

Abstract
Background
Genome wide association studies (GWAS) identified SLC44A2 as a novel susceptibility gene for venous thrombosis (VT) and previous work established that SLC44A2 contributed to clot formation upon vascular injury.
Objective
To further investigate the role of SLC44A2 in VT by utilizing SLC44A2 deficient mice (Slc44a2− / −) in two representative disease models.
Methods
Mice were included in a hypercoagulability model driven by siRNA‐mediated hepatic gene silencing of anticoagulants Serpinc1 (antithrombin) and Proc (protein C) and a flow restriction (stenosis) model induced by partial ligation of the inferior vena cava.
Results
In the hypercoagulability model, no effect in onset was observed in Slc44a2− / − animals; however, a drop in plasma fibrinogen and von Willebrand factor coinciding with an increase in blood neutrophils was recorded. In the neutrophil dependent stenosis model after 48 hours, Slc44a2− / − mice had significantly smaller thrombi both in length and weight with less platelet accumulation as a percentage of the total thrombus area. During the initiation of thrombosis at 6 hours post‐stenosis, Slc44a2− / − mice also had smaller thrombi both in length and weight, with circulating platelets remaining elevated in Slc44a2− / − animals. Platelet activation and aggregation under both static‐ and venous and arterial shear conditions were normal for blood from Slc44a2− / − mice.
Conclusions
These studies corroborate the original GWAS findings and establish a contributing role for SLC44A2 during the initiation of VT, with indications that this may be related to platelet‐neutrophil interaction. The precise mechanism however remains elusive and warrants further investigation.
Keywords: animal models of human disease, basic science research, thrombosis, vascular biology, vascular diseases
Essentials.
SLC44A2 does not affect onset in a murine model of venous thrombosis driven by hypercoagulability.
Mice lacking SLC44A2 have less severity of venous thrombosis in the stenosis model following 6 and 48 hours.
There is significantly less platelet accumulation in thrombi of SLC44A2 deficient mice at 48 hours post‐stenosis.
1. INTRODUCTION
Venous thrombosis (VT) is a major contributor to the global health burden with a number of well‐characterized genetic determinants identified, that are all linked to coagulation pathways. 1 A recent meta‐analysis of 12 genome wide association studies (GWAS) identified a novel susceptibility locus for VT within the SLC44A2 gene which had never been linked to coagulation and/or hemostasis before. 2 The association between SLC44A2 and thrombotic events has been next reported in a second GWAS study, confirming SLC44A2 as being a unique contributor to thrombotic disease. 3
The solute carrier family 44 member 2 protein (SLC44A2) 4 is a presumed choline transporter based on its sequence; however, functional evidence remains limited. 5 We previously characterized SLC44A2 deficient mice (Slc44a2− / −) by evaluating several parameters of hemostasis including thrombin generation, transcriptional profiling of coagulation related genes, plasma levels of coagulation factors, von Willebrand factor (VWF) antigen plasma levels, multimerization, and localization within vessels, in addition to recording responses to vascular injury. 6 , 7 We established that Slc44a2− / − have relatively normal hemostasis with the exception of a reduced level of circulating plasma VWF (~20%). These mice have also an impaired response to laser injury of the cremaster arterioles, with significantly less platelet accumulation measured at the site of injury in the C57BL/6 background.
In the present study we delineate the importance of SLC44A2 in VT by utilizing Slc44a2− / − mice in two different VT models; the hypercoagulability small interfering ribonucleic acid (siRNA)‐induced model and the flow restriction (stenosis) model. In the first model, mice are injected with siRNA targeting hepatic expression of the anticoagulants Proc (protein C) and Serpinc1 (antithrombin), thereby creating a state of hypercoagulability and resulting in the formation of pronounced blood clots within the large veins in and around the mandibular area of the head. 8 In the second model thrombus formation is induced by reduction in blood flow of the inferior vena cava (IVC) by approximately 90%, thereby activating the local endothelium and the recruitment of immune cells. 9
2. METHODS
2.1. Mice
Mice deficient for SLC44A2 (Slc44a2− / −) were previously generated 10 and introduced on a C57BL/6J background. 7 , 10 Slc44a2− / − and littermate controls (Slc44a2+ / +) were genotyped using ear biopsy DNA as described. 11 Experimental animal procedures were approved by local animal welfare committees at the Leiden University Medical Center and Aix‐Marseille University. All experiments were performed blinded for genotype.
2.2. Spontaneous thrombosis following silencing of antithrombin and protein C
Female Slc44a2− / − and Slc44a2+ / + mice 6 weeks of age were intravenously injected with siRNAs targeting antithrombin (siSerpinc1: #S62673; Ambion) and protein C (siProc: #S72192) complexed with invivofectamine 3.0 (Invitrogen) as previously described. 8 A dose of 80 nmol of siSerpinc1 and siProc per kg of body weight in study one and 60 nmol in study two was used. The endpoint was reached once 50% of all mice displayed previously described typical clinical features. 8 Blood was collected 24 hours pre‐injection via tail cut using dipotassium ethylenediaminetetraacetic (K2EDTA) acid coated vials (Sarstedt). Blood was also collected from the IVC with 11 µmol/L sodium citrate upon sacrifice and under anesthesia induced by subcutaneous injection of ketamine (100 mg/kg), xylazine (12.5 mg/kg), and atropine (125 µg/kg). Cell counts were assessed by SysmexXT‐2000iV (Sysmex Europe GMBH).
2.3. Thrombosis following stenosis of the IVC
Male Slc44a2− / − and Slc44a2+ / + mice 11 to 12 weeks of age underwent partial ligation of the IVC as previously described, 9 , 11 with all side branches below the renal veins completely ligated. A 50 µL blood sample was collected from the periorbital eye plexus directly before the collection surgery and stabilized with 0.5 mol/L K2EDTA. Cell counts were assessed by Sysmex XN 3000 instrument.
2.4. Plasma analysis
VWF antigen levels were determined by enzyme‐linked immunosorbent assay (ELISA) with anti‐human VWF (DAKO A082) as described. 12 Fibrinogen levels were measured by ELISA according to the manufacturer's protocol, with the exception of using 1 mol/L H2SO4 and half the reaction volume (MGF‐EIA, Stago). Extracellular DNA was quantified using the Quant‐iT PicoGreen DNA Assay Kit (ThermoFisher Scientific).
2.5. Liver analysis
RNA was isolated from liver using RNA‐Bee (Tel‐Test, Inc) and subsequently cDNA was synthesized according to manufacturer's protocol (SuperScript II Reverse Transcriptase, Thermo Fisher). Liver transcript levels of Slc44a2, Serpinc1, and Proc were determined by quantitative polymerase chain reaction (qPCR). 13 IVC RNA was isolated using the RNeasy microkit (Qiagen 74034). cDNA was synthesized and transcript levels of Slc44a2, Ccl2, Cxcl1, Cxcl5, Il6, Selplg, and Vcam1 were quantified by qPCR. ß‐actin (Actb) was used as reference gene (primers Table S1 in supporting information). Fibrin in liver (and also lungs) was determined by immunoblotting using the mouse monoclonal 59D8 anti‐fibrin antibody as previously described. 14
2.6. Histology
Following removal, thrombi were measured, weighed, and frozen in optimum cutting temperature (OCT; Tissue Tek). Serial cryo sections from the medial region of the thrombi were made at 10 µm. Immunohistochemical staining was performed as described 11 using anti‐Ly6G (Biolegend #127602; 1:1000 dilution), anti‐citrullinated histone H3 (CitH3; Abcam #ab5103; 1:300 dilution), anti‐VWF (DAKO #A0082; 1:4000 dilution; Abcam # ab11713; 1:50 dilution), anti‐glycoprotein Ib (GPIb; Emfret #R300; 1:2000 dilution) and anti‐tissue factor (TF; in‐house resource, 1:2000 dilution) antibodies. The corresponding secondaries were anti‐Rat IgG horseradish peroxidase (HRP) conjugated (Abcam #ab205720; 125 µg/mL) or anti‐Rabbit IgG HRP conjugated (DAKO # P039901‐2; 125 µg/mL) and signal was visualized using Vector NovaRED (Vector Laboratories #SK4800). Immunofluorescence staining was performed as before 11 using the fluorophore‐conjugated secondaries anti‐rabbit 488 (#A‐11008), anti‐rat 568 (#A‐11077), anti‐sheep 647 (#A‐21448) (Invitrogen, all at 1:750 dilution). Microscopic images were taken using the Panoramic MIDI Slide Scanner and Caseviewer software (3Dhistech). Quantifications were calculated using the Fiji ImageJ program. 15 Per mouse, when a thrombus was available, one stained section from the medial region was used for comparison.
2.7. Reactive oxygen species production
Citrated blood was collected via tail cut and leukocyte activation was measured ex‐vivo in 25 µL blood incubated with anti‐CD11b (BD Pharmingen; clone M1/70), anti‐B220 (eBioscience; clone RA3‐6B2), and anti‐Ly6G (eBioscience, clone 1A8) for 30 minutes on ice. Erythrocytes were then lysed using 0.155 mol/L NH4Cl, 0.01 mol/L KHCO3, 0.1 mmol/L ethylenediaminetetraacetic acid (EDTA) for 10 minutes at 20°C and remaining cells were incubated with DHR‐123 (ThermoFischer, #D23086, 1:1000, 10 minutes, 37°C) and stimulated or not with phorbol myristate acetate (PMA; 2 µg/mL, Sigma P8139, 10 minutes, 37°C). Fluorescence was measured on an LSR II flow cytometer (BD Bioscience).
2.8. Platelet aggregation
Citrated blood was collected via IVC and centrifuged at 313 g for 3 minutes at room temperature using a soft break. Platelet rich plasma layer (PRP) and one third of the erythrocytes layer was collected and separately centrifuged at 704 g for 15 seconds using soft break. Platelet counts were determined by Sysmex and adjusted with platelet poor plasma (PPP). PRP was further diluted into HEPES:Tyrode pH 7.3 and rested for 30 minutes before measurements. PRP was then incubated with 1.2 mmol/L thrombin receptor activating peptide 4 (TRAP4; Bachem, #4035529‐005) or 24 µmol/L adenosine diphosphate (ADP; HART biological). Aggregation was measured for 15 minutes on a Thrombo‐Aggregometer (SD Medical, #TA‐8V) against PPP.
2.9. Platelet perfusion
Platelet adhesion and activation following perfusion over coverslip coated with VWF‐binding peptide (50 µg/mL, Dept. of Biochemistry, Cambridge University) or collagen type I (50 µg/mL, HORM collagen, Takeda) was assessed as described before. 16 Citrated whole blood was re‐calcified with 7.5 mmol/L CaCl2 and 3.75 mmol/L MgCl2 in the presence of D‐phenylalanyl‐prolyl‐arginyl chloromethyl ketone (PPACK; 53 µmol/L, Calbiochem) and heparin (5 U/mL), labelled with 0.5 µg/mL DiOC6 (AnaSpec). Analysis of fluorescence images was performed with pre‐defined scripts in Fiji software. 15 , 17
2.10. Platelet activation by flow cytometry
Citrated tail blood was diluted 25 times in Tyrode HEPES pH 7.45 (5 mmol/L HEPES, 136 mmol/L NaCl, 2.7 mmol/L KCl, 0.42 mmol/L NaH2PO4, 2 mmol/L MgCl2, 0.1% glucose and 0.1% bovine serum albumin) in the presence of PPACK (20 µmol/L) and fragmin (20 U/mL, Pfizer). The blood was activated for 10 minutes with various concentrations of cross‐linked collagen‐related peptide (CRP‐XL, from Cambridge University), 2‐methylthio‐adenosine‐5'‐diphosphate (2‐MeSADP, BioConnect), or the protease activated receptor 4 (PAR4) agonist AYPGKF. Platelets were labelled with anti‐GPIIbIIIa (JON/A; Emfret, PE, 1:10 dilution) and anti‐P‐selectin (CD62P; Emfret, FITC, 1:10 dilution), and activation measured with an Accuri C6 flow cytometer (Becton Dickinson).
2.11. Statistics
For phenotype free survival, differences were calculated using the Mantel‐Cox log rank test method. Spearman's correlation was used to compute r and determine correlation between immunohistochemical stains. Reactive oxygen species (ROS) production and platelet aggregation were evaluated by t‐test. Changes in platelet binding under perfusion and platelet activation with flow cytometry were determined by two‐way analysis of variance. Statistical testing for the remaining readouts were calculated using the Mann‐Whitney rank‐sum test. All calculations were performed using the Prism statistical program, version 8 (GraphPad).
3. RESULTS
3.1. SLC44A2 does not alter onset in a model of hypercoagulability driven VT
In order to determine whether SLC44A2 had an effect on thrombus formation driven by hypercoagulability, we induced a hypercoagulable state in SLC44A2 deficient mice (Slc44a2− / −) and wild type control (Slc44a2+ / +) through siRNA‐mediated knockdown of hepatic expression of the anticoagulants antithrombin (Serpinc1) and protein C (Proc). Venous thrombosis and related features in this model are same for male and female mice, but well characterized particularly for female mice. 8 , 18 We thus used female mice here. After 24 hours, mice from both groups began to present typical features coinciding with thrombotic coagulopathy, ie, edema of mandibular area and bleeding around the eye. At the time of sacrifice, 32 hours post‐siRNA treatment, 50% of Slc44a2+ / + (6/12) and 92% of Slc44a2− / − mice (11/12) had developed the observable phenotype (Figure 1A), but this difference in onset was not significant. Both groups lost approximately 8% of their original body weight (Figure S1 in supporting information). To validate the incidence over a more protracted timeline, we repeated the study using a lower dose of siRNA. Again no significant difference could be observed with 43% of Slc44a2+ / + (3/7) and 29% of Slc44a2− / − mice (2/7) developing the phenotype 32 hours post injection (Figure 1B). All mice were affected at the collection point of 46 hours. Upon sacrifice, knockdown of hepatic Serpinc1 and Proc was confirmed and was 70% and 98% lower, respectively, as compared to untreated control mice. This was comparable between genotypes (Figure S2 in supporting information), although Proc levels were lower (P = .0024) in Slc44a2− / −, measured at 1% of control versus 3.6% in Slc44a2+ / +.
Figure 1.
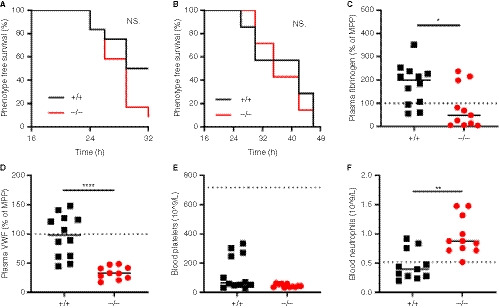
Hypercoagulability mediated venous thrombosis through siRNAs targeting Serpinc1 and Proc in SLC44A2 deficient mice. A, Phenotype free survival in SLC44A2 deficient mice (Slc44a2− / −, −/−) and littermate wild type controls (Slc44a2+ / +, +/+) shown as percentage following injection with 80nmol/kg siRNA (n = 12 per group). B, Phenotype free survival following injection with 60nmol/kg siRNA (n = 6 per group). The following blood parameters were measured after treatment with 80nmol/kg siRNA: (C) Plasma fibrinogen levels 32 hours post‐treatment expressed as a percentage of MPP (mouse pool plasma), (D) plasma VWF (von Willebrand factor) levels 32 hours post‐treatment expressed as a percentage of MPP (mouse pool plasma), (E) blood platelet counts at time of sacrifice. For reference platelet levels of Slc44a2+ / + before siRNA treatment are represented by the dotted line (mean cell counts). F, Circulating neutrophil levels at time of sacrifice. For reference platelet levels of Slc44a2+ / + mice before siRNA treatment are represented by the dotted line (mean cell counts). Statistical analysis for phenotype free survival determined using Mantel‐Cox test. Solid line represents median value. Statistical differences were evaluated using Mann‐Whitney rank‐sum test (NS = non‐significant; *signifies P < .05; **signifies P < .01; ****signifies P < .0001)
In additional to macrovascular thrombosis in the head the deposition of fibrin in the lungs and liver typically occurs in this model. 8 , 18 Accordingly, fibrin was detected in both the lungs and liver of the Slc44a2+ / + mice and was comparable to Slc44a2− / − mice (Figure S3 in supporting information). Fibrinogen levels in the plasma were lowered in the Slc44a2− / − mice as compared to Slc44a2+ / + (P = .0129, Figure 1C). We confirmed the previously described reduction in circulating VWF of ~20% in Slc44a2− / − mice before siRNA treatment (P = .0211, Figure S4 in supporting information). 7 Remarkably, upon sacrifice following thrombosis, this difference in VWF was even more pronounced as measured antigen levels were 66% lower in Slc44a2− / − mice as compared to Slc44a2+ / + (P < .0001, Figure 1D). No differences in blood cell counts prior to injection and following siRNA treatment were observed. Platelet consumption is associated with thrombosis in this model and even though four Slc44a2+ / + mice did not develop an observable thrombus, corresponding with higher amounts of blood platelets, there was no significant difference in platelet levels following treatment between genotypes (Figure 1E). Interestingly, a significant increase in blood neutrophils (P = .0017, Figure 1F) was recorded in the Slc44a2− / − mice upon sacrifice. However the formation of thrombi in this model was previously demonstrated to occur independently of VWF or neutrophils. 18 Thus, even with such changes, SLC44A2 does not affect thrombosis incidence in this model.
3.2. Slc44a2− / − mice have reduced thrombosis following 48 hours of stenosis
We continued by using a model demonstrated to be dependent on neutrophils and VWF: the flow restriction (stenosis) model of deep vein thrombosis (DVT). 11 , 19 , 20 Male mice, littermates of the female mice used in the previous model, were used in respect of the “3R’s” rule for more ethical use of animals and to maintain the same environmental exposure and breeding sources for mice included in the different VT models. 21 In this model the blood flow was reduced by approximately 90%, with blood stasis activating the local endothelium and driving inflammation‐mediated thrombosis. 9 Following 48 hours, 100% of the Slc44a2+ / + mice (10/10) and 80% of the Slc44a2− / − mice (12/15) developed a thrombus (Figure 2A). The thrombi formed in Slc44a2− / − mice were significantly smaller both in length (−43%, P = .0120) and weight (−52%, P = .0099; Figure 2B‐C). Mice heterozygous for SLC44A2 (Slc44a2+ / −) were found to have 100% incidence (7/7) with median thrombus measurements for length and weight falling between those of the Slc44a2+ / +and Slc44a2− / − groups (Figure S5 in supporting information), suggesting a dose‐dependent effect of SLC44A2 on VT. No differences in thrombi neutrophil density were visible (Ly6G staining, Figure 2D) and the levels of circulating blood neutrophils at the time of sacrifice were also similar (Figure 2E). To determine whether neutrophil extracellular trap release (NETosis) was affected, we evaluated CitH3 staining into the obtained thrombi. A trend was observed for lower CitH3 in the Slc44a2− / − mice (P = .0908; Figure 2F) and the positive correlation found between the neutrophil and CitH3 staining (r = .7708; P = .0252) in Slc44a2+ / + was absent in Slc44a2− / − animals (r = .2207; P = .5143; Figure 2G) suggesting reduced NETosis within thrombi in absence of SLC44A2. However, no signs of systemic decrease in plasma DNA levels could be found 48 hours after stenosis (Figure 2H). Additionally, ex‐vivo production of reactive oxygen species (ROS) by stimulated neutrophils was not different between neutrophils deficient or not for SLC44A2 (Figure 2I). Together these data demonstrate a role for SLC44A2 in stenosis driven thrombosis and suggest a possible effect on NETosis at the site of thrombosis.
Figure 2.
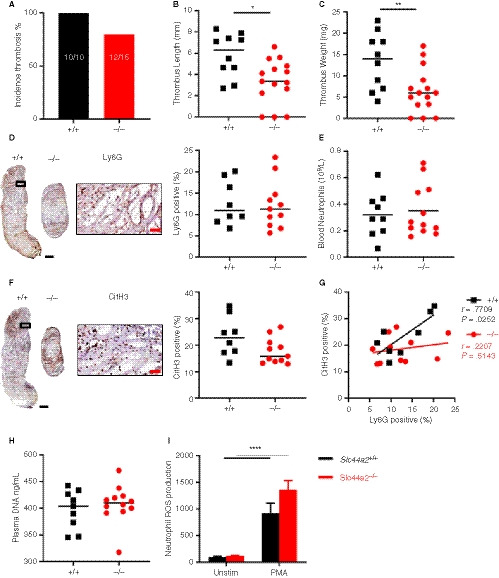
Venous thrombosis in SLC44A2 deficient mice following 48 hours stenosis of the inferior vena cava (IVC). A, Incidence of thrombosis after 48 hours in wild type (Slc44a2+ / +, +/+) (n = 10) or SLC44A2 deficient mice (Slc44a2− / −,−/−) (n = 15), shown as percentage (%). B, Length and (C) weight of thrombi formed at 48 hours. D, Immunohistochemical (IHC) staining of neutrophil marker Ly6G in a representative thrombus from a Slc44a2+ / + and Slc44a2− / −, with high magnification from Slc44a2+ / + (location is indicated by the black box; left) and quantification of positive area as a percentage of total thrombus area (right, +/+ n = 8, −/− n = 11). E, Blood neutrophil counts at time of sacrifice (48 hours; +/+ n = 9, −/− n = 12). F, IHC staining of the NETosis marker citrullinated histone H3 (CitH3) in a representative thrombus of a Slc44a2+ / + and Slc44a2− / −, with high magnification from Slc44a2+ / + (location is indicated by the black box; left) and quantification of positive area as a percentage of total thrombus area (right, +/+ n = 8, −/− n = 11). G, Correlation plot between LY6G and CitH3 staining (+/+, n = 8; −/−, n = 11). H, Plasma DNA level at time of sacrifice (48 hours; ng/mL; +/+, n = 9; −/−, n = 11). I, Reactive oxygen species (ROS) produced by Slc44a2+ / + or Slc44a2− / − neutrophils ex vivo following activation of whole blood with 2 µg/mL phorbol 12‐myristate 13‐acetate (PMA), quantified by FACS analysis, expressed in median fluorescence index (MFI; n = 5). (Note on numbers: blood samples that became coagulated or thrombi that were damaged during sectioning were not included for subsequent analysis.) Black bars equal 500 µm. Red bars equal 50 µm. Solid line represents median value. Statistical differences for ROS production were evaluated by t‐test and for remaining biological readouts, the Mann‐Whitney rank‐sum test (*signifies P < .05; **signifies P < .01; ****signifies P < .0001)
3.3. Slc44a2− / − mice have less platelet accumulation after 48 hours stenosis
Previously, we determined that Slc44a2− / − have a reduced level of circulating plasma VWF antigen when compared to Slc44a2+ / +. 7 To investigate whether the VWF release from the endothelium was reduced in SLC44A2 deficient mice, this discharge was stimulated using intraperitoneal injection of 2 mg/kg lipopolysaccharides (LPS), a dose inducing Weibel‐Palade body release. VWF plasma levels were increased following LPS injection; however, in time we observed no differences in VWF levels between Slc44a2+ / + and Slc44a2− / − mice (Figure S6 in supporting information). This suggests that smaller thrombi in Slc44a2− / − mice are not likely due to reduced levels of available VWF following stenosis. We also found that VWF distribution within thrombi was comparable between the two genotypes by constituting approximately 30% of the thrombus area (Figure 3A) and that plasma VWF at 48 hours post‐stenosis was not significantly different between groups (P = .2030; Figure 3B). When we focused on platelets, platelet counts could be correlated to thrombus size both in Slc44a2+ / + and Slc44a2− / − mice as platelet accumulation is a major determinant of thrombus size at 48 hour‐stenosis (Figure 3D). Remarkably though, the proportion of platelet marker GPIb‐positive area was significantly reduced (P = .0259) in thrombi from Slc44a2− / − mice (Figure 3C). As VWF and GPIb are established binding partners, 22 we evaluated the relationship of the two proteins on serial sections. VWF was strongly associated with GPIb in Slc44a2+ / + mice (r = .9004; P = .0023); however, this association was absent in Slc44a2− / − (r = −.3471; P = .2956; Figure S7 in supporting information). We then verified these relationships on the same thrombus section by confocal microscopy. We measured a clear association between VWF and GPIb in the Slc44a2+ / + group (r = .9048; P = .0046) and none within the Slc44a2− / − (r = .2818; P = .4023) (Figure 3E). Moreover, the area of VWF that co‐localized with GPIb was reduced in the Slc44a2− / − animals (−36%, P = .0506; Figure 3F). As this indicated a possible altered interaction between GPIb and VWF, we evaluated the binding potential of platelets to VWF under venous flow (150 s−1) ex vivo using slides coated with a murine VWF binding peptide and perfused with whole blood. We observed a comparable increase of platelet binding with a final mean surface area coverage of 15.4% (5.6%‐22.2%) by Slc44a2+ / + platelets and 19.7% (17.2%‐25.6%) by Slc44a2− / − platelets after 6 minutes (P = .9122; Figure 3G). When we further dissect between firmly adherent and translocating platelets we also did not observe differences, indicating that also stable binding of platelets is unaffected by SLC44A2 (Figure 3H). To evaluate whether platelets under arterial flow have altered adherence, whole blood was perfused over collagen at 1000 s−1, also here no differences were observed (Figure S8 in supporting information). Based on these findings, platelet accumulation is reduced in thrombi from Slc44a2− / − mice at 48 hours and this is not related to VWF availability. Additionally, perfusion studies revealed that platelet‐VWF interactions did not seem to be affected in Slc44a2− / − mice.
Figure 3.
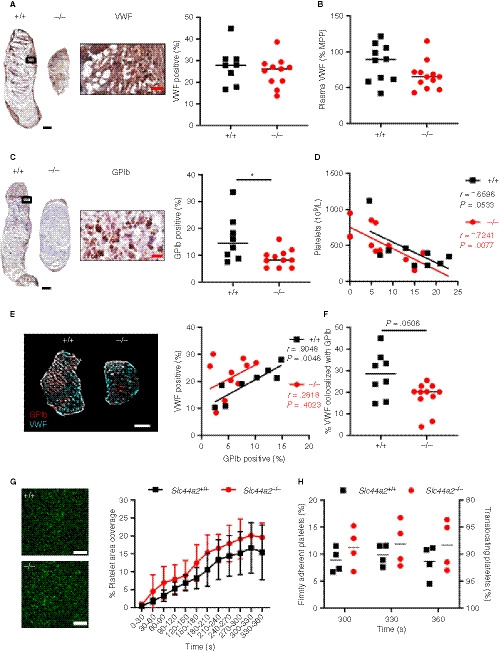
von Willebrand factor (VWF) and platelet characteristics of SLC44A2 deficient mice following 48 hours stenosis and ex vivo under flow conditions. A, Immunohistochemical (IHC) staining of VWF in thrombus of a representative wild type (Slc44a2+ / +, +/+) and SLC44A2 deficient mice (Slc44a2− / −, −/−), with high magnification from Slc44a2+ / + (location is indicated by the black box; left) and quantification (right) of positive area as a percentage of total thrombus area in Slc44a2+ / + (n = 8) or Slc44a2− / − mice (n = 11). B, Plasma VWF levels after 48 hours stenosis expressed as a percentage of MMP (mouse pool plasma; +/+ n = 9, −/− n = 12). C, IHC staining of glycoprotein Ib (GPIb) in thrombus of a representative Slc44a2+ / + and Slc44a2− / −, with high magnification from Slc44a2+ / + (location is indicated by the black box; left) and quantification (right) of positive area as a percentage of total thrombus area (+/+ n = 8, −/− n = 11). D, Correlation plot between thrombus weight and circulating blood platelets (+/+ n = 9, −/− n = 12). E, Immunofluorescent co‐stain of GPIb (red) and VWF (cyan) on thrombus sections (left) with correlation plot (right; n = 8 +/+; 11 −/−). F, Percentage of VWF positive area colocalized with GPIb. G, Representative images of DiOC6‐labeled platelets of a wild type (Slc44a2+/+, +/+) control (up) and a SLC44a2 deficient mouse (Slc44a2− / −, −/−; down) on a surface coated with VWF‐binding peptide. Scale bar is 20 µm. H, Percentage of stable platelet area coverage over a 30 second time period in field view within heparinized and D‐phenylalanyl‐prolyl‐arginyl chloromethyl ketone (PPACK) treated whole blood flowing over slides coated with a murine VWF binding peptide at venous shear rate (150 s−1) over time (n = 5 per group). (Note on numbers: blood samples that became coagulated or thrombi that were damaged during sectioning were not included for subsequent analysis.) I, Percentage platelets displaying a firm or transient interaction with VWF‐binding peptide determined by a method described by Meyer dos Santos et al. 17 Black and white bars equal 500 µm. Red bar equals 50 µm. Statistical differences between for platelet perfusion were evaluated by two‐way analysis of variance and for remaining biological readouts, the Mann‐Whitney rank‐sum test. Coefficient r calculated using Spearman's correlation (*signifies P < .05)
3.4. Thrombosis in Slc44a2− / − mice is reduced at 6 hours post‐stenosis
To gain further insight into the role of SLC44A2 during the initiation phase of thrombosis, thrombosis was measured 6 hours post IVC ligation. At this timepoint 100% of the Slc44a2+ / + (6/6) and 62.5% of the Slc44a2− / − mice formed a thrombi (5/8) (Figure 4A). The thrombi from the Slc44a2− / − mice were again smaller both in length (−83%, P = .0007) and weight (−99%, P = .0013; Figure 4B‐C). As before, we observed that Slc44a2+ / − mice (incidentally included) fell in between with 75% incidence (3/4) and median values of length and weight halfway in between those of the Slc44a2+ / + and Slc44a2− / − (Figure S9 in supporting information). An independent experiment executed by a different operator, in a different facility, substantiated these findings, with significant effects of genotype on thrombus weight (P = .0020) and length (P = .0010), and again no effect on incidence (Figure S10 in supporting information). Evaluation of the thrombi revealed large variability within each group for GPIb staining with high and low subsets for both Slc44a2+ / + and Slc44a2− / −, leading to no significant differences with regard to platelet density (Figure S11A in supporting information). Similarly, no differences could be detected with regard to VWF staining (Figure S11B). We found again a correlation between the VWF and GPIb staining in Slc44a2+ / + mice (r = .8857; P = .0333) that persisted this time in Slc44a2− / − (r = .9000; P = .0833; Figure 4D). The percentage of VWF that colocalized with GPIb was also comparable between the groups (Figure 4E). These data indicate that the early binding of platelets to VWF is unaltered. Additional quantification determined no differences in thrombus density in neutrophils (Ly6G staining) and NETs (CitH3 staining; Figure S11C‐D). The CitH3‐positive area was found to be strongly correlated with GPIb levels in Slc44a2+ / + mice (r = .9429; P = .0167), but not in the Slc44a2− / − animals (r = .5000; P = .4500; Figure 4F), which was not observed at the 48‐hour timepoint (Figure S12 in supporting information).
Figure 4.
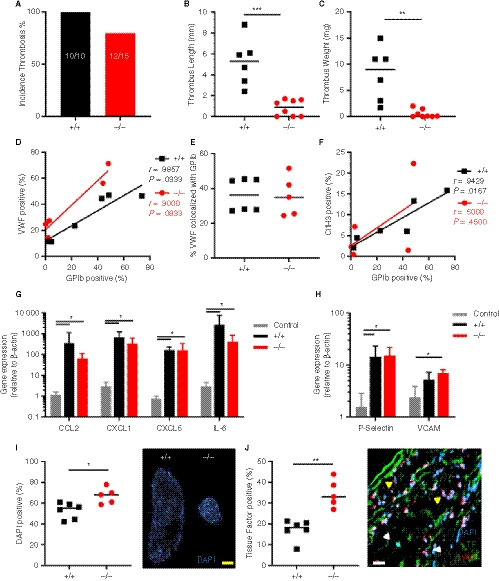
Thrombosis in SLC44A2 deficient mice following 6 hours stenosis of the inferior vena cava (IVC). A, Incidence of thrombosis after 6 hours in mice wild type (+/+, n = 6) or SLC44A2 deficient mice (−/−, n = 8), shown as percentage. B, Length and (C) weight of thrombi formed at 6 hours. D, Correlation plot of GPIb and VWF immunostaining on thrombus sections (+/+, n = 6; −/−, n = 5). E, Percentage of VWF positive area colocalized with GPIb. F, Correlation plot of GPIb and citrullinated histone H3 immunostaining on thrombus sections. G, Transcript levels of inflammatory molecules produced by the local IVC in Slc44a2+ / + (+/+, n = 6) or Slc44a2− / − (−/−, n = 5) for SLC44A2 collected at 6 hours post‐stenosis as compared to non‐ligated control C57BL/6J mice (n = 3); chemokine (C‐C motif) ligand 2 (Ccl2), chemokine (C‐X‐C motif) ligand 1 (Cxcl1), Cxcl5, interleukin‐6 (Il6). H, IVC expression of mRNA transcripts encoding adhesion molecules; P‐Selectin and vascular cell adhesion molecule 1 (VCAM1) following 6 hours stenosis. Bars represent mean values with standard deviation (error bars). I, Nuclear stain (DAPI) of leukocytes in thrombi from Slc44a2+ / + and Slc44a2− / − mice (top left) with quantification as a percentage of total thrombus area (n = 6 +/+; 5 −/−). J, Quantification of immunofluorescent (IF) staining of tissue factor (TF; left) as a percentage of total thrombus area. Representative image of IF co‐stain by confocal microscopy of neutrophil marker Ly6G, TF, and nuclear stain (DAPI; right panel). Yellow arrows indicate Ly6G positive cells and white arrows indicate TF positive cells. Yellow bar equals 500 µm and white bar equals 20 µm. Statistical differences were evaluated using Mann‐Whitney rank‐sum test (*signifies P < .05; **signifies P < .01; ***signifies P < .001)
Stenosis is followed by an acute response from the local endothelium marked by upregulation of inflammatory cytokines and adhesion markers. 9 Transcript levels of cytokines CCL2, CXCL1, CXCL5, and IL‐6 in the IVC 6 hours post‐stenosis were increased compared to control untreated IVC (P < .05), which was in line with previous reports. 9 However, there were no differences in transcript levels between genotypes (Figure 4G). The same observations were made for the transcripts coding for the adhesion molecules P‐selectin and VCAM‐1, which are central to immune cell interactions with the endothelium (P < .05; Figure 4H). These data indicate a normal inflammatory response by the endothelium in Slc44a2− / − mice after stenosis.
In this model endothelial activation is followed by recruitment of immune cells to the site of stenosis. Leukocytes incorporated into the thrombi can be visualized using a nuclear stain because platelets and erythrocytes are enucleated cells. Interestingly, when the thrombi were stained with 4′,6‐diamidino‐2‐phenylindole (DAPI), the density of DAPI‐positive leukocytes was significantly elevated in the Slc44a2− / − mice (%, P = .0303; Figure 4I). This was not observed at 48 hours post‐stenosis (Figure S13 in supporting information). As we established there was no difference in Ly6G positive cell composition (Figure S11C); this observation suggests that the increased leukocytes are likely not neutrophils. TF production by leukocytes is another critical step in this model and we observed a two‐fold increase in TF immunostaining density in thrombi from the Slc44a2− / − animals (P = .0043; Figure 4J). Interestingly, the nucleated cells that co‐localized with TF were mainly Ly6G negative (Figure 4J) while all nucleated cells were Ly6C positive (Figure S14 in supporting information) indicating that these are likely monocytes, which was previously reported to be the main source of TF in this model. 9 Together these data demonstrate that SLC44A2 is important during the initiation of thrombosis, but not for endothelial activation in this VT model.
3.5. Platelet activation, aggregation, and neutrophil interactions is unaltered in Slc44a2− / − platelets
We established that SLC44A2 deficiency has a pronounced effect on stenosis‐driven thrombosis, coinciding with reduced platelet accumulation after 2 days without any noticeable effect on platelet/VWF colocalization. We hypothesized that SLC44A2 may be involved after platelet adhesion to the vessel wall in subsequent stages of platelet activation. We observed that the consumption of platelets from circulation had not yet occurred in the Slc44a2− / − mice at 6 hours post‐stenosis. As a result the levels of circulating platelets differ between Slc44a2+ / + and Slc44a2− / − after stenosis (P = .0061; Figure S15 in supporting information). The upregulation of membrane protein P‐selectin (CD62P) is a key step during platelet activation, 23 whereas a feature of later stages of platelet activation is the binding of platelets to fibrinogen via the αIIbβ3 integrins. When we look at the percentage positive platelets for CD62P exposure as well as αIIbβ3 activation after stimulation with different concentrations of ADP, PAR4 agonist and collagen‐related peptide CRP‐XL, we observed a significantly lowered exposure in Slc44a2− / − platelets under some conditions (Figure S16 in supporting information). When we, however, analyzed the median fluorescence intensity of both CD62P and αIIbβ3 we did not find an effect of SLC44A2 (Figure 5A‐B). To determine if SLC44A2 had an effect on platelet‐platelet interactions, we measured platelet aggregation using PRP following activation using TRAP‐4 (strong stimulus) and ADP (weak stimulus). We did not detect any significant differences in platelet aggregation in Slc44a2− / − (Figure 5C‐D). This is in line with our previous observation, using washed platelets, in which platelet aggregation induced by thrombin or collagen was unaffected. 7 Together, this is suggestive of a normal response by Slc44a2− / − platelets.
Figure 5.
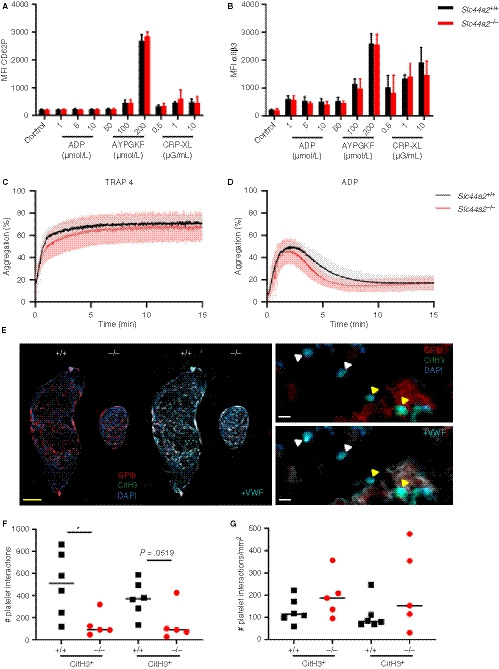
Characterization of platelets in SLC44A2 deficient mice. Percentage of cells positive for (A) exposed P‐selectin (CD62P) or (B) activated integrin αIIbβ3 following activation with ADP (1, 5, 10 µmol/L), protease‐activated receptor 4 (PAR4) agonist AYPGKF (50, 100, 200 µmol/L) or collagen related peptide (CRP‐XL; 0.5, 1 or 10 µg/mL) as determined by flow cytometry and quantified as the percentage of cells staining positive (%) in wild type (Slc44a2+ / +, N = 5) or SLC44A2 deficient mice (Slc44a2− / −, N = 5). Platelet aggregation following stimulation with (C) 1.2 mmol/L thrombin receptor activating peptide 4 (TRAP‐4) or (D) 24 µmol/L adenosine diphosphate (ADP) in Slc44a2+ / + and Slc44a2− / −. E, Immunofluorescent co‐stain of thrombi from Slc44a2+ / + and Slc44a2− / − of nuclei (DAPI; blue), platelet marker GPIb (red), and citrullinated histone H3 (CitH3; green) with and without additional visualization of staining with VWF (cyan; left). Higher magnification indicates leukocyte‐platelet interactions (yellow triangles) and leukocytes positive for CitH3 without platelet interactions (white triangles) with (right bottom) and without (right top) additional visualization of VWF. Yellow bar represents 500 µm, white bar represents 10 µm. F, Quantification of nucleated cells in contact with platelets either positive or negative for CitH. G, Number of platelet interactions with leukocytes per mm2 of thrombus. Statistical differences for flow cytometry were calculated using two‐way analysis of variance (shown as mean with standard deviation [SD]) and for aggregometry, t‐test (shown with mean and SD). For platelet‐leukocyte quantifications, statistical differences were evaluated using Mann‐Whitney rank‐sum test (*signifies P < .05; **signifies P < .01)
In addition to the primary role of platelets in hemostasis, they can also activate neutrophils and stimulate NETosis 24 as neutrophils and NETs can activate and recruit platelets. 20 , 25 , 26 To evaluate whether SLC44A2 influenced murine platelet‐neutrophil interactions, we perfused recalcified whole blood at venous shear rates (150 s−1) over slides coated with a VWF capturing peptide. Under these conditions, we did not observe neutrophil binding to the platelets that adhered to the slides; however, the rapid formation of fibrin and coagulation of the blood also made the analysis challenging. We then used a different approach and analyzed platelet‐leukocyte interactions within the thrombi of the mice obtained 6 hours after stenosis. Here we could clearly distinguish leukocytes adjacent to platelets and record if they were either positive or negative for CitH3 (Figure 5E, left panel). We noticed that areas positive for GPIb strongly overlapped with VWF expression (Figure 5E, right panel) as we saw earlier (Figure 4D). Quantification of the total number of platelet‐leukocyte interactions revealed that more platelets were found interacting with CitH3 positive cells in Slc44a2+ / + mice than in Slc44a2− / − mice (Figure 5F); however, this difference was not significant when thrombus size was taken into account (Figure 5G).
4. DISCUSSION
Genomic studies can be a powerful tool for identifying novel factors that contribute to pathophysiology. 27 , 28 SLC44A2 is the first gene outside of the coagulation cascade found to be associated with VT risk. 2 , 3 VT is influenced both by inflammation and coagulation. 29 During inflammation, endothelial cells and neutrophils will play a major role in the initiation phase of thrombosis, whereas hypercoagulability will mostly influence the propagation phase. 9 Here we used SLC44A2 deficient mice in two independent models of VT. We have substantiated the genomic data from the GWAS and established a functional role for SLC44A2 in stenosis‐, but not in hypercoagulability‐, driven thrombosis. The absence of a role for SLC44A2 in thrombosis driven by hypercoagulability supports the notion that SLC44A2 does not interfere with the coagulation system. This is in agreement with our previous finding that SLC44A2 does not influence mouse hemostasis (including coagulation) under normal conditions 7 and also with the observation that variation in SLC44A2 did not associate with hemostasis phenotypes included in the GWAS. 2 For stenosis‐driven thrombosis we observed that SLC44A2 deficiency affects thrombus size after 6 and 48 hours of blood flow restriction in mice. This is interesting because in contrast to the hypercoagulability model, the stenosis model is inflammation driven, indicating a role for SLC44A2 in inflammation‐regulated thrombosis. This suggests that neutrophil recruitment and/or activation could be altered in Slc44a2− / − mice. Our data do not permit us to conclude on a possible reduction in neutrophil adhesion to the vessel wall in absence of SLC44A2 but they could still suggest a possible reduction in NETosis and platelet accumulation in this model. The percentage of platelets activated ex vivo was slightly reduced under specific conditions; the median fluorescence index (MFI) of platelet activation markers however was not affected, leading us to conclude that the overall platelet activation is unaffected. Moreover, aggregation and thrombus formation in vitro/ex vivo were found to be normal for blood from SLC44A2 deficient mice, which is suggestive of normal hemostatic function of Slc44a2− / − platelets. Despite the normal hemostatic function of platelets, altogether, these studies establish a contributing role for SLC44A2 during the initiation of VT.
The function of SLC44A2 is poorly understood, although there is emerging data that describe ways it may be modulating thrombosis. SLC44A2 is suggested to be a binding partner of VWF and this interaction on neutrophils led to agglutination when in the presence of plasma containing anti‐SLC44A2 antibodies (HNA3A), thereby inducing transfusion related acute lung injury (TRALI). 30 Additionally, findings presented at the European Congress on Thrombosis and Hemostasis 2018 demonstrated that a specific allelic variant of SLC44A2, also associated with VT, plays a role in neutrophil binding to VWF which leads to NETosis after priming with TNFα. 31 Most recently, it was shown that platelets primed by VWF display activated integrin αIIbβ3 (but not CD62P), which can then bind neutrophil SLC44A2 and mediate NETosis under venous flow. 32 Based on these findings, the role of SLC44A2 is supposedly limited to neutrophil biology and NET release; however, the suggested binding partner differs. One theory suggests a direct interaction with VWF and the other with platelet integrin αIIbβ3 following adherence to VWF. Combining all these findings would implicate SLC44A2 as a binding partner of two elements involved in thrombosis, one being VWF and the other platelet integrin αIIbβ3, with both binding to SLC44A2 on neutrophils.
Neutrophils are known to be a major driver of venous thrombosis in the stenosis model. 9 Therefore it is tempting to speculate that SLC44A2 on neutrophils is responsible for the underlying effects of this protein on thrombus formation. In the current study, upon stenosis we observe a trend for less CitH3 within the Slc44a2− / − thrombi at 48 hours, which is in line with the notion that SLC44A2 deficient neutrophils are less active in the production of NETs. However, we demonstrated that neutrophil activation is still occurring in these mice both in vivo and ex vivo, as determined by CitH3 staining, plasma DNA levels, and ROS production. It is possible that the observed reduced platelet incorporation into Slc44a2− / − thrombi at 48 hours is due to slightly less NETosis as this process promotes platelet aggregation through the interaction of NET bound cathepsin G with platelet P2Y12 and αIIbβ3 receptors. 26 , 33
Alternatively, platelet activation can also drive NET production through the presentation of high mobility group box 1 (HMGB1) 34 or CD62P. 35 This would support our observation of increased CitH3 expression with increasing platelet levels in thrombi following 6 hours stenosis in the Slc44a2+ / + mice. Importantly, though, we did observe direct contact of platelets with CitH3 positive leukocytes within the thrombi of both groups at 6 hours, implying that platelets deficient for SLC44A2 can still activate neutrophils, even when they are also lacking SLC44A2. Interestingly, the lost correlation of GPIb and VWF at 48 hours is in line with the previous finding of reduced platelet accumulation following laser injury of the cremaster arterioles. 7 This may, however, still be related to neutrophil activation as it was demonstrated to also contribute to clot formation in this model. 36
To better address the importance of SLC44A2 in platelets versus neutrophils, a more dynamic system would be useful such as intra‐vital microscopy combined with live cell imaging of the cell types following stenosis. 9 Furthermore, as SLC44A2 is expressed by many cell types central to thrombosis including endothelial cells, leukocytes, platelets, and erythrocytes, cell transfer experiments or cell‐specific knock‐outs may also be necessary when dissecting out the contributions of certain compartments or cell types. In particular neutrophil and platelet specific Slc44a2 knock‐outs would be of interest for future investigations as our data, in addition to the findings from others, 30 , 31 , 32 points toward SLC44A2 involvement on these two cell types as being relevant to VT pathophysiology. With the existence of mice carrying a conditional allele for Slc44a2, these studies are certainly feasible. We do not find evidence that SLC44A2 is important for endothelial activation following stenosis. Notably, in the present study we used female mice for the hypercoagulability model and male mice for the stenosis model, which was in part because it allowed comparison with previous published studies using these models. Future work using a cell‐specific approach may include both sexes, allowing detection of possible sex‐specific effects, which was not possible in the present study.
In conclusion, by utilizing a murine representation of DVT, we were able to corroborate the recent genomic studies identifying SLC44A2 as a susceptibility gene for VT and establish that SLC44A2 is key during the initiation of thrombosis with indications that this may be related to platelet‐neutrophil interaction, either directly and/or indirectly. The precise mechanism however remains elusive and warrants further investigation.
CONFLICTS OF INTEREST
The authors declare no competing financial interests.
AUTHOR CONTRIBUTIONS
Experimental design: JT, DMC, GZ, SD, HCdB, JMEMC, HHV, PEM, BJMvV, CXM, GMT. Performed experiments and analyzed data: JT, DMC, GZ, SD, MFAK, HCdB, JMEMC, BJMvV, CXM, GMT. Wrote the paper: JT, BJMvV, CXM, GMT. All authors commented on manuscript drafts.
Supporting information
Supinfo
ACKNOWLEDGMENTS
The authors thank Dr Pavan K. Kommareddi, Thankam S. Nair, and Dr Thomas E. Carey (Kresge Hearing Research Institute, Department of Otolaryngology/Head and Neck Surgery, University of Michigan, Ann Arbor, MI, USA) for the floxed Slc44a2 mouse embryos used to generate the mice on a C57BL/6J background and Dr Charles Esmon (University of Oklahoma Health Sciences Center, Oklahoma City, OK, USA) for providing the 59D8 fibrin antibody. Also, we thank Lieke van den Heijkant for assisting with the hypercoagulability model.
Tilburg J, Coenen DM, Zirka G, et al. SLC44A2 deficient mice have a reduced response in stenosis but not in hypercoagulability driven venous thrombosis. J Thromb Haemost. 2020;18:1714–1727. 10.1111/jth.14835
Chrissta X. Maracle and Grace M. Thomas contributed equally
Manuscript handled by: Roger Preston
Final decision: Roger Preston, 26 February 2020
Funding Information
This work was supported by Trombosestichting Nederland (#2015‐4), the Landsteiner Foundation for Blood Transfusion Research (#1503), the Dutch Heart Foundation (2015T79), the Netherlands Organization for Scientific Research (NWO Vidi 91716421), a European Molecular Biology Organization Short‐Term Fellowship (EMBO #7468), a European Society of Cardiology First Contact Initiative grant, and by a French ANR grant (ANR‐17‐CE14‐0003‐01).
REFERENCES
- 1. Raskob GE, Angchaisuksiri P, Blanco AN, et al. Day ISCfWT. Thrombosis: a major contributor to global disease burden. Arterioscler Thromb Vasc Biol. 2014;34:2363‐2371. [DOI] [PubMed] [Google Scholar]
- 2. Germain M, Chasman DI, de Haan H, et al. Meta‐analysis of 65,734 individuals identifies TSPAN15 and SLC44A2 as two susceptibility loci for venous thromboembolism. Am J Hum Genet. 2015;96:532‐542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hinds DA, Buil A, Ziemek D, et al. Genome‐wide association analysis of self‐reported events in 6135 individuals and 252 827 controls identifies 8 loci associated with thrombosis. Hum Mol Genet. 2016;25:1867‐1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nair TS, Kozma KE, Hoefling NL, et al. Identification and characterization of choline transporter‐like protein 2, an inner ear glycoprotein of 68 and 72 kDa that is the target of antibody‐induced hearing loss. J Neurosci. 2004;24:1772‐1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Iwao B, Yara M, Hara N, et al. Functional expression of choline transporter like‐protein 1 (CTL1) and CTL2 in human brain microvascular endothelial cells. Neurochem Int. 2016;93:40‐50. [DOI] [PubMed] [Google Scholar]
- 6. Tilburg J, Michaud SA, Maracle CX, et al. Plasma Protein Signatures of a Murine Venous Thrombosis Model and Slc44a2 Knockout Mice Using Quantitative‐Targeted Proteomics. Thromb Haemost. 2020; 120:423–426. [DOI] [PubMed] [Google Scholar]
- 7. Tilburg J, Adili R, Nair TS, et al. Characterization of hemostasis in mice lacking the novel thrombosis susceptibility gene Slc44a2. Thromb Res. 2018;171:155‐159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Safdar H, Cheung KL, Salvatori D, et al. Acute and severe coagulopathy in adult mice following silencing of hepatic antithrombin and protein C production. Blood. 2013;121:4413‐4416. [DOI] [PubMed] [Google Scholar]
- 9. von Bruhl ML , Stark K, Steinhart A, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med. 2012;209:819‐835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kommareddi P, Nair T, Kakaraparthi BN, et al. Hair cell loss, spiral ganglion degeneration, and progressive sensorineural hearing loss in mice with targeted deletion of Slc44a2/Ctl2. J Assoc Res Otolaryngol. 2015;16:695‐712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brill A, Fuchs TA, Savchenko AS, et al. Neutrophil extracellular traps promote deep vein thrombosis in mice. J Thromb Haemost. 2012;10:136‐144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tjernberg P, Vos HL, Castaman G, Bertina RM, Eikenboom JC. Dimerization and multimerization defects of von Willebrand factor due to mutated cysteine residues. J Thromb Haemost. 2004;2:257‐265. [DOI] [PubMed] [Google Scholar]
- 13. Cleuren AC, Van der Linden IK, De Visser YP, Wagenaar GT, Reitsma PH, Van Vlijmen BJ. 17alpha‐Ethinylestradiol rapidly alters transcript levels of murine coagulation genes via estrogen receptor alpha. J Thromb Haemost. 2010;8:1838‐1846. [DOI] [PubMed] [Google Scholar]
- 14. Visser YP, Walther FJ, el Laghmani H, Laarse A, Wagenaar GT. Apelin attenuates hyperoxic lung and heart injury in neonatal rats. Am J Respir Crit Care Med. 2010;182:1239‐1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schindelin J, Arganda‐Carreras I, Frise E, et al. Fiji: an open‐source platform for biological‐image analysis. Nat Methods. 2012;9:676‐682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. de Witt SM , Swieringa F, Cavill R, et al. Identification of platelet function defects by multi‐parameter assessment of thrombus formation. Nat Commun. 2014;5:4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Meyer dos Santos S, Klinkhardt U, Schneppenheim R, Harder S. Using ImageJ for the quantitative analysis of flow‐based adhesion assays in real‐time under physiologic flow conditions. Platelets. 2010;21:60‐66. [DOI] [PubMed] [Google Scholar]
- 18. Heestermans M, Salloum‐Asfar S, Streef T, et al. Mouse venous thrombosis upon silencing of anticoagulants depends on tissue factor and platelets, not FXII or neutrophils. Blood. 2019;133:2090‐2099. [DOI] [PubMed] [Google Scholar]
- 19. Brill A, Fuchs TA, Chauhan AK, et al. von Willebrand factor‐mediated platelet adhesion is critical for deep vein thrombosis in mouse models. Blood. 2011;117:1400‐1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Martinod K, Demers M, Fuchs TA, et al. Neutrophil histone modification by peptidylarginine deiminase 4 is critical for deep vein thrombosis in mice. Proc Natl Acad Sci USA. 2013;110:8674‐8679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Alvarado CM, Diaz JA, Hawley AE, Wrobleski SK, Sigler RE, Myers DD Jr. Male mice have increased thrombotic potential: sex differences in a mouse model of venous thrombosis. Thromb Res. 2011;127:478‐486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bockenstedt P, Greenberg JM, Handin RI. Structural basis of von Willebrand factor binding to platelet glycoprotein Ib and collagen. Effects of disulfide reduction and limited proteolysis of polymeric von Willebrand factor. J Clin Invest. 1986;77:743‐749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Stenberg PE, McEver RP, Shuman MA, Jacques YV, Bainton DF. A platelet alpha‐granule membrane protein (GMP‐140) is expressed on the plasma membrane after activation. J Cell Biol. 1985;101:880‐886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zucoloto AZ, Jenne CN. Platelet‐neutrophil interplay: insights into neutrophil extracellular trap (NET)‐driven coagulation in infection. Front Cardiovasc Med. 2019;6:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Darbousset R, Thomas GM, Mezouar S, et al. Tissue factor‐positive neutrophils bind to injured endothelial wall and initiate thrombus formation. Blood. 2012;120:2133‐2143. [DOI] [PubMed] [Google Scholar]
- 26. Fuchs TA, Brill A, Duerschmied D, et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci USA. 2010;107:15880‐15885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Visscher PM, Wray NR, Zhang Q, et al. 10 Years of GWAS discovery: biology, function, and translation. Am J Hum Genet. 2017;101:5‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tregouet DA, Morange PE. What is currently known about the genetics of venous thromboembolism at the dawn of next generation sequencing technologies. Br J Haematol. 2018;180:335‐345. [DOI] [PubMed] [Google Scholar]
- 29. Branchford BR, Carpenter SL. The role of inflammation in venous thromboembolism. Front Pediatr. 2018;6:142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bayat B, Tjahjono Y, Berghofer H, et al. choline transporter‐like protein‐2: new von Willebrand factor‐binding partner involved in antibody‐mediated neutrophil activation and transfusion‐related acute lung injury. Arterioscler Thromb Vasc Biol. 2015;35:1616‐1622. [DOI] [PubMed] [Google Scholar]
- 31. Zirka G, Robert P, Legendre P, et al. HNA‐3a Expressed by CTL‐2 is a crucial epitope for neutrophil adhesion and activation on VWF European Congress on Thrombosis and Haemostasis (ECTH). Marseille, France, 2018. [Google Scholar]
- 32. Constantinescu‐Bercu A, Frontini M, Salles‐Crawley I, Woollard K, Crawley J. Activated αIIbβ3 on platelets mediates flow‐dependent NETosis via SLC44A2 bioRxiv 373670. 2019. [DOI] [PMC free article] [PubMed]
- 33. Elaskalani O, Abdol Razak NB, Metharom P. Neutrophil extracellular traps induce aggregation of washed human platelets independently of extracellular DNA and histones. Cell Commun Signal. 2018;16:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Maugeri N, Campana L, Gavina M, et al. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J Thromb Haemost. 2014;12:2074‐2088. [DOI] [PubMed] [Google Scholar]
- 35. Etulain J, Martinod K, Wong SL, Cifuni SM, Schattner M, Wagner DD. P‐selectin promotes neutrophil extracellular trap formation in mice. Blood. 2015;126:242‐246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang Y, Gao H, Shi C, et al. Leukocyte integrin Mac‐1 regulates thrombosis via interaction with platelet GPIbalpha. Nat Commun. 2017;8:15559. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supinfo