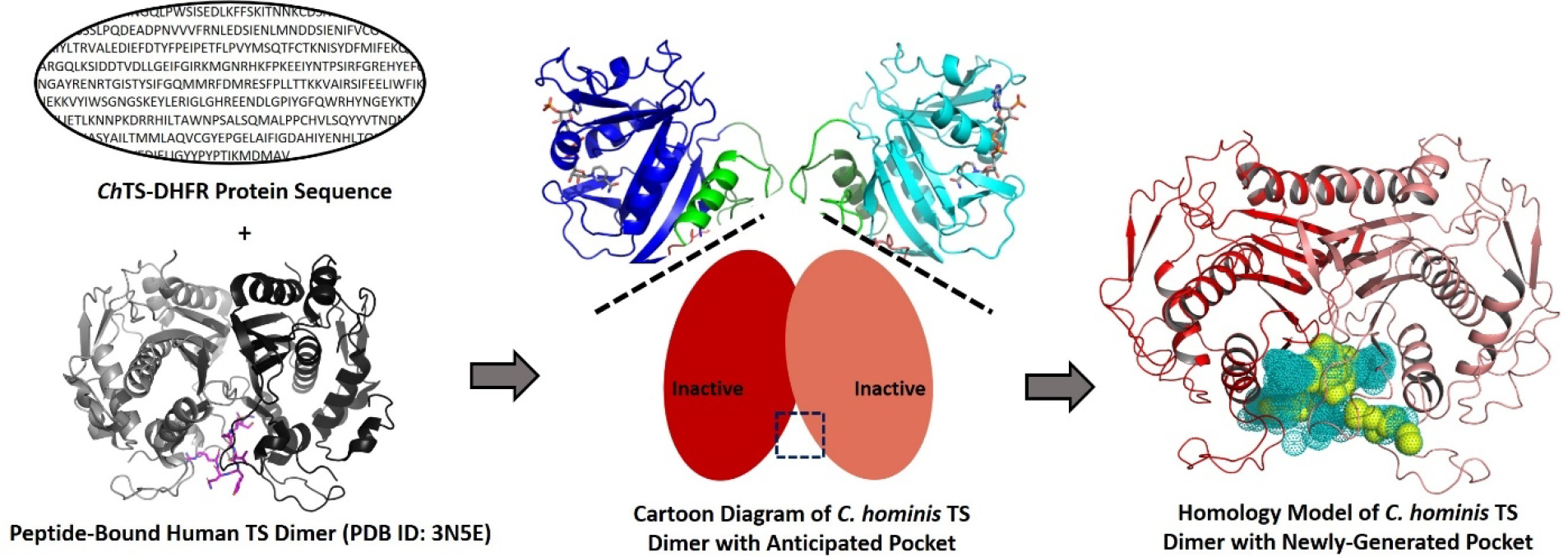
Homology model of C. hominis TS created using the amino acid sequence of ChTS and the peptide-bound human TS structure. The newly-generated binding pocket is shown as dots (teal) and spheres (yellow). The hydrophobic parts of the pocket, including the buried hydrophobic cavity, are highlighted as spheres in yellow.
An alignment of backbone atoms between the peptide-bound human TS and C. hominis homology model structures reveals good agreement between the two (0.165 Å RMSD), which is expected given the >50% sequence identity (Supplemental Figure 1). The major difference is that residues Phe-142, Gly-143, and Cys-180 in human TS are residues Tyr-349, Asn-350, and Thr-387, respectively, in the C. hominis enzyme (Figure 2). We took advantage of these differences by targeting ChTS-specific residues at the TS/TS interface using high-throughput virtual screening. Docking was performed in successive standard precision (SP) and extra precision (XP) modes in Glide using the 20,000 compound Maybridge Diversity library. This library was selected for having a range of chemotypes with drug-like properties that are available commercially. From 26 compounds matching our filter criteria, ten were obtained from the Yale Center for Molecular Discovery for screening and additional compounds were obtained from commercial sources where available.
Figure 2.
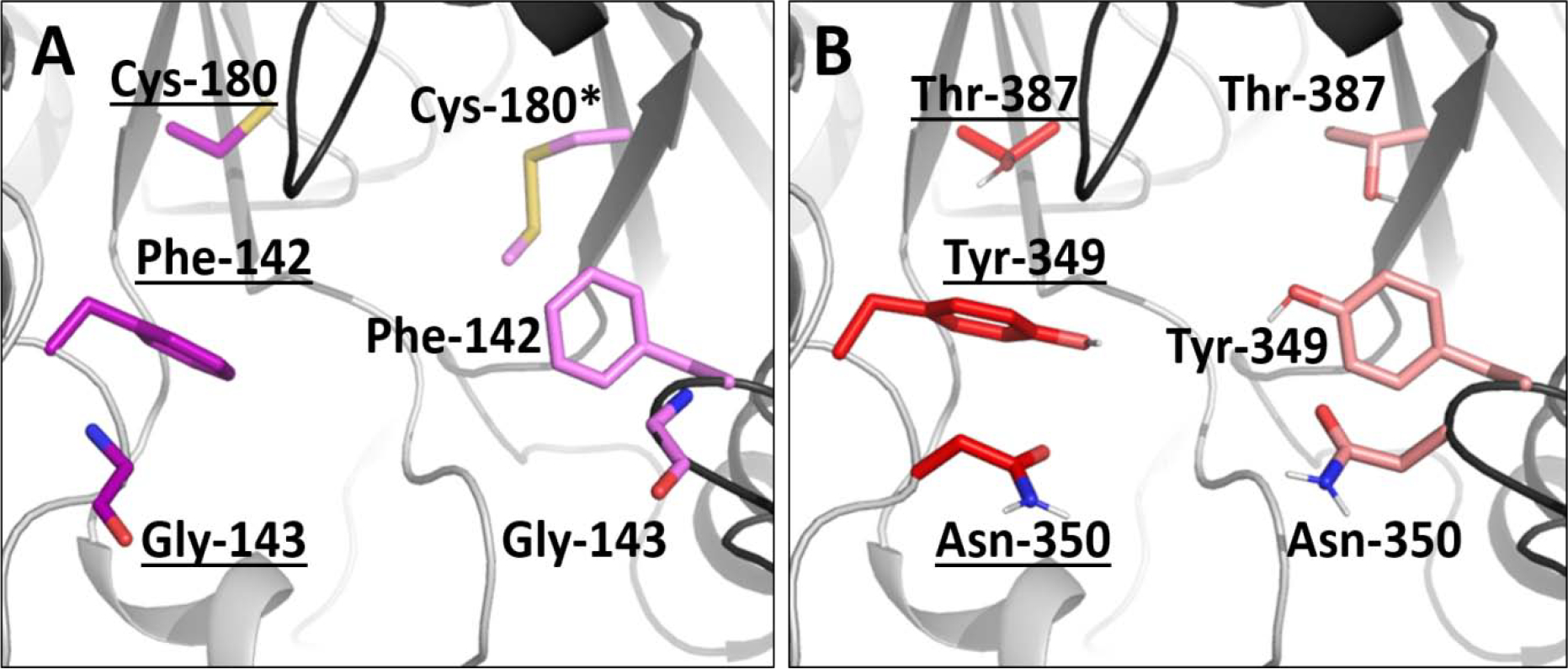
Differences between human TS (A, PDB ID: 3N5E) and our ChTS homology model (B) at the TS/TS interface. Residues Phe-142, Gly-143, and Cys-180 in human TS (A, purple and hot pink) correspond to residues Tyr-349, Asn-350, and Thr-387, respectively, in the C. hominis enzyme (B, red and light pink). We took advantage of these differences in our virtual screen to search for Ch-specific inhibitors. Note that Cys-180* was mutated to S-methylthiocysteine in the human structure to help with crystallization.
Our assay consisted of monitoring ChTS-catalyzed turnover of radiolabeled CH2H4F to H2F by HPLC and radioactivity flow detection in the presence of compound. As a first step, ChTS-DHFR was incubated with compound for 30 minutes before adding dUMP and CH2H4F to start the reaction. Since dUMP binding is known to stabilize the active conformation of ChTS, incubating the compound with enzyme for 30 minutes before adding dUMP would make the inactive conformation of the enzyme more accessible to binding. Results from the inhibition assay are shown in Figure 3A, and included in Table 1 along with Glide XP scores. Four compounds, 2, 6, 7, and 10, were determined to significantly inhibit ChTS activity in the presence of 500 μM compound [(76 ± 9)%, (83 ± 1)%, (46.0 ± 0.8)%, and (55 ± 1)%, respectively]. Notably, the three compounds with the highest percent inhibition, 2, 6, and 10, have the same methylenedioxyphenyl-aminophenoxypropanol chemotype (Figure 3b).
Figure 3.

(A) ChTS Inhibition for the ten compounds selected from our virtual screen. The y-axis denotes ChTS percent activity in the presence of 500 μM compound. (B) Two-dimensional structure depicting the common chemotype of three methylenedioxyphenyl-aminophenoxypropanol compounds, 2, 6, and 10, identified by our virtual screen and determined to be inhibitors of ChTS activity.
Table 1.
Experimental evaluation of top 10 virtual screen compounds ranked by GLIDE XP
| Compound No. | Maybridge Identification Code | GLIDE XP Score | Chemical Structure | % Inhibition of ChTS Activity at 500 μM |
|---|---|---|---|---|
| 1 | KM 10219 | −10.211 | 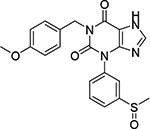 |
< 5 |
| 2 | HTS 03850 | −10.186 |  |
76 ± 9 |
| 3 | GK 02743 | −10.170 |  |
No inhibition |
| 4 | KM 10022 | −10.104 | 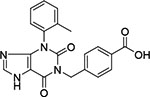 |
< 5 |
| 5 | JFD 02033 | −10.000 | 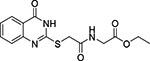 |
No inhibition |
| 6 | HTS 00950 | −9.999 | 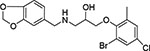 |
83 ± 1 |
| 7 | CD 11057 | −9.977 | 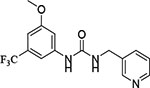 |
46.0 ± 0.8 |
| 8 | HTS 02399 | −9.863 |  |
No inhibition |
| 9 | BTB 15245 | −9.845 | 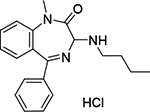 |
15 ± 2 |
| 10 | HTS 00967 | −9.792 |  |
55 ± 1 |
We examined the binding modes of 2, 6, and 10 to identify possible structure activity relationships. In all three, the common chemotype is solvent-exposed and directed away from the center of the pocket towards the exposed surface of one of the monomers (Supplemental Figure 2). The major differences in binding are with respect to functional group substitutions on the phenyl ring of the anisole moiety, which is directed towards the interior of the pocket. For example, 2 contains a thiadiazole ring at the C-4 position which enables it to penetrate deeper into the buried hydrophobic cavity compared to 6 and 10, which do not have a ring substitution on the anisole ring (Supplemental Figure 2). However, compound 6, which demonstrates the highest level of inhibition among the three, has chloro and bromo groups in the buried anisole ring, allowing it to penetrate the cavity with a much larger volume.The higher entropic contribution to the binding free energy resulting from this difference, as well as greater enthalpic contributions from halogen bonding to interface pocket residues, may help explain why 6 exhibits better inhibition. The predicted binding mode for 6 is shown in Figure 4.
Figure 4.
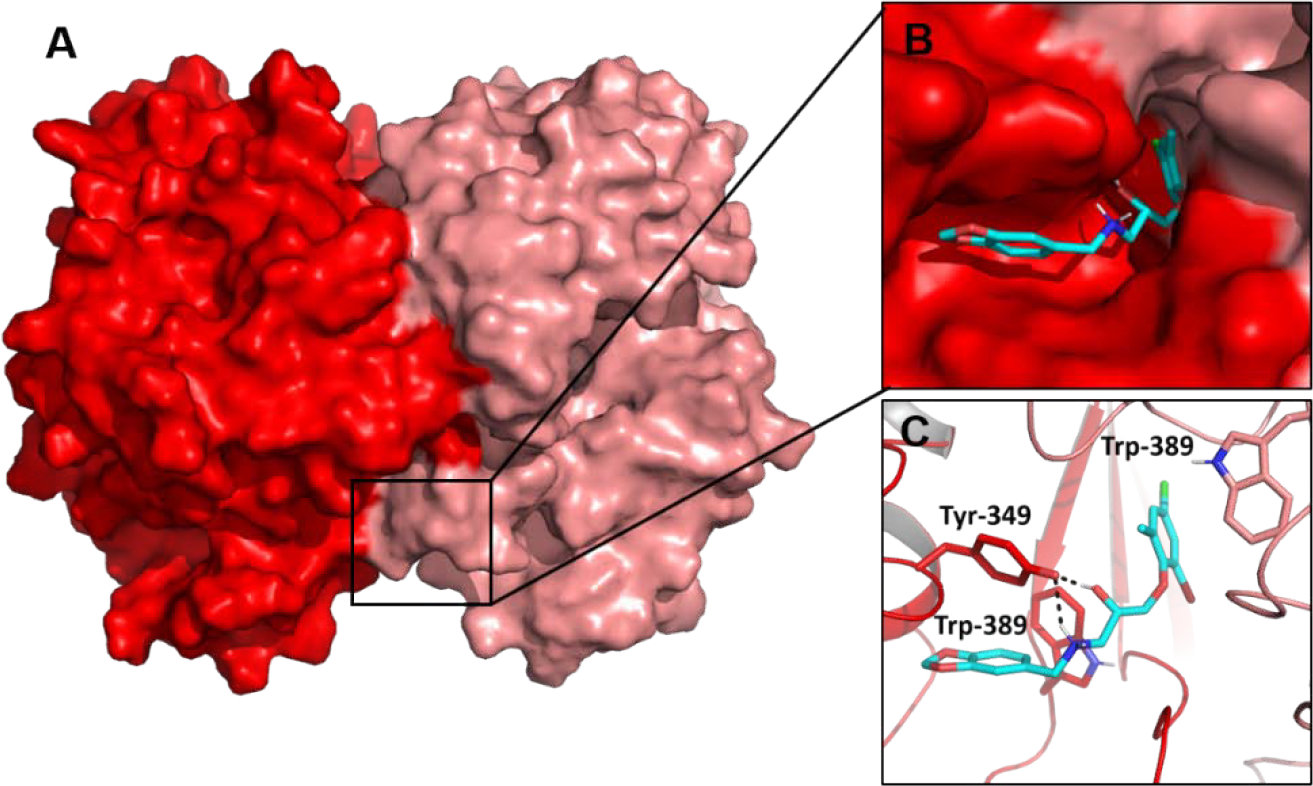
Docked model of Cpd 6 (HTS 00950, cyan) bound to the ChTS/TS interface. Cpd 6 was identified by our virtual screen and determined to be our best inhibitor of ChTS activity.
To further evaluate the chemical space of ChTS-TS interface inhibitors, we performed a structure activity relationship (SAR) study using commercially-available compounds containing the same methylenedioxyphenyl-aminophenoxypropanol chemotype as 2, 6, and 10 from our virtual screen. As illustrated in Table 2, several of the derivative compounds displayed significant inhibitory activity at 500 μM. Compound 14, which is similar to compound 2 (Figure 5), has a hydrophobic benzyl substituent at the C-4 position of the anisole moiety and displayed the greatest inhibitory activity at (87 ± 1)%. Compound 18 is most similar to compound 6 (Figure 5) and contains a C-4 chloride substituent in combination with another halogen substituent in the anisole moiety for an inhibition activity of (83 ± 1) %. Also noteworthy are the similarities between compounds 10 and 17 (Table 2), which contain hydrophobic methyl substituents in the anisole moiety and displayed ~50 % inhibition. Among the least inhibitory were compounds which contained an o-methoxy group at the C-2 position (Table 2). Interestingly, removal of the o-methyl group in 6 to produce 19 (Table 2) resulted in a ~3-fold increase in inhibitory activity, suggesting that polar atoms in the anisole ring are detrimental to protein-ligand binding. Finally, converting the anisole moiety to either a phenylthiol or thiophenylthiol moiety, as in 11 and 12 (Table 2), resulted in moderate levels of inhibitory activity, (34 ± 4) % and (43 ± 11) %, respectively, thus providing a useful reference point for evaluating the effect of hydrophobicity of anisole substituents on inhibition. Taken together, these results suggest that large, hydrophobic substituents on the anisole ring are important for ligand interactions with hydrophobic residues in the buried cavity of the ChTS-TS interface. Based on their potency in the ChTS inhibition assay, compounds 2, 6, 14, and 18 were selected for further characterization.
Table 2.
SAR of methylenedioxyphenyl-aminophenoxypropanol derivative compounds
| Compound No. | Vendor and Identification Code | Chemical Structure | % Inhibition of ChTS Activity at 500 μM |
|---|---|---|---|
| 2 | Maybridge HTS 03850 |  |
76 ± 9 |
| 6 | Maybridge HTS 00950 | 83 ± 1 | |
| 10 | Maybridge HTS 00967 |  |
55 ± 1 |
| 11 | Maybridge HTS 00807 | 34 ± 4 | |
| 12 | Maybridge HTS 00959 | 43 ± 11 | |
| 13 | Maybridge HTS 02908 | < 5 | |
| 14 | Enamine Z31367869 | 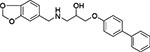 |
87 ± 1 |
| 15 | Enamine Z235247810 | 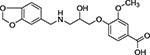 |
< 5 |
| 16 | Enamine Z55679604 | 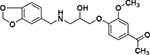 |
20 ± 6 |
| 17 | Enamine Z57990126 | 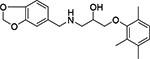 |
48 ± 9 |
| 18 | Chemspace PB31445275 | 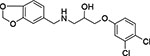 |
83 ± 1 |
| 19 | Chemspace PB31445326 | 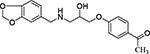 |
55 ± 6 |
| 20 | Chemspace PB31445328 | 26 ± 4 | |
| 21 | Chemspace PB31445377 | 7 ± 1 |
Figure 5.
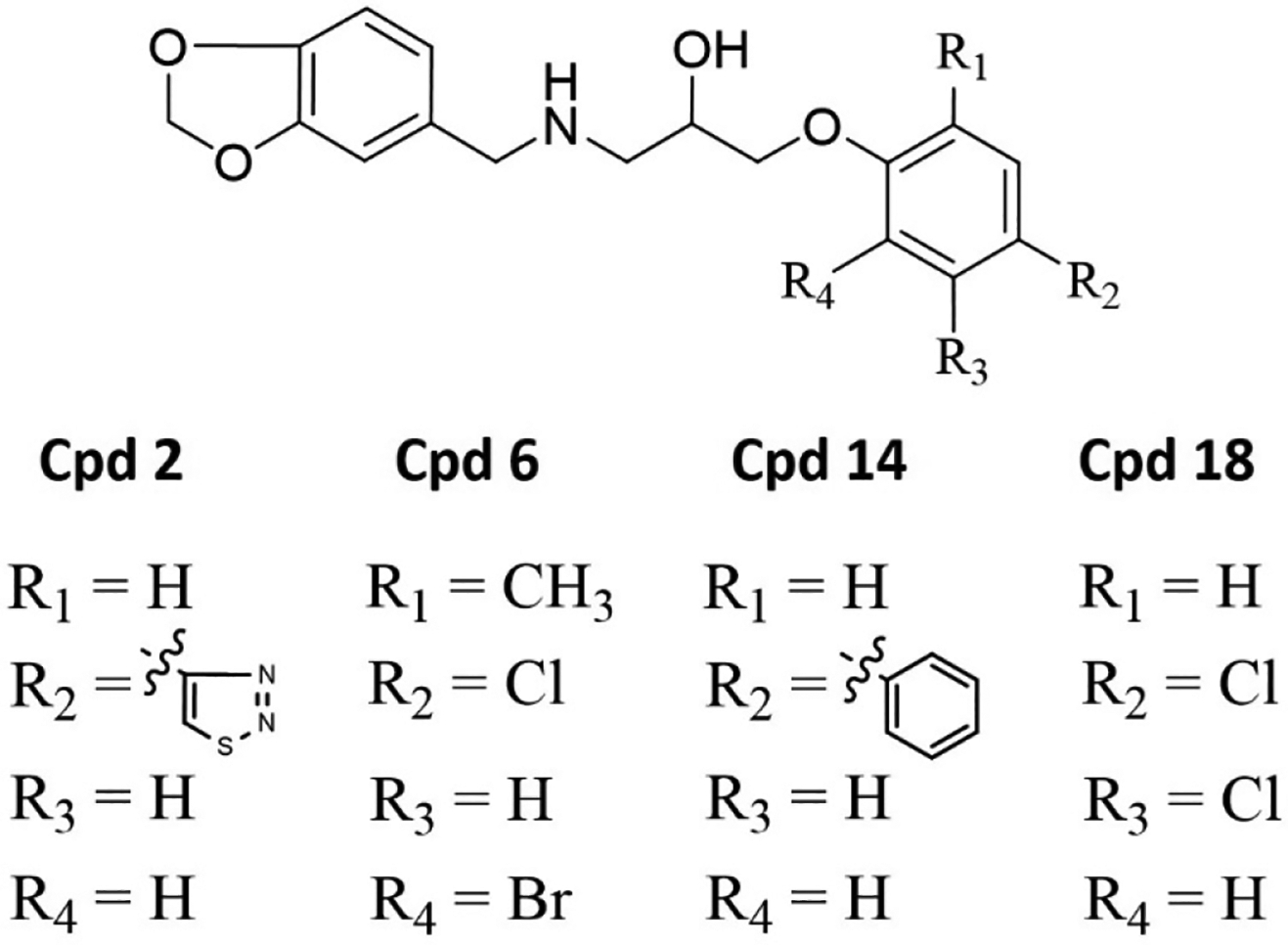
Similarities and differences between the four methylenedioxyphenyl-aminophenoxypropanol derivatives with greater than 75% inhibition of ChTS activity at 500 μM.
Keeping in mind that ChTS-DHFR is a bifunctional enzyme, we next tested whether the compounds might also be inhibiting ChDHFR activity to assess whether the inhibition is selective for ChTS activity and also to rule out nonspecific effects that may be due to compound aggregation. The evaluation of DHFR activity was accomplished using a previously-described radiometric assay to monitor the conversion of radiolabeled H2F to H4F by HPLC and determine the % ChDHFR inhibition.[26, 39]
We found that 2 had negligible inhibition on ChDHFR activity (6 ± 10)%, while 6 and 14 had a small inhibitory effect, (19 ± 5)% and (26 ± 6)%, respectively (Figure 6). Compound 18 displayed the highest inhibition of ChDHFR activity at (60 ± 4)% (Figure 6). This data suggests 2, 6, and 14 are specific for ChTS. Furthermore, this data suggests that 2, 6, and 14 inhibit ChTS activity through a mechanism independent from colloidal aggregation, while the inhibition of ChTS by 18 may result from some amount of compound aggregation. Compound 6 displayed the best balance of potency and specificity for ChTS and was therefore selected for further characterization.
Figure 6.
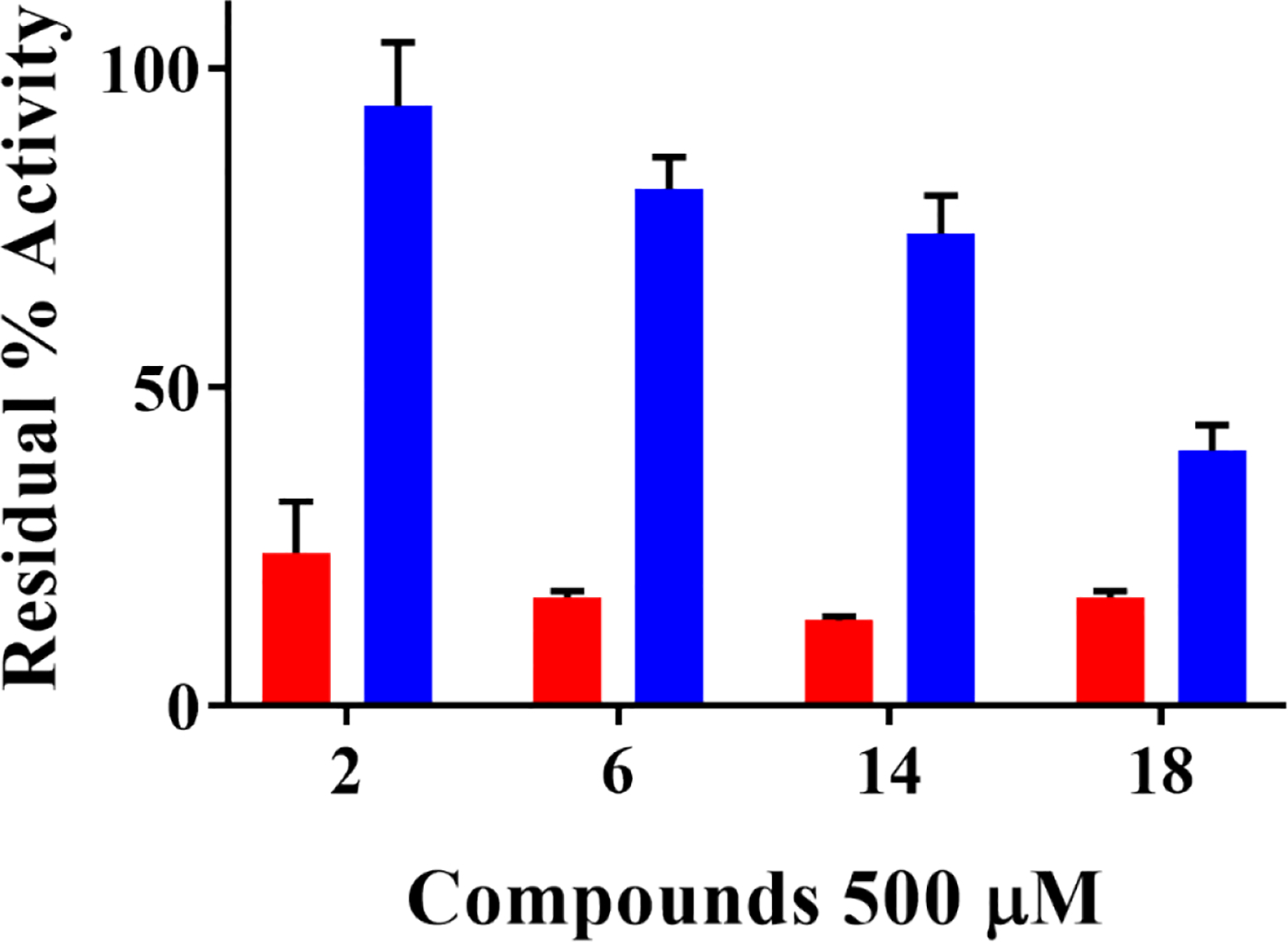
Selective inhibition of ChTS activity over ChDHFR activity by compounds 2, 6, 14, and 18. Residual ChTS percent activity upon inhibition is shown in red. Residual ChDHFR percent activity is shown in blue.
Whereas our initial screen consisted of pre-incubating the enzyme with compound before adding dUMP, we next tested whether changing the order of addition of dUMP to stabilize the active conformation of ChTS might influence the pattern of inhibition initially observed. In addition, for both the dUMP-apo and dUMP-liganded conditions, we tested multiple concentrations of compound to test whether the inhibition observed was dose-dependent. As a first step, ChTS-DHFR was mixed with dUMP and allowed to incubate for 5 minutes before adding inhibitor and later CH2H4F to start the reaction. The prediction is that compound 6 would have better inhibition against the dUMP-apo form of the enzyme representing the inactive conformation.
From the order-of-addition and dose-response experiments, it appears that 6 displays a preference for the inactive conformation of the enzyme (Figure 7). This was evident by the increase in IC50 from 172 ± 11 μM in the dUMP-apo condition to an IC50 of 304 ± 16 μM in the dUMP-liganded condition. These results suggest that compound 6 preferentially binds to the inactive conformation of ChTS and that inhibition from both compounds is dose-dependent. Finally, although our virtual screen protocol was designed to target residues that are unique to ChTS, it is possible that compound 6 still retains some ability to bind the human enzyme. We determined the specificity of 6 for ChTS versus human TS. The compound was incubated with the TS enzyme in the absence of dUMP to evaluate its potential inhibitory effects. While 6 inhibited the activity of human TS by ~50% at 500 uM, it displayed about a two-fold selectivity for ChTS (Figure 8).
Figure 7.

Preferential binding of compound 6 to the inactive conformation of ChTS. ChTS activity was measured at varying concentrations of 6. The plot in (A) denotes dose-dependent inhibition when 6 was mixed with ChTS in the absence of dUMP (inactive conformation). The plot in (B) denotes dose-dependent inhibition when 6 was mixed with ChTS in the presence of dUMP (active conformation).
Figure 8.

Selective inhibition of ChTS activity over hTS activity by compound 6. Residual ChTS percent activity upon inhibition is shown in red; Residual hTS percent activity is shown in blue.
Finally, to gain a better understanding of the important interactions between our inhibitors and the ChTS-TS interface pocket, we performed crystallization experiments. While we were unsuccessful in obtaining a co-crystal structure with any of the virtual screen inhibitors, we very recently solved a structure of ChTS-DHFR (PDB ID 6WEP) in which dUMP and folate are not bound to the ChTS active site, similar to human TS structures 3N5E and 4FGT. The 6WEP structure contains two protein molecules within the asymmetric subunit and crystallized as a P212121 space group with a resolution of 2.79 Å. Since this is the first time a TS-apo structure has been solved for TS-DHFR from C. hominis, we explored the possibility that the conformation of the interface region may be more open and therefore better accommodate ChTS interface inhibitors.
We compared the ChTS-apo structure with a ChTS-DHFR crystal structure of similar resolution containing dUMP and a folate analogue bound to the TS active site (PDB ID 6PF7; Supplemental Figure 3A). Overall, both structures are very similar, though the ChTS-TS interface region of the ChTS-apo structure appears to be in a slightly more open conformation relative to the structure with dUMP bound. However, the overlay in Supplemental Figure 3B revealed that the conformation observed in the ChTS-apo structure is significantly less open than the ChTS homology model we generated in Figure 1. This difference is understandable considering that the homology model in Figure 1 was prepared using a crystal structure of human TS in which the ChTS-TS interface region was “locked” in an open conformation by covalent linkage with a peptide inhibitor. Taken together, these results suggest that incorporating a sulfhydryl group or other cysteine reactive moiety to the anisole ring of our methylenedioxyphenyl-aminophenoxypropanol derivatives within the size constraints of the pocket may be a promising strategy to stabilize the ChTS-TS interface in an open conformation and allow for co-crystallization.
In summary, we have discovered novel inhibitors of C. hominis TS activity from the Maybridge Diversity Library using a combined homology modeling and virtual screen approach targeting the ChTS/TS dimer interface. This is the first time this approach has been used to target a non-active site pocket of the TS domain from ChTS-DHFR. Three commercially-available analogues from our screen, 2, 6, and 14 demonstrated greater specific for ChTS over ChDHFR. Because ChDHFR activity was not significantly affected, the inhibition observed for 2, 6, and 14 is unlikely to be due to compound aggregation. Compound 6 from this group was further characterized and found to preferentially bind to the dUMP-apo/inactive conformation of ChTS, with a ~2-fold preference over the dUMP-liganded form. In studies previously conducted by our lab a 2.6-to-2.7-fold preference for the inactive conformation was observed with small-molecule inhibitors of the TS/TS interface in bifunctional TgTS-DHFR.[38] Similarly, crystal structures of human TS with a peptide bound at the TS/TS interface (PDB IDs 3N5E and 4FGT) do not have dUMP bound and TS is in the inactive conformation.[35, 36] Since a preference for the dUMP-apo/inactive conformation is characteristic of TS/TS interface-mediated inhibition in TgTS, human TS, and Lactobacillus casei TS,[40] our results suggest that 6 inhibits ChTS by binding to the TS/TS interface.
Interestingly, the best inhibitors from our study share the same methylenedioxyphenyl-aminophenoxypropanol chemotype. In the absence of co-crystallographic information, it is not fully clear what role this chemotype plays in binding the TS/TS interface pocket in C. hominis. However, the fact that this chemotype was successfully enriched in 3 out of 10 compounds resulting from our screen and further validated with 11 additional compounds in our SAR analysis suggests that it plays a role in ChTS inhibition and is worth pursuing with further optimization. Co-crystallization studies are currently underway, with preliminary results supporting the development of covalent inhibitors that bind to and stabilize the pocket formed in the open conformation of the ChTS-TS interface.
Supplementary Material
Acknowledgements
This work is supported by NIH Grant (AI083146) to K.S.A., NIH Grant (GM32136) to W.L.J. This work is based upon research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences (NIGMS) from the NIH [P41 GM103403]. Crystals screening was conducted with supports in the Yale Macromolecular X-ray Core Facility [1S10OD018007-01]. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DEAC02-06CH11357. This research used FMX beamline of the National Synchrotron Light Source II, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract No. DE-SC0012704. The Life Science Biomedical Technology Research resource is primarily supported by the NIH, NIGMS through a Biomedical Technology Research Resource P41 grant (P41 GM111244), and by the DOE Office of Biological and Environmental Research (KP1605010). We would like to thank James B. Herrington at the Yale Center for Molecular Discovery for his generosity in providing us with screening compounds.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Behera B, Mirdha BR, Makharia GK, Bhatnagar S, Dattagupta S, Samantaray JC, Parasites in patients with malabsorption syndrome: a clinical study in children and adults, Dig Dis Sci, 53 (2008) 672–679. [DOI] [PubMed] [Google Scholar]
- [2].Checkley W, White AC Jr., Jaganath D, Arrowood MJ, Chalmers RM, Chen XM, Fayer R, Griffiths JK, Guerrant RL, Hedstrom L, Huston CD, Kotloff KL, Kang G, Mead JR, Miller M, Petri WA Jr., Priest JW, Roos DS, Striepen B, Thompson RC, Ward HD, Van Voorhis WA, Xiao L, Zhu G, Houpt ER, A review of the global burden, novel diagnostics, therapeutics, and vaccine targets for cryptosporidium, Lancet Infect Dis, 15 (2015) 85–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Kotloff KL, Nataro JP, Blackwelder WC, Nasrin D, Farag TH, Panchalingam S, Wu Y, Sow SO, Sur D, Breiman RF, Faruque AS, Zaidi AK, Saha D, Alonso PL, Tamboura B, Sanogo D, Onwuchekwa U, Manna B, Ramamurthy T, Kanungo S, Ochieng JB, Omore R, Oundo JO, Hossain A, Das SK, Ahmed S, Qureshi S, Quadri F, Adegbola RA, Antonio M, Hossain MJ, Akinsola A, Mandomando I, Nhampossa T, Acacio S, Biswas K, O’Reilly CE, Mintz ED, Berkeley LY, Muhsen K, Sommerfelt H, Robins-Browne RM, Levine MM, Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): a prospective, case-control study, Lancet, 382 (2013) 209–222. [DOI] [PubMed] [Google Scholar]
- [4].Miller CN, Panagos CG, Mosedale WRT, Kvac M, Howard MJ, Tsaousis AD, NMR metabolomics reveals effects of Cryptosporidium infections on host cell metabolome, Gut Pathog, 11 (2019) 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Dayao DA, Sheoran A, Carvalho A, Xu H, Beamer G, Widmer G, Tzipori S, An immunocompetent rat model of infection with Cryptosporidium hominis and Cryptosporidium parvum, Int J Parasitol, (2019). [DOI] [PubMed] [Google Scholar]
- [6].Kissinger JC, Evolution of Cryptosporidium, Nat Microbiol, 4 (2019) 730–731. [DOI] [PubMed] [Google Scholar]
- [7].Martucci WE, Udier-Blagovic M, Atreya C, Babatunde O, Vargo MA, Jorgensen WL, Anderson KS, Novel non-active site inhibitor of Cryptosporidium hominis TS-DHFR identified by a virtual screen, Bioorg Med Chem Lett, 19 (2009) 418–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].McAteer J, Jernigan S, Mao C, Gonzalez MD, Watson RJ, Liverman R, Tobin DAM, Dishman MH, Shane A, Yildirim I, Cryptosporidiosis among solid organ transplant recipient attendees at a summer camp, Pediatr Transplant, (2019) e13649. [DOI] [PubMed] [Google Scholar]
- [9].Reinoso R, Becares E, Smith HV, Effect of various environmental factors on the viability of Cryptosporidium parvum oocysts, J Appl Microbiol, 104 (2008) 980–986. [DOI] [PubMed] [Google Scholar]
- [10].Fayer R, Trout JM, Jenkins MC, Infectivity of Cryptosporidium parvum oocysts stored in water at environmental temperatures, J Parasitol, 84 (1998) 1165–1169. [PubMed] [Google Scholar]
- [11].Peng X, Murphy T, Holden NM, Evaluation of the effect of temperature on the die-off rate for Cryptosporidium parvum oocysts in water, soils, and feces, Appl Environ Microbiol, 74 (2008) 7101–7107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Duhain GL, Minnaar A, Buys EM, Effect of chlorine, blanching, freezing, and microwave heating on Cryptosporidium parvum viability inoculated on green peppers, J Food Prot, 75 (2012) 936–941. [DOI] [PubMed] [Google Scholar]
- [13].Bouzid M, Hunter PR, Chalmers RM, Tyler KM, Cryptosporidium pathogenicity and virulence, Clin Microbiol Rev, 26 (2013) 115–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Goodgame RW, Kimball K, Ou CN, White AC Jr., Genta RM, Lifschitz CH, Chappell CL, Intestinal function and injury in acquired immunodeficiency syndrome-related cryptosporidiosis, Gastroenterology, 108 (1995) 1075–1082. [DOI] [PubMed] [Google Scholar]
- [15].Ribeiro Machado F, Gonzaga Vaz Coelho L, Chausson Y, Greco DB, Fat malabsorption assessed by 14C-triolein breath test in HIV-positive patients in different stages of infection: is it an early event?, J Clin Gastroenterol, 30 (2000) 403–408. [DOI] [PubMed] [Google Scholar]
- [16].Sciarretta G, Bonazzi L, Monti M, Furno A, Mazzoni M, Mazzetti M, Gritti F, Malaguti P, Bile acid malabsorption in AIDS-associated chronic diarrhea: a prospective 1-year study, Am J Gastroenterol, 89 (1994) 379–381. [PubMed] [Google Scholar]
- [17].Borad A, Ward H, Human immune responses in cryptosporidiosis, Future Microbiol, 5 (2010) 507–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Grayson ML, Kucers’ the use of antibiotics: a clinical review of antibacterial, antifungal, antiparasitic and antiviral drugs, Seventh edition ed., CRC Press, Boca Raton, 2017. [Google Scholar]
- [19].O’Neil RH, Lilien RH, Donald BR, Stroud RM, Anderson AC, Phylogenetic classification of protozoa based on the structure of the linker domain in the bifunctional enzyme, dihydrofolate reductase-thymidylate synthase, J Biol Chem, 278 (2003) 52980–52987. [DOI] [PubMed] [Google Scholar]
- [20].Mukerjee A, Iyidogan P, Castellanos-Gonzalez A, Cisneros JA, Czyzyk D, Ranjan AP, Jorgensen WL, White AC Jr., Vishwanatha JK, Anderson KS, A nanotherapy strategy significantly enhances anticryptosporidial activity of an inhibitor of bifunctional thymidylate synthase-dihydrofolate reductase from Cryptosporidium, Bioorg Med Chem Lett, 25 (2015) 2065–2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Anderson AC, Two crystal structures of dihydrofolate reductase-thymidylate synthase from Cryptosporidium hominis reveal protein-ligand interactions including a structural basis for observed antifolate resistance, Acta Crystallogr Sect F Struct Biol Cryst Commun, 61 (2005) 258–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kumar VP, Cisneros JA, Frey KM, Castellanos-Gonzalez A, Wang Y, Gangjee A, White AC Jr., Jorgensen WL, Anderson KS, Structural studies provide clues for analog design of specific inhibitors of Cryptosporidium hominis thymidylate synthase-dihydrofolate reductase, Bioorg Med Chem Lett, 24 (2014) 4158–4161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Czyzyk DJ, Valhondo M, Deiana L, Tirado-Rives J, Jorgensen WL, Anderson KS, Structure activity relationship towards design of cryptosporidium specific thymidylate synthase inhibitors, Eur J Med Chem, 183 (2019) 111673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Landau MJ, Sharma H, Anderson KS, Selective peptide inhibitors of bifunctional thymidylate synthase-dihydrofolate reductase from Toxoplasma gondii provide insights into domain-domain communication and allosteric regulation, Protein Sci, 22 (2013) 1161–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Martucci WE, Rodriguez JM, Vargo MA, Marr M, Hamilton AD, Anderson KS, Exploring novel strategies for AIDS protozoal pathogens: alpha-helix mimetics targeting a key allosteric protein-protein interaction in C. hominis TS-DHFR, Medchemcomm, 4 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Ruiz V, Czyzyk DJ, Valhondo M, Jorgensen WL, Anderson KS, Novel allosteric covalent inhibitors of bifunctional Cryptosporidium hominis TS-DHFR from parasitic protozoa identified by virtual screening, Bioorg Med Chem Lett, 29 (2019) 1413–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Anderson AC, O’Neil RH, DeLano WL, Stroud RM, The structural mechanism for half-the-sites reactivity in an enzyme, thymidylate synthase, involves a relay of changes between subunits, Biochemistry, 38 (1999) 13829–13836. [DOI] [PubMed] [Google Scholar]
- [28].Johnson EF, Hinz W, Atreya CE, Maley F, Anderson KS, Mechanistic characterization of Toxoplasma gondii thymidylate synthase (TS-DHFR)-dihydrofolate reductase. Evidence for a TS intermediate and TS half-sites reactivity, J Biol Chem, 277 (2002) 43126–43136. [DOI] [PubMed] [Google Scholar]
- [29].Perry KM, Pookanjanatavip M, Zhao J, Santi DV, Stroud RM, Reversible dissociation and unfolding of the dimeric protein thymidylate synthase, Protein Sci, 1 (1992) 796–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Berger SH, Berger FG, Lebioda L, Effects of ligand binding and conformational switching on intracellular stability of human thymidylate synthase, Biochim Biophys Acta, 1696 (2004) 15–22. [DOI] [PubMed] [Google Scholar]
- [31].Almog R, Waddling CA, Maley F, Maley GF, Van Roey P, Crystal structure of a deletion mutant of human thymidylate synthase Delta (7–29) and its ternary complex with Tomudex and dUMP, Protein Sci, 10 (2001) 988–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Sayre PH, Finer-Moore JS, Fritz TA, Biermann D, Gates SB, MacKellar WC, Patel VF, Stroud RM, Multi-targeted antifolates aimed at avoiding drug resistance form covalent closed inhibitory complexes with human and Escherichia coli thymidylate synthases, J Mol Biol, 313 (2001) 813–829. [DOI] [PubMed] [Google Scholar]
- [33].Phan J, Steadman DJ, Koli S, Ding WC, Minor W, Dunlap RB, Berger SH, Lebioda L, Structure of human thymidylate synthase suggests advantages of chemotherapy with noncompetitive inhibitors, J Biol Chem, 276 (2001) 14170–14177. [DOI] [PubMed] [Google Scholar]
- [34].Chen D, Jansson A, Sim D, Larsson A, Nordlund P, Structural analyses of human thymidylate synthase reveal a site that may control conformational switching between active and inactive states, J Biol Chem, 292 (2017) 13449–13458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Cardinale D, Guaitoli G, Tondi D, Luciani R, Henrich S, Salo-Ahen OM, Ferrari S, Marverti G, Guerrieri D, Ligabue A, Frassineti C, Pozzi C, Mangani S, Fessas D, Guerrini R, Ponterini G, Wade RC, Costi MP, Protein-protein interface-binding peptides inhibit the cancer therapy target human thymidylate synthase, Proc Natl Acad Sci U S A, 108 (2011) E542–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Tochowicz A, Santucci M, Saxena P, Guaitoli G, Trande M, Finer-Moore J, Stroud RM, Costi MP, Alanine mutants of the interface residues of human thymidylate synthase decode key features of the binding mode of allosteric anticancer peptides, J Med Chem, 58 (2015) 1012–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Lovelace LL, Minor W, Lebioda L, Structure of human thymidylate synthase under low-salt conditions, Acta Crystallogr D Biol Crystallogr, 61 (2005) 622–627. [DOI] [PubMed] [Google Scholar]
- [38].Sharma H, Landau MJ, Sullivan TJ, Kumar VP, Dahlgren MK, Jorgensen WL, Anderson KS, Virtual screening reveals allosteric inhibitors of the Toxoplasma gondii thymidylate synthase-dihydrofolate reductase, Bioorg Med Chem Lett, 24 (2014) 1232–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Atreya CE, Anderson KS, Kinetic characterization of bifunctional thymidylate synthase-dihydrofolate reductase (TS-DHFR) from Cryptosporidium hominis: a paradigm shift for ts activity and channeling behavior, J Biol Chem, 279 (2004) 18314–18322. [DOI] [PubMed] [Google Scholar]
- [40].Prasanna V, Bhattacharjya S, Balaram P, Synthetic interface peptides as inactivators of multimeric enzymes: inhibitory and conformational properties of three fragments from Lactobacillus casei thymidylate synthase, Biochemistry, 37 (1998) 6883–6893. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.