

Abstract
Objectives
Erythrocytosis is characterized by the expansion of erythrocyte compartment including elevated red blood cell number, hematocrit, and hemoglobin content. Familial erythrocytosis (FE) is a congenital disorder with different genetic background. Type 1 FE is primary FE caused by mutation in erythropoietin receptor gene (EPOR). Type 2‐5 FE are secondary FEs caused by mutations of genes involved in oxygen sensing pathway important for erythropoietin (EPO) regulation. In the present study, we summarized associations between EPOR and EPO gene variations with development of FE and searched for genetic variants located within regulatory regions.
Methods
Publications reporting EPOR and EPO sequence variants associated with FE or clinical features of erythrocytosis were retrieved from PubMed and WoS. In silico, sequence reanalysis was performed using Ensembl genomic browser, release 89 to screen for variants located within regulatory regions.
Results
To date, 28 variants of the EPOR and seven variants of the EPO gene have been associated with erythrocytosis or upper hematocrit. Sequence variants were also found to be present within regulatory regions.
Conclusions
Role of variants in regulatory regions of the EPO gene should be further investigated.
Keywords: erythropoietin, erythropoietin receptor, familial erythrocytosis, regulatory regions, sequence variants
1. INTRODUCTION
Erythrocytosis comprises a heterogeneous group of disorders characterized by the expansion of the erythrocyte compartment in the peripheral blood, clinically reflected by an increased hematocrit, hemoglobin or red blood cell (RBC) number.1 Erythrocytosis is classified as congenital or acquired and its mechanism may be primary or secondary.2, 3, 4, 5
Primary erythrocytosis results from a molecular defect in the hematopoietic progenitor cells, leading to cell hypersensitivity to erythropoietin (EPO) and consequently in serum EPO levels below normal. The primary cause of increased RBC number may be polycythemia vera (PV) (acquired) or familial erythrocytosis (FE) (congenital). The mutation of a Janus kinase 2 (JAK2) gene is indicative for PV. The erythropoietin receptor (EPOR) mutation is the indicator for primary FE type 1 (ECYT1), also known as congenital erythrocytosis (CE) or primary congenital familial polycythemia.6, 7 Secondary erythrocytosis results from aberrant regulation of erythropoiesis promoting substances acting on hematopoietic progenitors, mainly EPO.4 Secondary congenital erythrocytosis is characterized by inappropriately normal or high serum EPO levels due to defects in the oxygen sensing pathway (ECYT2‐5) or variants effecting hemoglobin oxygen affinity (ECYT6‐7)8 (OMIM).9 Defects in oxygen sensing pathway can be associated with pathogenic variants in one of the factors regulating EPO production; including von Hippel‐Lindau tumor suppressor (VHL) indicative for ECYT2, egl‐9 family hypoxia‐inducible factor 1 (EGLN1, synonym PHD2) indicative for ECYT3, and endothelial PAS domain protein 1 (EPAS1, synonym HIF2A) indicative for ECYT4. Recently, the EPO gene was added to the OMIM database as new cause for FE (indicative for ECYT5).9, 10
Glycoprotein hormone EPO is the main regulator of red blood cell production. EPOR dimerization and activation by EPO leads to signaling cascade triggering several genes responsible for proliferation, survival, and differentiation of erythroid progenitor cells, leading to a tight control of RBC production in the bone marrow.11 Hormone EPO and its receptor EPOR can therefore be involved in development of FE.
Erythropoietin receptor gene is located on 19p13.2. The 2459 bp long primary transcript variant 1 encodes 508 amino acids (aa) EPOR precursor, resulting in 484 aa long mature receptor. The mature receptor consists of an extracellular, a transmembrane, and a cytoplasmic region. Soluble (EPOR‐S) and truncated (EPOR‐T) isoforms associated with cancer have also been identified.12 Multiple EPOR mutations associated with ECYT1 have been described, lacking a signal termination due to truncation of the inhibitory domain SHP‐1/SOCS‐3. All patients with EPOR mutations display suppressed serum EPO levels, but still produce erythroid colonies due to constitutive receptor activation. EPOR mutations have been found in 15% of all hereditary erythrocytosis cases.13
Erythropoietin gene is located at 7q22.1. Single 1340 bp long splice variant encodes a 193 aa erythropoietin precursor, resulting in 165 aa long mature hormone. EPO production is increased in hypoxia resulting from anemia or decreased cellular oxygen tension. Under normal oxygen tension, HIF‐alfa is hydroxylated by EGLN1 (PHD2) and targeted by VHL for ubiquitin‐mediated degradation, resulting in no EPO production. Under hypoxic conditions, hydroxylase activity is inhibited, therefore EGLN1 proteins are unable to hydroxylate HIF‐alfa allowing it to escape VHL protein recognition and subsequent degradation. HIF‐alfa can therefore form an active transcriptional complex with HIF‐beta, aryl hydrocarbon receptor nuclear translocator (ARNT). Hypoxia‐inducible factor (HIF) complex translocates to nucleus and binds to the hypoxia response element (HRE) of the target genes and upregulates expression of more than 200 genes, including EPO.2 Among members of HIF‐alfa gene family, EPAS1 (HIF2A) is involved in regulation of EPO production.14
Tissue‐specific expression of EPO gene in adult kidneys or fetal livers is dependent on far upstream cis elements and an enhancer element downstream from the poly‐adenylation signal.2 EPO gene expression is also regulated via stimulatory hepatocyte nuclear factor 4 alpha (HNF4A), inhibitory GATA binding protein 2 (GATA2), and NF‐kappa‐B (NFKB).15, 16, 17 The motif 5′‐RCGTG‐3′ has been reported as the core sequence for a HIF binding site.14, 16, 18, 19
One of the main obstacles in the research field presents the fact that terminology regarding genetic variants is heterogeneous and dispersed across publications and databases. Additionally, identification of regulatory regions was performed on various previous genomic assemblies and needs to be updated according to the genomic locations from latest genomic browsers. The main aim of the present study was therefore to: (a) summarize the literature and databases for associations between EPOR and EPO genes and familial erythrocytosis or its clinical signs and (b) to perform in silico sequence reanalysis of the EPO gene to identify variants overlapping regulatory sites.
2. MATERIAL AND METHODS
Literature databases (PubMed and Web of Science; WoS) were screened for gene variations in EPO and EPOR genes in association with FE and its clinical characteristics: increased RBC mass, elevated hemoglobin, and hematocrit. The literature was retrieved using the keywords such as erythropoietin, erythropoietin receptor, mutations, polymorphism, erythrocytosis, polycythemia, hematocrit, and elevated red blood cell number. The time span of the literature search was from 1/1993 to 4/2018. Nucleotide sequence, genomic coordinates, and variants were extracted from the Ensembl, release 89.20 Obtained data were complemented with additional relevant genomic information. Reference SNP ID number (rs ID) if available, synonyms and clinical data were obtained from: The Single Nucleotide Polymorphism database (dbSNP),21 The Human Gene Mutation Database (HGMD),22 www.erythrocytosis.org and Leiden Open Variation Database (LOVD), release 3.0.23 Gene names were unified according to the HUGO Gene Nomenclature Committee (HGNC) database.24 Collected genomic data were organized and unified according to the human genome variation society (HGVS) recommendations.25 Locations of regulatory regions were extracted from the published literature and compared with genomic location of sequence variants deposited in the Ensembl browser.
3. RESULTS
The review revealed that 25 genetic variants of the exon 8 of EPOR gene associated with FE are deposited in the LOVD database. Literature review revealed three additional variants reported in1, 26, 27 (Table 1). Mutations result in premature termination and, consequently, these mutant receptors lack portions of the C‐terminal EPOR cytoplasmic domain responsible for negative regulation.
Table 1.
Genetic variants of the EPOR gene causing truncation of EPOR protein in familial erythrocytosis
| dbSNP HGVS name | Var_pub_as | Location of the variant in the EPOR gene | Geographic origin/ethnic origin | Number of patients reported | Reference | Remarks |
|---|---|---|---|---|---|---|
|
c.1252_1255del p.Gly418Profs*34 |
g.5938_5941del | Exon 8 | Denmark | One family, four affected members |
Petersen et al26 PMID: 15142125 |
58 aa truncation confirmed in three members |
|
c.1273G>T p.Glu425* |
G5959T | Exon 8 | na | One family, four affected members |
Kralovics and Prchal1 PMID: 11559951 |
84 aa truncation |
|
c.1362C>G p.Tyr454* |
/ | Exon 8 | France | One patient (30‐y‐old female) |
Chauveau et al27 PMID: 26010769 |
55 aa truncation, shortest known truncation |
EPOR, erythropoietin receptor; Var_pub_as, variant as reported originally; listed only when different from Variant/DNA.
Out of 11 tested sequence variants of the EPO gene, seven were associated with erythrocytosis or upper‐limit hematocrit (Table 2).4, 10, 28, 29 Four tested sequence variants were not associated with FE or clinical signs.19, 28 The first pathogenic variant of the EPO gene is located in the 5′ UTR region and it was reported by the WGS500 project. It was identified in two independent families with erythrocytosis and is sufficiently substantiated by segregation data to assume its disease causing effect, most probably due to increased EPO regulation.29 Three variants are located within exons 2 and 4 in patients with FE4; however, the causative for FE needs to be confirmed. Pathogenic variant c.32delG located in exon 2 causative for FE was recently identified. This pathogenic variant introduces a frameshift in exon 2 and interrupts EPO mRNA translation from primary transcript variant 1. Increased transcription of EPO mRNA is initiated from an alternative promoter located in intron 1.10 Two sequence variants located within regulatory regions; rs1617640 and rs551238 were identified in blood donors with significantly increased hematocrit,28 however their causative for FE needs to be reviewed.
Table 2.
Genetic variants of the EPO gene associated with erythrocytosis or hematocrit level
| dbSNP HGVS name (reference SNP ID number, if available) | Var_pub_as | Location of the variant in the EPO gene | Geographic origin/ethnic origin | Number of patients reported | Reference | Remarks |
|---|---|---|---|---|---|---|
| NM_000799.2:c.‐1306C>A (rs1617640) | G>T | Upstream | Jordan | 298 healthy male blood donors; divided into: Ht level greater or equal to 48 (181), and Ht between 42% and 47.5% (117) |
Khabour et al28 PMID: 23142128 |
G allele was found at significant higher frequency among upper‐Ht group; unconfirmed |
| NM_000799.2:c.‐136G>A | / | 5′UTR | United Kingdom (Great Britain) | One family, three affected members and one family, five affected members |
Taylor29 PMID: 25985138 |
Co‐segregated with the disease in two independent families |
| NM_000799.2:c.32delG | / | exon 2 | Norway | One family, nine affected members |
Zmajkovic et al10 PMID: 29514032 |
Frameshift in exon 2, excess production of EPO from alternative promoter located in intron 1 |
| NM_000799.2:c.19delC, p.P7fs | / | exon 2 | / | Female with affected father and parental grandmother. |
Camps et al4 PMID: 27651169 |
Identified as novel erythrocytosis‐associated gene variant in WGS500 and gene panel sequencing project; unconfirmed. |
| NM_000799.2:c.250G>C, p.G84R (rs137953994) | / | exon 4 | / | One male patient | ||
| NM_000799.2:c.296A>G, p.E99G | / | exon 4 | / | Two male patients | ||
| NM_000799.2:c.*772G>T (rs551238) | G3544T | 3′UTR enhancer | Jordan | 298 healthy male blood donors; divided into: Ht level greater or equal to 48 (181), and Ht between 42% and 47.5% (117) |
Khabour et al28 PMID: 23142128 |
G allele was found at significant higher frequency among upper‐Ht group; unconfirmed |
EPO, erythropoietin; Hb, hemoglobin; HRE, hypoxia response element in 3′ UTR enhancer; Ht, hematocrit; Var_pub_as, variant as reported originally; listed only when different from Variant/DNA.
We also analyzed if retrieved EPO gene sequence variants are located within regulatory regions. Genetic locations of binding regions for EPAS1 (HIF2A), ARNT (HIF1B), and HNF4A binding sites were retrieved from publications16, 18, 19 and mapped to the sequence according to the latest genome assembly. Downstream from the EPAS1 and ARNT binding site (HIF binding site, HBS), the EPO 3′ enhancer contains a series of CA repeats (HIF ancillary sequence, HAS) and two tandem consensus steroid hormone response elements 5′—YGACCY—3′ separated by 2 bp, binding sites for HNF4A (nuclear receptor half site, DR‐2). The analysis revealed that seven variants associated with erythrocytosis or hematocrit level (Table 2) are not located within regulatory regions (Figure 1). We additionally screened Ensembl database and retrieved two SNPs located within regulatory regions (rs796952255 and rs539229161; Figure 1). These two SNPs have unknown function and were also not yet tested in association with FE.
Figure 1.
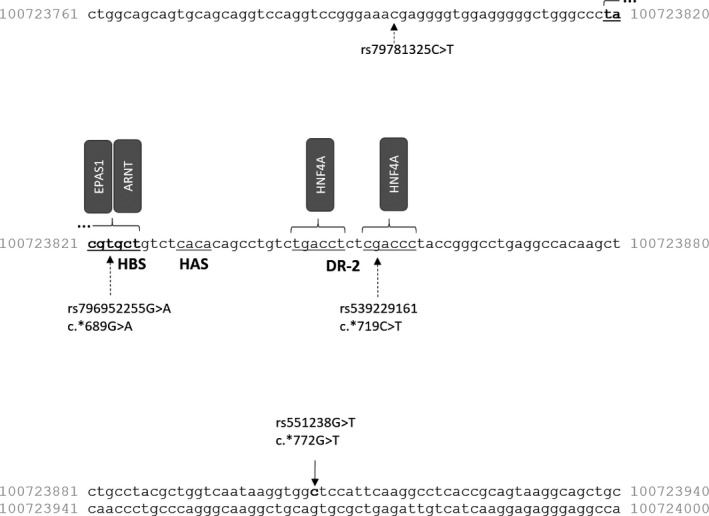
Nucleotide sequence of the EPO gene 3′ enhancer, with marked sequence variants and regulatory regions. EPO, erythropoietin HAS, HIF ancillary sequence; HBS, HIF binding site; DR‐2, nuclear receptor half site
4. DISCUSSION
Most pathogenic genetic variants of EPOR the gene are causative for FE due to truncation of protein cytoplasmic region responsible for receptor negative regulation by SHP‐1 and SOCS‐3 binding to EPOR at 454 aa and 454‐456 aa, respectively.30, 31, 32 For example, variant c.1362C>G at 454 aa causes deletion of the last 55 aa,27 while the variant rs281860299 at 382 aa results in deletion of the last 127 aa.33
In contrast, EPO variants are spread all over the gene and several disease causing mechanisms seem to be involved. EPO variant c.‐136G>A is pathogenic due to increased EPO regulation29 and variant c.32delG due increased EPO production from alternative promoter.10 Variants in promoter and enhancer regions of EPO gene have been associated with elevated hematocrit suggesting involvement in FE, since both regulatory regions have an important role in EPO expression. Variant rs1617640 and rs551238 of EPO gene have also been associated with diabetic retinopathy34 and hematopoietic disorder myelodysplastic syndrome.35
Identification of variants within EPO regulatory regions is of interest since they could contain variants that constitutively activate the EPO gene. Sequence analysis of the 3′HRE region revealed four variants; however, none of which affected the HIF‐1‐binding site, although one was present within HNF‐4 consensus region.19
Screening of the Ensembl database revealed two variants with unknown function located within regulatory regions. Variant rs796952255 is located in HBS, also termed as hypoxia response element (HRE). Variants located within HRE were previously termed as HRE‐SNPs.36 Additionally, variant rs539229161 is located within HNF4A binding region (DR‐2).
In conclusion, sequence variants of the EPO gene show potential for further functional analyses based on their location within previously reported regulatory regions, which might enable generating more targeted hypotheses for testing in the future and enable more efficient biomarker development.
AUTHOR CONTRIBUTION
DV and TP preformed analysis of the literature and regulatory sites and drafted the paper, ND and TK designed the study and edited the final version of the manuscript.
ACKNOWLEDGEMENTS
This work was supported by the Slovenian Research Agency (ARRS) through the Research programmes L3‐9279, P1‐0390 and P4‐0220. We thank Daniel Kriz for language editing.
Vočanec D, Prijatelj T, Debeljak N, Kunej T. Genetic variants of erythropoietin (EPO) and EPO receptor genes in familial erythrocytosis. Int J Lab Hem. 2019;41:162–167. 10.1111/ijlh.12949
Vočanec and Prijatelj contributed equally to the work.
[The copyright line for this article was changed on 29 December 2018 after original online publication].
Contributor Information
Nataša Debeljak, Email: natasa.debeljak@mf.uni-lj.si.
Tanja Kunej, Email: tanja.kunej@bf.uni-lj.si.
REFERENCES
- 1. Kralovics R, Prchal JT. Genetic heterogeneity of primary familial and congenital polycythemia. Am J Hematol. 2001;68(2):115‐121. [DOI] [PubMed] [Google Scholar]
- 2. Bunn HF. Erythropoietin. Cold Spring Harb Perspect Med. 2013;3(3):a011619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hussein K, Percy M, McMullin MF. Clinical utility gene card for: familial erythrocytosis. Eur J Hum Genet. 2012;20(5):4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Camps C, Petousi N, Bento C, et al. Gene panel sequencing improves the diagnostic work‐up of patients with idiopathic erythrocytosis and identifies new mutations. Haematologica. 2016;101(11):1306‐1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bento C, Percy MJ, Gardie B, et al. Genetic basis of congenital erythrocytosis: mutation update and online databases. Hum Mutat. 2014;35(1):15‐26. [DOI] [PubMed] [Google Scholar]
- 6. Arcasoy MO, Karayal AF, Segal HM, Sinning JG, Forget BG. A novel mutation in the erythropoietin receptor gene is associated with familial erythrocytosis. Blood. 2002;99(8):3066‐3069. [DOI] [PubMed] [Google Scholar]
- 7. Bento C, McMullin M, Percy M, Cario H. Primary familial and congenital polycythemia. GeneReviews®. 2016. http://www.ncbi.nlm.nih.gov/books/NBK395975/ [PubMed] [Google Scholar]
- 8. McMullin MF. The classification and diagnosis of erythrocytosis. Int J Lab Hematol. 2008;30(6):447‐459. [DOI] [PubMed] [Google Scholar]
- 9. Amberger JS, Bocchini CA, Schiettecatte F, Scott AF, Hamosh A. OMIM.org: Online Mendelian Inheritance in Man (OMIM®), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 2015;43(D1):D789‐D798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zmajkovic J, Lundberg P, Nienhold R, et al. A gain‐of‐function mutation in EPO in familial erythrocytosis. N Engl J Med. 2018;378(10):924‐930. [DOI] [PubMed] [Google Scholar]
- 11. de la Chapelle A, Träskelin AL, Juvonen E. Truncated erythropoietin receptor causes dominantly inherited benign human erythrocytosis. Proc Natl Acad Sci U S A. 1993;90(10):4495‐4499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Debeljak N, Sytkowski AJ. EpoR. UCSD‐Nature Molecule Pages. 2007; A000863. [Google Scholar]
- 13. Braunstein EM, Moliterno AR. Back to biology: new insights on inheritance in myeloproliferative disorders. Curr Hematol Malig Rep. 2014;9(4):311‐318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Haase VH. Regulation of erythropoiesis by hypoxia‐inducible factors. Blood Rev. 2013;27(1):41‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jelkmann W. Regulation of erythropoietin production. J Physiol. 2011;589(Pt 6):1251‐1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ebert BL, Bunn HF. Regulation of the erythropoietin gene. Blood. 1999;94(6):1864‐1877. [PubMed] [Google Scholar]
- 17. Imagawa S, Yamamoto M, Miura Y. Negative regulation of the erythropoietin gene expression by the GATA transcription factors. Blood. 1997;89(4):1430‐1439. [PubMed] [Google Scholar]
- 18. Weidemann A, Johnson RS. Nonrenal regulation of EPO synthesis. Kidney Int. 2009;75(7):682‐688. [DOI] [PubMed] [Google Scholar]
- 19. Percy MJ, McMullin MF, Lappin TR. Sequence analysis of the 3′ hypoxia‐responsive element of the human erythropoietin gene in patients with erythrocytosis. Biochem Mol Med. 1997;62(1):132‐134. [DOI] [PubMed] [Google Scholar]
- 20. Yates A, Akanni W, Amode MR, et al. Ensembl 2016. Nucleic Acids Res. 2016;44(D1):D710‐D716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sherry ST, Ward MH, Kholodov M, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29(1):308‐311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Stenson PD, Mort M, Ball EV, Shaw K, Phillips A, Cooper DN. The human gene mutation database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum Genet. 2014;133(1):1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fokkema IF, Taschner PE, Schaafsma GC, Celli J, Laros JF, denDunnen JT LOVD vol 2.0: the next generation in gene variant databases. Hum Mutat. 2011;32(5):557‐563. [DOI] [PubMed] [Google Scholar]
- 24. Yates B, Braschi B, Gray KA, Seal RL, Tweedie S, Bruford EA. Genenames.org: the HGNC and VGNC resources in 2017. Nucleic Acids Res. 2017;45(D1):D619‐D625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. den Dunnen JT, Dalgleish R, Maglott DR, et al. HGVS recommendations for the description of sequence variants: 2016 update. Hum Mutat. 2016;37(6):564‐569. [DOI] [PubMed] [Google Scholar]
- 26. Petersen KB, Hokland P, Petersen GB, et al. Erythropoietin receptor defect: a cause of primary polycythaemia. Br J Haematol. 2004;125(4):537‐538. [DOI] [PubMed] [Google Scholar]
- 27. Chauveau A, Luque Paz D, Lecucq L, et al. A new point mutation in EPOR inducing a short deletion in congenital erythrocytosis. Br J Haematol. 2016;172(3):475‐477. [DOI] [PubMed] [Google Scholar]
- 28. Khabour OF, Bani‐Ahmad MA, Hammash NM. Association between polymorphisms in erythropoietin gene and upper limit haematocrit levels among regular blood donors. Transfus Clin Biol. 2012;19(6):353‐357. [DOI] [PubMed] [Google Scholar]
- 29. Taylor JC, Martin HC, Lise S, et al. Factors influencing success of clinical genome sequencing across a broad spectrum of disorders. Nat Genet. 2015;47(7):717‐726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Klingmüller U, Lorenz U, Cantley LC, Neel BG, Lodish HF. Specific recruitment of SH‐PTP1 to the erythropoietin receptor causes inactivation of JAK2 and termination of proliferative signals. Cell. 1995;80(5):729‐738. [DOI] [PubMed] [Google Scholar]
- 31. Hörtner M, Nielsch U, Mayr LM, Heinrich PC, Haan S. A new high affinity binding site for suppressor of cytokine signaling‐3 on the erythropoietin receptor. Eur J Biochem. 2002;269(10):2516‐2526. [DOI] [PubMed] [Google Scholar]
- 32. Sasaki A, Yasukawa H, Shouda T, Kitamura T, Dikic I, Yoshimura A. CIS3/SOCS‐3 suppresses erythropoietin (EPO) signaling by binding the EPO receptor and JAK2. J Biol Chem. 2000;275(38):29338‐29347. [DOI] [PubMed] [Google Scholar]
- 33. Al‐Sheikh M, Mazurier E, Gardie B, et al. A study of 36 unrelated cases with pure erythrocytosis revealed three new mutations in the erythropoietin receptor gene. Haematologica. 2008;93(7):1072‐1075. [DOI] [PubMed] [Google Scholar]
- 34. Yang X, Deng Y, Gu H, et al. Candidate gene association study for diabetic retinopathy in Chinese patients with type 2 diabetes. Mol Vis. 2014;20:200‐214. [PMC free article] [PubMed] [Google Scholar]
- 35. Ma W, Kantarjian H, Zhang K, et al. Significant association between polymorphism of the erythropoietin gene promoter and myelodysplastic syndrome. BMC Med Genet. 2010;11:163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Slemc L, Kunej T. Transcription factor HIF1A: downstream targets, associated pathways, polymorphic hypoxia response element (HRE) sites, and initiative for standardization of reporting in scientific literature. Tumor Biol. 2016;37(11):14851‐14861. [DOI] [PubMed] [Google Scholar]