

Abstract
Self-synthesizing transposons are integrative mobile genetic elements (MGEs) that encode their own B-family DNA polymerase (PolB). Discovered a few years ago, they are proposed as key players in the evolution of several groups of DNA viruses and virus–host interaction machinery. Pipolins are the most recent addition to the group, are integrated in the genomes of bacteria from diverse phyla and also present as circular plasmids in mitochondria. Remarkably, pipolins-encoded PolBs are proficient DNA polymerases endowed with DNA priming capacity, hence the name, primer-independent PolB (piPolB). We have now surveyed the presence of pipolins in a collection of 2,238 human and animal pathogenic Escherichia coli strains and found that, although detected in only 25 positive isolates (1.1%), they are present in E. coli strains from a wide variety of pathotypes, serotypes, phylogenetic groups and sequence types. Overall, the pangenome of strains carrying pipolins is highly diverse, despite the fact that a considerable number of strains belong to only three clonal complexes (CC10, CC23 and CC32). Comparative analysis with a set of 67 additional pipolin-harboring genomes from GenBank database spanning strains from diverse origin, further confirmed these results. The genetic structure of pipolins shows great flexibility and variability, with the piPolB gene and the attachment sites being the only common features. Most pipolins contain one or more recombinases that would be involved in excision/integration of the element in the same conserved tRNA gene. This mobilization mechanism might explain the apparent incompatibility of pipolins with other integrative MGEs such as integrons. In addition, analysis of cophylogeny between pipolins and pipolin-harboring strains showed a lack of congruence between several pipolins and their host strains, in agreement with horizontal transfer between hosts. Overall, these results indicate that pipolins can serve as a vehicle for genetic transfer among circulating E. coli and possibly also among other pathogenic bacteria.
Subject terms: Microbial genetics, Mobile elements, Bacterial genes
Introduction
Mobile genetic elements (MGE), comprising bacteriophages, transposons, plasmids, and insertion sequences, contribute to the great plasticity of the bacterial genome, resulting in an extremely large pangenomes that, in the case of Escherichia coli, can amount to more than 16,000 genes1. Thus, MGEs dynamics is the main source of horizontal gene transfer, which leads to the spread of antimicrobial resistance (AR) among both E. coli and other commensals, thereby enlarging the spectrum of resistance (the resistome) among circulating strains2.
Pipolins constitute a recently reported new group of integrative MGEs widespread among diverse bacterial phyla and also identified in mitochondria as circular plasmids3. The hallmark feature of the pipolins is a gene encoding for a replicative family B DNA polymerases (PolB) with an intrinsic de novo primer synthesis capacity, called primer-independent PolBs (piPolBs), hence their name (piPolB-encoding elements). Preliminary phylogenetic analyses indicated that piPolBs would form a third major branch of PolB, besides the protein-primed PolBs (pPolBs), found in a wide range of viruses and plasmids, and RNA-primed PolBs (rPolBs), which are the principal replicative enzymes of archaea and eukaryotes, some archaea and many dsDNA viruses3.
Because of the fact that pipolins encode the major protein required for their replication (i.e., piPolB), they are included in the proposed class of self-synthesizing (or self-replicating) MGEs, which also includes two other superfamilies of elements integrated in various cellular genomes4. The first superfamily comprises eukaryotic virus-like transposable elements, called Polintons (also known as Mavericks), which besides a putative pPolB, encode retrovirus-like integrases and a set of proteins hypothesized to be involved in the formation of viral particles5–7. The second superfamily of PolB-encoding elements, denoted as casposons, is present in a wide range of archaea and also in a few bacteria8. Similar to the aforementioned self-replicating MGEs, most pipolins are integrated within bacterial chromosomes, although they are also occasionally detected as episomal plasmids. However, unlike polintons and casposons, the integrated pipolins encode for one or more integrases of the tyrosine recombinase superfamily, which could be responsible for pipolin excision and/or integration3.
The widespread and patchy distribution of pipolins among bacteria is in agreement with an ancient origin and horizontal dispersal of this MGE group. However, pipolins from the same or related species seem closely related, as was the case for pipolins from E. coli3. The great majority of pathogenic E. coli strains encode for virulence-associated cassettes and antibiotic resistance genes, which are usually carried by MGEs, such as pathogenicity islands (PAIs), plasmids, integrons, etc. 9–11. However, the annotation of pipolins from E. coli as well as from proteobacteria did not lead to the identification of any antibiotic resistance genes or virulence factors3.
Therefore, whereas reported evidence of mobility of polintons and casposons is limited and based on metagenomic data12, pipolins provide the opportunity to analyze the occurrence, diversity, and dynamics of self-replicative MGEs in well-characterized commensal and pathogenic bacteria, not only in genomic or metagenomics data, but also in circulating field isolates and pathogenic variants. In this work, we surveyed the presence of pipolins in a wide collection of pathogenic strains from the Spanish E. coli reference laboratory (LREC). We found that pipolins, although not very abundant, are widespread among a great variety of human and animal strains belonging to different pathotypes, serotypes and sequence types (STs).
Whole-genome sequencing of the LREC pipolin-harboring E. coli strains allowed us to characterize in detail the pipolins’ hosts and also the genetic structure and phylogeny of pipolins. Most pipolins contain att-like terminal direct repeats and they are integrated in the same tRNA gene. Nevertheless, they encode a great diversity of proteins, many of which are orphans with unpredictable function. The comparison of the new genome assemblies from LREC dataset with a number of detected E. coli pipolin-harboring strains from the NCBI GenBank database further confirmed our results. Finally, pangenome and cophylogeny analyses among pipolins and host strains, demonstrated that, except for strains from the same clonal complex, there is an overall lack of phylogenetic congruence between pipolins and host strains. Therefore, our results support that pipolins are a novel group of active mobile elements that might serve as a platform for horizontal gene transference among diverse pathogenic bacteria.
Results and discussion
Limited prevalence of pipolins among E. coli isolates from animal and human sources
The first objective of this study was to investigate the occurrence of pipolins in E. coli strains causing intestinal and extraintestinal infections in humans and animals.
We performed a survey of pipolin distribution among 2,238 strains from the LREC collection, using a 587 nt fragment of the piPolB coding sequence as a marker. We detected 25 pipolin-harboring isolates, indicating that pipolins are not particularly abundant (1.1%) among pathogenic E. coli causing intestinal (13/931; 1.4%) and extraintestinal infections (12/1,307; 0.9%) (Table 1). Interestingly, however, pipolins are present in strains isolated from humans, swine and avian, and belonging to a wide range of pathotypes (Table 2 and Table S1). Twenty-four of the 25 LREC pipolin-harboring strains were isolated in Spain from 2005 to 2016.
Table 1.
Pipolin identification survey among E. coli strains from diverse origins and pathotypes.
| Source | Infection | Strains | Pipolins (%) |
|---|---|---|---|
| Human | Intestinal | 608 | 8 (1.3%) |
| Human | Extraintestinal | 1,061 | 9 (0.8%) |
| Swine | Intestinal | 323 | 5 (1.5%) |
| Avian | Extraintestinal | 246 | 3 (1.2%) |
| Total | 2,238 | 25 (1.1%) | |
Table 2.
Features of the 25 pipolin-harboring E. coli genomes from LREC dataset.
| Name | PhyG1 | ST2 | CC | Pathotype | Virulence genes (PCR)3 | Virulence genes4 | Plasmids5 | Prophages6 | Complete Integrons5 | CRISPR/Cas (Type)6 |
|---|---|---|---|---|---|---|---|---|---|---|
| 3-373-03_S1_C2 | A | 5,293 | 206 | – | iutA, iucD, | gad, iha | IncFII, IncB/O/K/Z, Col(BS512) | – | – | 9/3 (I–E) |
| LREC237 | D* | 524 | 32 | aEPEC | chuA, ompT, fimH, eae | astA, cif, eae, ehxA, espA, espB, espF, espJ, etpDP, gad, katP, nleA, nleB, tccP, tir | IncFIB/IncX1, ColRNAI, NT | 4P, 1 M, 1S, 2U | CALINP | 9/1 (I–E) |
| LREC239 | C | 88 | 23 | – | iutA, iucD, fyuA, ompT, fimH | espPP, gad, iha, ireA, iss, lpfA, mchB, mchC, mchF | IncFIB/IncFIC, NT, Col(MG828) | 1P, 1S, 3 M, 1U | In0P | 12/1 (I–E) |
| LREC240 | B1 | 156 | 156 | APEC | iutA, iucD, hlyF, iroN, iss, fimH | gad, iroN, iss, lpFA, mchF, tsh | IncFIB/IncFIC, NT | 1S | 2 CALINP | 5/1 (I–E) |
| LREC241 | A | 48 | 10 | ETEC | fyuA, fimH, ltcA, sta1 | gad, ltcAP, sta1P | NT, Col(MGD2), NT | 2P, 2S, 1 M, 2U | – | 9/4 (I–E) |
| LREC242 | A | 746 | 10 | ETEC | fimH, fanA, sta1 | astAP, fanAP, gad, sta1 | IncFIB(K)/IncFIA, IncFIB/IncFIC/IncFII, NT, NT, NT, NT, Col8282, NT, NT | 1 M, 2S | – | 9/7 (I–E) |
| LREC243 | A | 3,011 | 10 | ETEC | fimH, fasA, sta1 | astA, cbaP, cmaP, fasAP, gad, sta1P | IncX1/IncR, NT, NT | 2P, 1U | 3 CALINP, CP | 10/2 |
| LREC244 | A | 10 | 10 | STEC | fimH, stx2 | fedF, stx2A, stx2B | IncFIB/IncR, NT, IncX4, NT, Col156, NT, NT | 4 M, 5P, 3 M/P, 1S, 3U | In0P | 7/3 |
| LREC245 | A | 10888 N | NONE | – | fimH | gad | IncFIB, NT, IncX1, NT, Col8282, Col156, NT | 1 M, 1S | CALINP, In0P, CP | 10/5 (I–E) |
| LREC246 | C | 10889 N | 23 | – | vat, fyuA, ompT, iroN, hlyA, fimH | iha, iroNP, iss, lpfA, tshP | IncFIB | 1P, 3 M, 2S | – | 6/1 (I–E) |
| LREC247 | D* | 137 | 32 | aEPEC | iutA, iucD, chuA, ompT, fimH, eae | astA, cif, eae, ehxAP, espA, espB, espF, espJ, etpDP, iha, iss, katPP, nleA, nleB, nleC, tccP, tir | IncFIB, NT, NT, Col8282 | 3P, 1 M, 2S, 1S/P, 1U | – | 8/2 (I–E) |
| LREC248 | A | 10,850 | 10 | NTEC | iutA, iucD, cnf, fimH | cdtB, cnf1, espPP, ihaP, ireA, iss, saaP | IncFIB, Col8282, Col156, NT | 1P, 4 M, 5S, 1U | – | 8/0 |
| LREC249 | D* | 32 | 32 | aEPEC | iutA, iucD, chuA, ompT, fimH, eae | astA, cif, eae, ehxA, espAP, espB, espI, espP, iha, iss, katPP, nleA, nleB, nleC, tccP, tir, toxBP | IncFIB | 3P, 2 M, 3S, 2U | – | 7/2 (I–E, I–D) |
| LREC250 | D* | 137 | 32 | aEPEC | iutA, iucD, chuA, ompT, fimH, eae | astA, cif, eae, ehxAP, espA, espB, espF, espJ, etpDP, iha, iss, katPP, nleB, nleC, tccP, tir, toxBP | IncFIB, NT, Col(MG828) | 3P, 1S, 1U | – | 7/2 (I–E) |
| LREC251 | D* | 32 | 32 | STEC | chuA, ompT, fimH, stx2, eae | astA, cif, eae, ehxAP, espAP, espB, espJ, espP, gad, iha, iss, nleB, nleC, stx2A, stx2B, tccP, tir, toxBP | IncFIB | 1P, 3 M, 2S, 1U | – | 7/1 (I–E) |
| LREC252 | A | 48 | 10 | – | fimH | – | – | – | 12/4 (I–E) | |
| LREC253 | A | 347 | NONE | – | fimH | astA | – | 4 M, 1U | – | 6/3 (I–E) |
| LREC254 | B1 | 359 | 101 | APEC | iutA, iucD, hlyF, iroN, iss, fimH | iroNP, issP, lpfA, mchFP, tshP | IncFIB, NT, IncX1, NT | 2 S/P | – | 0 |
| LREC255 | C | 88 | 23 | APEC | iutA, iucD, fyuA, ompT, hlyF, iroN, iss, fimH | gad, ireA, iss, lpfA | IncY, NT, NT, NT | 1P, 4S | – | 10/2 (I–E, I–D) |
| LREC256 | C | 88 | 23 | – | iutA, iucD, fyuA, ompT, hlyA, fimH | cbaP, cmaP, gad, iha, iss, katPP, lpfA, sepA | IncFIB | 1P, 4 M, 2S | 1 | 8/1 (I–E) |
| LREC257 | C | 88 | 23 | ExPEC/ APEC | iuA, iucD, papAH, papC, fyuA, ompT, hlyF, iroN, iss, fimH | astA, ireA, iroNP, issP, lpfA, mchFP | IncFIB, NT, NT, Col(MG828) | 4 M, 1U | – | 9/4 (I–E, I–D) |
| LREC258 | A | 46 | 46 | APEC | iutA, iucD, hlyF, iroN, iss, fimH | gad, iroN, iss, mchF | IncFIB, IncFIB, NT, NT | 2S, 1S/P, 1U | 1 | 8/2 (I–E) |
| LREC259 | C | 10890 N | 23 | APEC | iutA, iucD, fyuA, ompT, hlyF, iroN, iss, fimH | astA, gad, ireA, iroN, iss, lpfA, mchF | IncFIB NT, NT | 3 M, 1U | 1 | 9/2 (I–E) |
| LREC260 | A | 10 | 10 | – | fimH | iss | IncY, NT, NT, Col440II, ColRNAI | 4 M, 1S, 1 M/P | – | 7/0 |
| LREC261 | A | 8,233 | NONE | – | fimH | gad | IncFIB, NT, NT, Col440II | 2 M, 1S | – | 6/2 (I–E) |
| LREC262 | B1 | 1,049 | 155 | – | fyuA, fimH | gad, lpfA | NT, NT | 1S | – | 3/1 (I–E) |
For reference, the strain 3-373-03_S1_C2 was included in the analysis. See Table S1 for more detailed analysis.
1PhyG: phylogenetic groups, where ”*” indicates strains with discrepancies between the assignation obtained with the quadruplex PCR of Clermont et al. (2014)14 and the in silico assignation using ClermonTyping tool, showing phylogroup E by PCR, but phylogroup D in silico.
2New sequence types (ST) are indicated with N.
3Virulence genes determined by conventional PCR as detailed in Methods.
4Virulence genes identified with VirFinder database (P indicates plasmid location): astA, EAST-1 heat-stable toxin; cba, Colicin B; cdtB, Cytolethal distending toxin B; cif, Type III secreted effector; cma, Colicin M; cnf1, Cytotoxic necrotizing factor; eae, Intimin; ehxA, Enterohaemolysin; espA, Type III secretion system; espB, Secreted protein B; espF, Type III secretion system; espI, Serine protease autotransporters of Enterobacteriaceae; espJ, Prophage-encoded type III secretion system effector; espP, Extracellular serine protease plasmid-encoded; etpD, Type II secretion protein; fanA, Involved in biogenesis of K99/F5 fimbriae; fasA, Fimbrial 987P/F6 subunit; fedF, Fimbrial adhesin AC precursor; gad, Glutamate decarboxylase; iha, Adherence protein; ireA, Siderophore receptor; iroN, Enterobactin siderophore receptor protein; iss, Increased serum survival; katP, Plasmid-encoded catalase peroxidase; lpfA, Long polar fimbriae; ltcA, Heat-labile enterotoxin A subunit; mchB, Microcin H47 part of colicin H; mchC, MchC protein; mchF, ABC transporter protein MchF; nleA, Non-LEE encoded effector A; nleB, Non-LEE encoded effector B; nleC, Non-LEE-encoded effector C; saa, STEC autoagglutinating adhesin; sepA, Serine protease autotransporters of Enterobacteriaceae; sta1, Heat-stable enterotoxin ST-Ia; stx2A, Shiga toxin 2 subunit A; stx2B, Shiga toxin 2 subunit B; tccP, Tir cytoskeleton coupling protein; tir, Translocated intimin receptor protein; toxB, Toxin B; tsh, Temperature-sensitive hemagglutinin.
5Plasmids are enumerated according to their compatibility group. NT, not typed.
6Prophages (analyzed with Phigaro56 and Phaster57): P, Podoviridae; M, Myoviridae; S, Siphoviridae; U, Unknown; a slash (/) indicate an ambiguous family assignment.
7Integrons: Integrons were analyzed with IntegronFinder56 as indicated in Materials and Methods. C, complete, Int0, integron lacking attC site, CALIN, integron lacking functional integrase gene. P indicates plasmid location.
8Number of Crispr units and associated proteins (Cas), as well as the element type determined with CRISPRCasFinder58 are indicated.
In order to ascertain the representativity of the pipolin-harboring strains from LREC collection, we performed a TBLASTN search against the E. coli nucleotide database at NCBI GeneBank using the 3-373-03_S1_C2 piPolB amino acid sequence as a query. This search yielded 76 hits, corresponding to highly conserved piPolB-encoding ORFs or fragments from 67 E. coli strains (identity above 85%), many of them many of them only reported as a genome draft, without further characterization (Table S2). A combined phylogeny of piPolB coding sequences from LREC and GeneBank datasets (Supplementary Fig. S1), shows that our collection of strains carrying pipolins spans all the available diversity of E. coli pipolins. Therefore, the low frequency of pipolins detected in our collection is in agreement with a low prevalence of this element among circulating E. coli strains. The source of 52 of the 67 GeneBank pipolin-harboring strains analyzed is known: 20 human, 7 swine, 7 environment, 6 wild animals (5 mouse and 1 reptile), 4 bovine, 4 poultry, 2 food, 1 companion animal and 1 marine mammal. The majority were isolated in Asia (27 strains) and North America (16 strains) from 2009 to 2018.
Diversity of pipolin-harboring E. coli strains
Although some of the strains in the LREC collection have been described in detail throughout the last years13–17, the pipolin-positive strains remained uncharacterized. We performed now a detailed molecular characterization, both by conventional methods and whole-genome sequence (WGS) analysis (Table 2 and Table S1). We found that pipolins were present in phylogroup A (11 LREC strains and the reference isolate 3-373-03-S1-C218), but also in B1 (3 strains) and C (6 strains). Five strains were typed as E by quadruplex PCR typing19 but later on reassigned as D after whole genome sequencing and in silico typing (see Methods for details). A similar distribution pattern was found within the GeneBank dataset, with pipolins in phylogroups A (46 strains), B1 (9 strains), C (2 strains) and D (10 strains).
The common presence of E. coli strains from phylogroup A in the dataset was somewhat expected, as this phylogroup is common among human and animal isolates and thus very abundant in most collections20,21. However, we were surprised by the absence of pipolins among B2 strains, despite the fact that this phylogroup is also very common in the LREC collection and, along with group D, it is responsible for most extraintestinal E. coli infections in human and animals11,19. The null prevalence of pipolins among B2 strains is opposite to the pattern of occurrence of some virulence-related MGEs, such as colibactin encoding pks islands, which are highly prevalent in B2 and D phylogroups but were not detected in strains carrying pipolins10,22. It is thus tempting to speculate whether there is an interference between pks islands and pipolins as they are also flanked by terminal direct repeats and found integrated into a constant tRNA23. Thus, the distribution of pipolins limited to phylogroups A, B1, C and D may be due to the restricted mobility beyond those groups. In line with this, these phylogenetic groups have been proposed to belong to different ancient lineages24, downplaying a strict vertical transmission of pipolins throughout the evolutionary diversification of E. coli phylogroups.
Regarding multilocus sequence typing (MLST), we have detected 18 different STs among 25 LREC pipolin-harboring strains, according to the Achtman scheme25 (Table 2), some of them are quite common in Enterobase26, like ST88, ST10, ST46, ST48 or ST746, but also very rare varieties (ST524, ST3011, ST8233 or ST10850) and three new type sequences (ST10888, ST10889 and ST10890). Interestingly, although some of them belong to the same clonal complex (CC10), in general strains from phylogroups A and B1 span a vast diversity of STs. In contrast, LREC pipolin-harboring strains from phylogenetic groups C and D seem more homogeneous and they could be assigned to the same clonal complex (CC23 and CC32, respectively). Clear clonality among pipolin-harboring strains is more evident when we analyzed the 67 strains of GenBank dataset (Table S2), as 61.2% of the strains belong to the above-mentioned clonal complexes. Moreover, among the GenBank strains, a fourth prevalent clonal complex was CC278, observed in 5 ST278 strains of serotype O178:H7 isolated from mice. The STs of 63 of the 67 GeneBank pipolin-harboring strains analyzed is known, encompassing a total of 29 different STs, 5 of them were especially prevalent: ST4 (7 strains), ST32 (6 strains), ST48 (11 strains), ST278 (5 strains) and ST1312 (4 strains).
Clonotypes and serotypes diversity is also in agreement with the presence of divergent strains (Table S1), particularly from phylogenetic groups A and B1, in which, for instance, present 14 different serotypes for 14 LREC strains. On the contrary, as in the case of STs and CCs, strains from phylogroups C and D showed overall more similar clonotypes and serotypes. Thus, the six strains of phylogroup C showed the H19 flagellar antigen and the five strains of phylogroup D showed the clonotype CH23-331 and the H28 flagellar antigen. A similar pattern can be observed among strains from the GenBank dataset, including a total of 38 different O:H serotypes and also a great diversity of clonotypes (Table S2).
Analysis of virulence genes among the 25 LREC pipolin-harboring strains also allowed us to identify diverse E. coli pathotypes, namely, extraintestinal pathogenic (ExPEC, 1 strain), avian pathogenic (APEC, 6 strains), Shiga toxin-producing (STEC, 2 strains), enterotoxigenic (ETEC, 3 strains), atypical enteropathogenic (aEPEC, 4 strains) and necrotoxigenic (NTEC, 1 strain). However, some other common pathotypes were not detected in the pipolin-carring LREC strains, like uropathogenic E. coli (UPEC), typical enteropathogenic E. coli (tEPEC), enteroinvasive E. coli (EIEC) and enteroaggregative E. coli (EAEC) (Table 2).
Antimicrobial resistance and virulence genes in LREC strains harboring pipolins
Antimicrobial resistance tests showed that pipolins are present in both antibiotic susceptible (11 strains) and antimicrobial resistant strains (14 strains) in the LREC collection (Table S1). Eleven strains exhibited a multidrug-resistant (MDR) phenotype. Besides, there were four extended-spectrum β-lactamase (ESBL)-producing strains, three cefoxitin-resistant strains and two colistin-resistant strains.
In line with these results, many antimicrobial resistance genes (ARGs) were found in the genome assemblies, including acquired resistance genes, point mutations and efflux/transporter genes (Table S1). We described genes conferring resistance to beta lactams (blaCTX-M-15, n = 1; blaCTX-M-14, n = 1; blaCTX-M-1, n = 2; blaTEM-1, n = 7), colistin (mcr-1.1, n = 2), tetracycline (tet(A), n = 6; tet(B), n = 3; tet(M), n = 3), aminoglycosides (aadA1, n = 3; aadA2, n = 4; aadA5, n = 1; aadA9, n = 1; aadA13, n = 1; ant(3′')-Ia, n = 2; aph(3′)-Ia, n = 2; aph(3′')-Ib, n = 5; aph(4)-Ia, n = 2; aph(6)-Id, n = 4; aac(3)-IV, n = 2 and aac(3)-IIa, n = 1 ), phenicols (catA1, n = 5 and cmlA1, n = 3), trimethoprim (dfrA1,n = 2 and dfrA12, n = 3), lincosamides (lnu(F), n = 2), macrolides (mph(A),n = 1; mph(B),n = 1 and mef(B), n = 1), quinolones (qnrS1, n = 1 and qnrB19, n = 1) and sulfonamides (sul1, n = 3; sul2, n = 2 and sul3, n = 3). Furthermore, we found chromosomally encoded point mutations in the gyrA and parC genes conferring resistance to quinolones in nine strains and also in the ampC promoter conferring resistance to beta lactams in three cefoxitin-resistant strains.
In summary, LREC pipolins-carrying strains, harbor a repertoire of ARGs, as expected of pathogenic E. coli strains, ruling out any correlation among ARGs and pipolins, in line with the diversity of phylogenetic groups, STs and pathotypes they belong to.
Pangenome and mobilome of E. coli strains harboring pipolins
We assembled the pangenome of the 25 LREC strains carrying pipolins plus the reference strain, 3-373-03_S1_C2 with Roary27, resulting in 10,178 different genes (Supplementary Fig. S2). Among those, 2,998 genes (29.45%) corresponded to the core-genome and were present in all strains, 327 were soft-core genes present in 95–99% of genomes and 2,259 were shell genes present in 15–95% genomes. As expected from such a diverse pangenome, almost half of the genes (4,594, 45.13%) were cloud-genes, found in less than 15% of strains. This diversity is even more evident when the pangenome of both (LREC and GenBank) datasets are analyzed together, with a total of 16,675 genes, only 934 genes comprise a core-genome and more than two-thirds of the genes in the cloud-genome (11,175, 67%). As such, the number of both total and unique genes associated with the cloud gene set increased consistently with the number of genomes (Supplementary Fig. S2, B–D). In conclusion, notwithstanding the clonality of several strains, pangenome analysis indicates that pipolins are present in a wide variety of E. coli strains.
Pangenome analysis and core-genome based phylogeny reconstruction of the LREC strains (Fig. 1), clustered pipolin-harboring strains in agreement with the assigned phylogenetic groups and clonal complexes, and this congruence is maintained for the phylogeny inferred from the core genome of all analyzed strains carrying pipolins (Supplementary Fig. S3). Similar results were obtained when the phylogeny of the strains was constructed by single-nucleotide polymorphism in EnteroBase26, although some of the strains are not available in this database (Supplementary Fig. S4).
Figure 1.

Maximum-likelihood tree generated from the core-genome data of new pipolin-harboring strains. Strain names are colored according to the phylogenetic groups as indicated. Previously described pipolin-harboring isolate 3-373-03_S1_C2 was included as a reference. The best-fit model was GTR + F + R2 for all considered criteria in ModelFinder62. Scale bar indicates substitution rate per site. The main features are indicated on the right: source, isolation date, pathotype, sequence type (ST) and clonal complex (CC). New ST combinations assigned at Enterobase are indicated with N.
Disregarding pipolins, when the mobilome of our strains was analyzed, we could identify the typical variety of plasmids and other mobile elements. PLACNETw28 allowed us to identify and assemble several plasmids in most of the strains (Table 2 and Table S1) and 79 elements could be extracted, which would correspond to at least a total of 86 plasmids, as some of the elements contained markers from more than one incompatibility group. All strains carried at least one plasmid moving in a range from 1 to 9, except for strains LREC252 and LREC253 (Table 2 and Table S1). Plasmids from IncF incompatibility group were the more prevalent including IncFIB (n = 20), IncFIA (n = 1), IncFII (n = 2) and IncFIC (n = 3) replicons. Followed by Col-like plasmids including Col8282 (n = 4), Col(MGD2) (n = 1), Col(BS512) (n = 1), Col(MG828) (n = 3), Col156(n = 3), Col440II(n = 2), and ColRNAI (n = 2). However, we also described the presence of other incompatibility groups like IncX1 (n = 3), IncY (n = 2), IncR (n = 2), IncX4 (n = 1) and IncB/O/K/Z (n = 1). Furthermore, 42 cryptic plasmids could not be affiliated with any category as they lack any known replication origin. Nonetheless, none of those correspond with an episomic pipolin and indeed piPolB-containing contig was detected as a single copy portion of the chromosome, which suggests that pipolin excision is negligible under standard growth conditions or undetectable by Illumina sequencing.
As expected, many of the reported antimicrobial and virulence factors were plasmid-borne genes, since conjugative plasmids, along with other MGEs, are the most successful genetic platforms allowing the horizontal transfer of antimicrobial resistance and virulence determinants among pathogenic E. coli isolates11,29,30.
Most of the strains contained genes from one or more prophages and, as expected, sequence arrays and genes from the CRISPR/Cas immunity system (Table 2). Strikingly, integrons seem quite uncommon, as we could detect complete integrons in 5 out of 25 LREC genomes (20%) and only 5 hits were detected in the pipolin-harboring genomes from Genbank (8.9%), whereas they are often reported to be usually highly prevalent and present in more than half of pathogenic E. coli strains31–34.
Mapping and extraction of new pipolins from LREC and GenBank datasets
Besides the presence of a piPolB gene, pipolins are characterized for the presence of att-like terminal direct repeats that might be involved in recombination-mediated excision/insertion, often in a tRNA site3. We extracted pipolins from both, LREC and GenBank pipolin-harboring strains, using a custom bioinformatics pipeline that entailed searching for piPolB gene or its gene fragments and terminal direct repeats to determine the element bounds (see Methods for details). Except for the strain LREC243, two att-repeats could be detected in all genomes. In the cases when att-repeats were located on the same contig or on a complete chromosome, the piPolB always sitting within the repeats, which confirmed the basic structure of all E. coli pipolins (Fig. 2 and Supplementary Fig. S5). This structure could be reconstructed also when piPolB and att-repeats were not on the same contig (see Methods). Reparably, all E. coli pipolins are integrated in the same point, at the Leu-tRNA gene, except for the pipolin from LREC252 strain that looks inconsistent with other pipolins. The att repeat that overlaps with the tRNA gene was represented and denoted as right end (attR).
Figure 2.

Genetic structure of new pipolins from LREC collection. Predicted protein-coding genes are represented by arrows, indicating the direction of transcription and colored following Prokka annotation as indicated in the legend. The greyscale on the right reflects the percent of amino acid identity between pairs of sequences. The image was generated by EasyFig software and re-annotated pipolins sorted according to the hierarchical clustering of the gene presence/absence matrix. Names of pipolin carrying strains are colored according to phylogroups as in Fig. 1.
In some genomes, three att-repeats were detected, as those pipolins seem to share the integration site and mechanism with some prophage, as previously detected for the enterotoxigenic Escherichia coli H10407 strain3. Indeed, comparison of the genetic structure of all pipolins (Supplementary Fig. S5), confirmed that a similar myovirus enterophage is present next to pipolins from eight strains, spanning phylogroups A (H10407, 2014EL-1346-6 and 99-3165), C (LREC239 and LREC246) and D (112,648, 122,715, 2015C-3125 and FWSEC0002). In addition, the presence of transposases and associated genes indicates that genetic islands and insertion sequences can as well contribute to the variability of pipolins, particularly in the case of stains LREC248 and LREC252, expanding also the pipolin gene repertoire (see below). The cohabitation of casposons with other MGEs is also common12, although in that case, the associated element seems to provide a vehicle for horizontal transfer. On the contrary, as pipolins possess att-like direct repeats and one or more recombinases (see below), they can be considered as self-transferable.
Altogether, mapping and extraction of the new E. coli pipolins, confirmed that, despite a great diversity, they share basic genetic structure and they can likely be mobilized using the same mechanism.
Pipolins annotation and pangenome analysis
The 92 extracted pipolins were reannotated with a custom pipeline using Prokka35 (see Methods), followed by Roary27 for pangenome analysis of E. coli pipolins, identifying a total of 272 genes. Remarkably, the core- and soft-core genomes are made up of a single gene cluster, the piPolB, and a XerC-like tyrosine-recombinase, respectively. In line with this, the shell genome contains only 38 genes, whereas 232 genes (85%) are cloud-genes, present in less than 15% of pipolins. Despite the great variety of different genes, some groups of pipolins share a similar gene composition and, as about 75% of genes are provided by about one-third of the pipolins (Supplementary Fig. S5). Although a certain level of synteny and modular organization can be detected (Fig. 2, Supplementary Fig. S5), genetic rearrangements, including inversions, duplications, and deletions, which often lead to gene exchange, are also frequent, as well as truncations and disruptions. Even truncated forms of piPolBs or XerC-like recombinases can also be detected, which might lead to impairment of replication or mobilization of pipolins. Overall, the genetic repertoire and structure of analyzed pipolins suggests that they can exchange genetic information among E. coli strains.
A detailed functional analysis of shell core genes is shown in Table S3. As mentioned above, besides piPolB, pipolins very often include one or more XerC and IntS (bacteriophage-type) tyrosine recombinases. When two complete recombinase genes are present, one of them is always close to an excisionase-like protein. A type-4 Uracil DNA glycosylase is also very frequent. Other proteins with DNA binding domains like mobilization proteins as well as components of restriction-modification systems are also common. Very few antimicrobial resistance genes or virulence genes are detected, mostly present in associated MGEs, like prophages, as in the case of LREC252 or LREC248 or insertion sequences, in the case of L53 or L37, among others (Fig. 2 and Supplementary Fig. S5).
In summary, a pipolin basic unit is composed of direct terminal repeats encompassing a piPolB gene and a variety of genes, most of them related to the metabolism of nucleic acids.
Cophylogeny of pipolins and host strains suggest pipolins horizontal transfer
Since the presence of the piPolB gene is the hallmark of pipolins and it constitute the only core gene, we performed a phylogenetic analysis of the new piPolB sequences from the new pipolin-harboring E. coli strains. Although some of the new annotated piPolB genes are partially truncated, particularly those from pipolins in phylogroup D strains, they have a high degree of identity, above 98.8% in the aligned regions. Phylogeny of the LREC piPolBs (Fig. 3) underlined again the similarity among pipolins in clonal strains that belong to phylogroups C and D, but sequences from phylogroups B1 and A were mixed together. A somewhat similar pattern was obtained for the phylogenies of XerC-like recombinases (Xer_C_2 group from Roary, see Table S3) and UDGs in the combined collection of pipolins (Supplementary Fig. S6). In order to assess the significance of different phylogenetic trees, we calculated the cophenetic correlation coefficient (CCC36,) among them as indicative of phylogenies clustering congruence. When comparing the piPolB and XerC phylogenies, the CCC was 0.29; for the comparison of piPolB and UDGs phylogenies it was 0.38; and a value of 0.27 was obtained for UDGs vs XerC comparison, indicating very low clustering similarity among different pipolins genes. Thus, increasing the number of genes in pipolins phylogeny, would increase the noise, to the detriment accuracy. Therefore, we considered only the phylogeny of piPolB coding sequence, as the only hallmark gene for all pipolins, for subsequent cophylogeny analyses. This can be considered also as a functional criterion, since piPolB is probably essential for episomal pipolin replication, being thus in agreement with conventional taxonomy of plasmids or other elements from the prokaryotic mobilome37,38.
Figure 3.
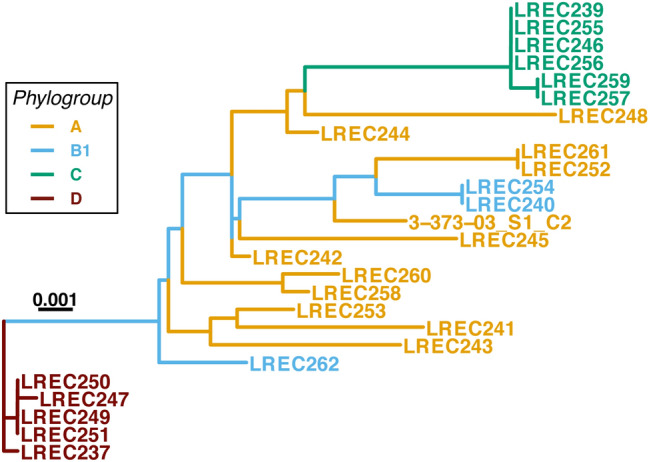
Maximum-likelihood tree of the new piPolB genes from the LREC dataset. As indicated, strain names are colored based on the phylogenetic group of strains. Previously described pipolin-harboring isolate 3-373-03_S1_C2 was included as a reference. The best-fit model was GTR + F + R2 for all considered criteria by ModelFinder62. Scale bar indicates the substitution rate per site.
The tanglegram in Fig. 4 allows us to visualize the cophylogeny between piPolBs and E. coli strains carrying pipolins. This plot reveals a complex association pattern, with numerous crisscrossing lines that suggest incongruence between the two phylogenies and, in line with this, the CCC is quite low, 0.14. Furthermore, we tested the cophylogeny between this group of pipolins and the host strains using PACo (Procrustean Approach to Cophylogeny)39. This method considers both clustering and relative distances, by using a procrustean approach in distance-based statistical shape analysis of phylogenetic trees that provide global-fit values () for the trees’ shape comparison. These analyses allow the detailed characterization of parasite-host and virus-host evolutionary interactions, under the null hypothesis that the topology of the host tree cannot predict the topology of the parasite tree40–42. The are inversely proportional to the topological congruence between the two phylogenetic trees39. In our case, the is 0.017, with a p-value of 0.46. Thus, in agreement with the very low CCC value, we cannot reject the null hypothesis, indicating that a significant portion of the pipolins tree topology does not depend on (i.e., cannot be predicted by) the host strains phylogeny. Moreover, when we analyzed the cophylogeny of pipolins and strains from each phylogenetic groups, we found that the Procrustes residuals of the pipolins and strains from phylogroup C and D, but not those from groups A or B1 (Supplementary Fig. S7A–D), were significantly smaller than the remainder of the interactions in the cophylogeny network, indicating that these interactions show significantly greater phylogenetic congruence than the rest. These lineages reflect the tree topologies of their host strains, indicating either co-evolutionary association or restricted horizontal transfer to highly related strains. Interestingly, some of the pipolins from strains from phylogroup D seem to have a truncated form of XerC_2 gene that only contains the Arm DNA binding domain present (see Fig. 3 and Supplementary Fig. S5, LREC247, LREC250 and LREC251), which could explain the cophylogeny of those pipolins with their host strains (i.e. low horizontal transfer), as they seem to lack any recombinase/integrase activity.
Figure 4.
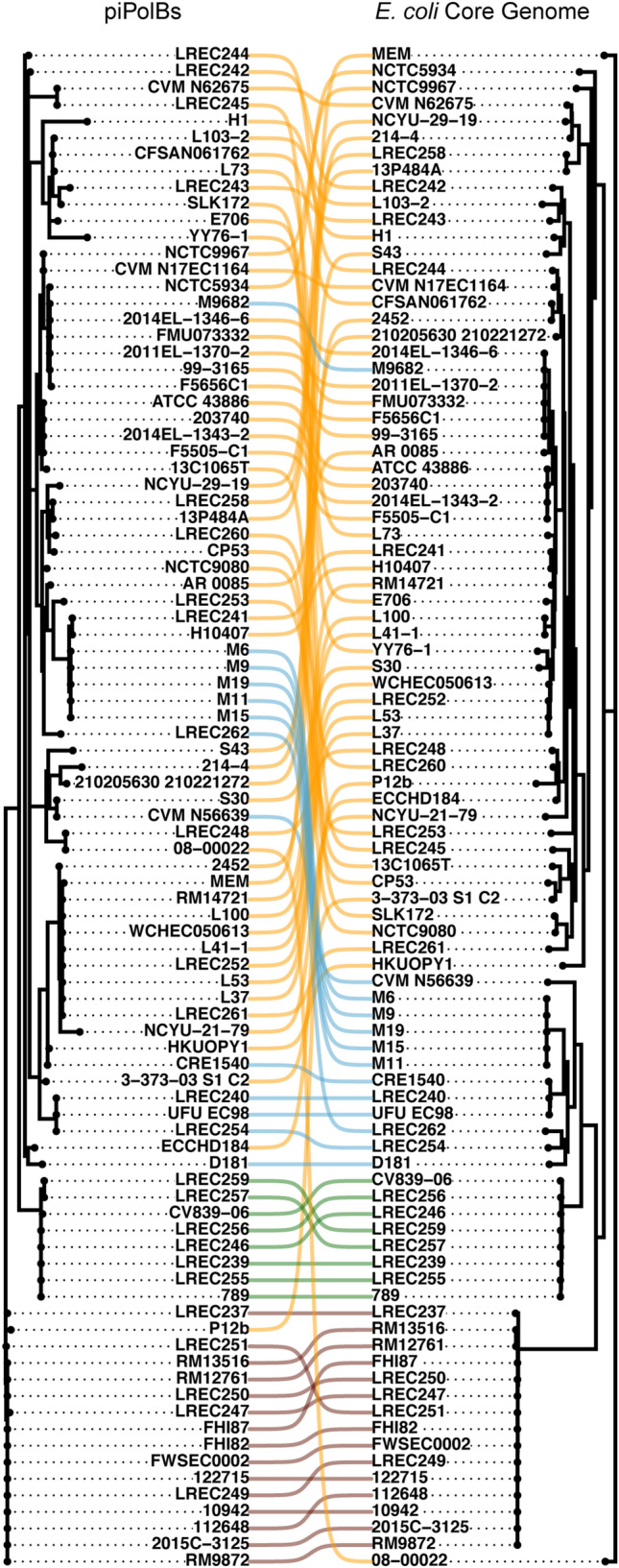
Cophylogeny of pipolins and host strains. Tanglegram representation of maximum-likelihood comparative phylogenies of piPolB and host strains core genome as hallmark of pipolins. Modelfinder Best-fit models were K3Pu + F + R2 and GTR + F + R7, respectively. Compared phylogenies are also displayed in Figures S1 and S3, respectively. Links between pipolins and E. coli strains are colored based on the phylogenetic groups.
Further, to identify the contribution of each E. coli strain to the overall cophylogeny structure, we evaluated the Jack-knifed squared residual values (Supplementary Fig. S7E). This analysis showed that an important proportion of pipolins from phylogroups C and D, but also for a few of the strains in phylogroup A, like L103-02, F5005-C1, ATCC-43886 or LREC258, among others, present low squared residual values, indicating congruent topologies between piPolBs and genomic trees. Interestingly, all the pipolins that present a tandem cohabitation with a prophage are within this group, in line with the previously hypothesized inactivation of pipolins as a consequence of the prophage insertion. On the other hand, pipolins with high squared residual values from phylogroups A and C would not have been evolutionary linked to their host strains, in agreement with the previous results.
Overall, we can conclude that the pipolins diversity is poorly congruent with the strains phylogeny and their distribution is rather indicative of a patchy distribution amongst a wide variety of pathogenic E. coli strains, as expected from horizontally transferred MGEs. This pattern may reflect the wide distribution of pipolins beyond E. coli, dispersed among major bacterial phyla, namely Actinobacteria, Firmicutes, and Proteobacteria, as well as in mitochondria3.
Conclusion and perspectives
Self-replicating integrative MGEs are highly diverse members of the prokaryotic and eukaryotic mobilome that seem to be involved in key evolutionary events, including the origin of the CRISPR-Cas systems from casposons and the evolution of several groups of eukaryotic dsDNA viruses from polintons. However, the information about their mobilization is limited and biased by the availability of genomic and metagenomic data in databases.
Pipolins represent a unique group of recently discovered, self-replicating integrative elements, which display broad distribution among bacteria. Their presence in mitochondria and phylogenetic analysis suggested ancient origin3, though their evolutionary history remains unclear. Here we have undertaken the first characterization of a predicted self-replicating MGE within a collection of circulating human and animal pathogenic strains of E. coli.
Despite the relatively low frequency of pipolins, our results confirmed that, rather than correlating with a certain phylogenetic group or pathotype, or the presence of a particular antimicrobial resistance, pipolins show a patchy distribution among a variety of circulating E. coli strains, both from a collection screening and a GenBank survey. We could detect clonality of several groups of isolates carrying pipolins, mainly belonging to the C and D phylogenetic groups. Moreover, their clonality reflects the phylogeny of their harbored pipolins, indicating that their mobility cannot be detected. However, mobilization of some of those pipolins seems impaired by the inactivation of XerC recombinase. On the contrary, the phylogeny of more diverse and numerous hosts from phylogroups A and B1 shows a lack of congruence with their pipolins, ruling out a monophyletic origin or strict vertical transmission of pipolins.
Analysis of the genetic structure and pangenome of pipolins from E. coli showed that they are more dynamic and flexible mobile elements than might be foreseeable from previous work. Pipolins encode a great diversity of genes, with an average of more than 7 different genes per element, with piPolB being the only shared gene in all pipolins. However, virulence and antimicrobial resistance genes were not detected in pipolins in our dataset, which may explain their low prevalence in pathogenic E. coli strains, but also raise questions about their biological role and evolution. Moreover, whereas diversity of plasmids is usual for pathogenic strains, our results suggest a possible interference between pipolins and other integrative MGEs, like pks islands or integrons, usually highly prevalent among pathogenic strains of Gram-negative bacteria.
Altogether, our results provide evidence for horizontal transfer of pipolins and suggest that pipolins are an active platform for horizontal gene transfer in E. coli, and they also pave the way for further analysis in other clinically relevant bacteria with pipolins.
Methods
Pipolin screening among E. coli strains
A total of 2,238 E. coli strains causing intestinal and extraintestinal infections in humans and animals from LREC collection were tested for the presence of pipolins by PCR using primers piPolB_FW (5′-GTTTTTTGACAAATTGCCCACTTG) and piPolB_RV (5′-CATATCAGAAAACACCGTCCG). Strains were cultured in a biosafety laboratory level 2 and handled in a microbiological safety cabinet Class II-A.
Conventional typing of LREC pipolin-harboring strains
The 25 LREC pipolin-harboring strains were first characterized by conventional typing. The determination of O and H antigens was carried out using the method previously described by Guinée et al.43 with all available O (O1 to O181) and H (H1 to H56) antisera. Isolates that did not react with any antisera were classified as O non-typeable (ONT) or HNT and those non motile were denoted as HNM. Assignment to the main phylogroups (A, B1, B2, C, D, E, F) was based on the PCR protocol of Clermont et al.44. The sequence types (STs) were established following the multilocus sequence typing (MLST) scheme of Achtman by gene amplification and sequencing of the seven housekeeping genes (adk, fumC, gyrB, icd, mdh, purA, and recA) according to the protocol and primers specified at the E. coli MLST web site (https://mlst.warwick.ac.uk/mlst/dbs/Ecoli)25. Clonotype identification was determined by fumC and fimH (CH) sequencing45.
Virulence factor (VF)-encoding genes of E. coli causing intestinal and extraintestinal infections were screened by PCR13,46. The virulence gene score was the number of virulence-associated genes detected. The isolates were designed presumptively as extraintestinal pathogenic E. coli (ExPEC) if positive for ≥ 2 of 5 markers, including papAH and/or papC, sfa/focDE, afa/draBC, kpsM II, and iutA47, as uropathogenic E. coli (UPEC) if positive for ≥ 3 of 4 markers, including chuA, fyuA, vat, and yfcV48, as avian pathogenic E. coli (APEC)49 if positive for ≥ 4 of 5 markers (hlyF, iutA, iroN, iss and ompT), and as necrotoxigenic E. coli (NTEC) if positive for cnf1, cnf2 or cnf3 genes50. In addition ten VF-encoding genes specific for pathotypes of diarrheagenic E. coli (DEC) were screened by PCR and the strains were designed as: typical enteropathogenic E. coli (tEPEC) (eae + , bfpA + , stx1−,stx2−), atypical enteropathogenic E. coli (aEPEC) (eae + , bfpA−, stx1−,stx2−), Shiga toxin-producing E. coli (STEC) (stx1 + and/or stx2 +), enterotoxigenic E. coli (ETEC) (eltA + and/or est +), enteroinvasive E. coli (EIEC) (ipaH +), and enteroaggregative E. coli (EAEC) (aatA + , aaiC + and/or aggR + 46.
Antimicrobial susceptibility screening of LREC pipolin-harboring strains
Antimicrobial susceptibility was determined by minimal inhibitory concentrations (MICs). Resistance was interpreted based on the recommended breakpoints of the CLSI51. Thirteen classes of antimicrobial agents were analyzed: penicillins (ampicillin, AMP), penicillins and β-lactamase inhibitors (amoxicillin-clavulanic acid, AMC; piperacillin-tazobactam, PTZ), non-extended spectrum 1st and 2nd generation cephalosporins (cefalotin, KF; cefazolin, CFZ; cefuroxime, CXM), extended-spectrum 3rd and 4th generation cephalosporins (cefotaxime, CTX; ceftazidime, CAZ; cefepime, FEP), cephalosporins and β-lactamase inhibitors (cefotaxime and clavulanic acid, CTXc; ceftazidime and clavulanic acid, CAZc), cephamycins (cefoxitin, FOX), carbapenems (imipenem, IMP; ertapenem, ETP), aminoglycosides (gentamicin, GEN; tobramycin, TOB), nitrofurans (nitrofurantoin, F), quinolones (nalidixic acid, NAL; norfloxacin, NOR; ciprofloxacin, CIP), folate pathway inhibitors (trimethoprim-sulphamethoxazole, SXT), phosphonic acids (Fosfomycin, FOS) and polymyxins (colistin, CL). E. coli multidrug resistant (MDR) was defined as resistance to one or more agents in three or more classes of tested drugs52.
Whole Genome sequence (WGS) and in silico characterization of E. coli strains carrying pipolins
WGS was carried out in an Illumina HiSeq1500 (2 × 100 or 2 × 150 bp) following standard protocols. Briefly, libraries for sequencing were prepared following the TruSeq Illumina PCR-Free protocol. Mechanical DNA fragmentation was performed with Covaris E220, and the final quality of the libraries assessed with Fragment Analyzer (Std. Sens. NGS Fragment Analysis kit 1–6,000 bp). The libraries were then sequenced, and reads were trimmed (Trim Galore 0.5.0) and filtered according to quality criteria (FastQC 0.11.7). Obtained sequences are available as a NCBI Bioproject PRJNA610160 (see Table S1 for Biosamples Ids and Enterobase Uberstrain codes for each strain). Strain 3–373-03_S2_C218 was sequenced and analyzed in parallel as a reference, but it was not included in the BioProject to avoid redundancy.
The reconstruction of the genomes and plasmids in the genomes was carried out using the methodology PLAsmid Constellation NETwork (PLACNETw)28. The assembled contigs, with genomic size ranging between 4.5 and 5.51 Mbp (mean size 5.08 Mbp), were annotated by Prokka35. Predicted CDS were analyzed using ABRicate53 for the presence of antibiotic resistance (ResFinder V2.1.), virulence genes (VirulenceFinder v1.5), plasmid replicon types (PlasmidFinder 1.3./PMLST 1.4.), and identification of clonotypes (CHTyper 1.0), sequence types (MLST 2.0) and serotypes (SerotypeFinder 2.0). PointFinder V3.2 was used in order to find antibiotic resistances encoded by chromosomal mutations (90% min. ID and 60% min. length thresholds)54. Phylogroups were predicted using the ClermonTyping online tool55. Moreover, to characterize the strains mobilome, prophages were searched with Phigaro56 and Phaster57; CRISPRCasFinder58 was used for the report of CRISPR/Cas cassettes and a custom database of IntI1, Intl2, Intl3, qacEdelta1 and sul1 genes was used for identification of integrons and subsequent integrity analysis with IntegronFinder59. For Pipolins-harboring strains from GenBank (see below), the presence of integrons was also analyzed by the same method using the chromosome sequence and then in Integrall database (60, updated on 1 April 2020), which allowed us the identification of one more integron, located in a plasmid. All predictions were called applying a select threshold for identification and a minimum length of 95 and 80%, respectively.
Pangenome analysis was performed with Roary27, which generated a codon aware alignment using Prank61. This alignment was then used for best-fit maximum likelihood-phylogenetic construction of phylogenetic tree IQTree Modelfinder62. For reference, single Nucleotide Polymorphism (SNP) tree were performed with EnteroBase26, which runs a number of pipeline jobs with The Calculation Engine (TCE) in the order refMasker, refMapper, refMapper_matrix and matrix_phylogeny.
Pipolins extraction and re-annotation
Pipolin-harboring E. coli genomes were retrieved from NCBI Genbank nucleotide collection using TBLASTN63 (October 30, 2019) and the piPolB amino acid sequence from E. coli 3-373-03_S1_C2 (Uniprot P0DPS1) as a query. Highly similar piPolBs from related Enterobacteriaceae (Citrobacter sp., Enterobacter sp., and Metakosakonia sp.) were also detected but we discarded them to facilitate the analysis. In total, 92 E. coli genomes (25 from LREC collection and 67 from GenBank) were employed in the subsequent analysis.
Pipolins from the obtained genomes were extracted for detailed characterization using a custom pipeline detailed as follows. Pipolin boundaries can be defined by att-like terminal direct repeats3. Based on that, the nucleotide BLAST was performed using one of the att-repeat sequences from the 3-373-03_S1_C2 isolate as a query against each of 92 E. coli genomes. In some cases, att-repeats and piPolB were located on different contigs, posing a challenge for us to understand the order and orientation of the contigs which parts of the contigs belong to a pipolin. We assumed that att-repeats should be headed in the same direction as they are direct repeats and that one of them could overlap with a tRNA gene on the opposite strand. For consistency, we referred to the latter att-repeat as attR and expected it always to be the rightmost att. According to these assumptions, we scaffolded the disrupted pipolin regions into a continuous sequence using a custom Python script. During scaffolding, different parts of a pipolin region were connected up by introducing the “assembly_gap” feature key of unknown length (DDBJ/ENA/GenBank Feature Table Definition, Version 10.9 November 2019). Comparative representation of the genetic structure of pipolins was generated by Easyfig64.
The extracted and scaffolded pipolin sequences were re-annotated by the Prokka pipeline35. This pipeline allows usage of different databases for protein annotation, among those we have been using Bacteria-specific UniProt (updated 16.10.2019), HAMAP (updated 16.10.2019) and Pfam-A (updated 08.2018). After the first try, ~ 50% of pipolin ORFs left unannotated and were classified as “hypothetical proteins”.
Since the pipolins annotation was quite incomplete, we attempted to improve the annotation of pipolin genes using HHpred65 for the most common pipolin ORFs, as defined by the Roary analysis. We considered the found hits as homologous if 1) the probability was > 90%, 2) E-value < 0.01, 3) secondary structure similarity was along the whole protein length, 4) there was a relationship among top hits, 5) only Bacteria, Archaea, and Viruses were allowed as the sources of the found hits. Using HHpred, functions were assigned to 6 more proteins. A list of these proteins was provided to Prokka as a trusted set of already annotated proteins. After the second re-annotation, only ~ 25% of proteins left unclassified.
Finally, pangenome analysis of pipolins gene content was carried out as detailed above and shell-core genes present in more than 15% of pipolins were analyzed by eggNog66 and KEGG orthology database functions with Blast Koala67.
Cophylogeny of pipolins and host strains
As mentioned above, the alignment of concatenated genes from the core-genome was used for the phylogeny of host strains. Phylogeny of piPolB, XerC and UDG pipolin genes was generated independently. When XerC recombinase gene appeared duplicated, so only the syntenic sequence with the UDG at the right end was included in the phylogeny reconstruction.
Phylogenetic analysis of gene sequences was carried out with Modelfinder62 upon PRANK codon aware alignment61.The obtained trees were then used for the comparative phylogenetic analyses with RStudio (Integrated Development for R. RStudio, Inc., Boston, MA https://www.rstudio.com/). Briefly, phylogenetic trees were handled and pruned when required with APE68 and tanglegrams for visual tree comparison were generated with Phytools69. We used the Dendextend package70 to calculate and represent the cophenetic distances of branches within a tree and the CCC (cophenetic correlation coefficient) between trees. Finally, we used PACo (Procrustean Approach to Cophylogeny)40 to investigate the phylogenetic congruence between trees.
Supplementary information
Acknowledgments
E. coli 3-373-03_S1_C2 isolate was kindly provided by Dr. Ellen Silbergeld (John Hopkins University, USA). We also thank Dr. Mart Krupovic (Pasteur Institute, France) for inspiring discussions and critical reading of the manuscript. Technical assistance of María Pilar Bea-Escudero is also acknowledged. This research was funded by the Spanish Ministry of Science, Innovation and Universities, Grant Number PGC2018-093723-A-I00 (AEI and FEDER, UE) to M.R.R., and by institutional grants from Fundación Ramón Areces and Banco de Santander to the Centro de Biología Molecular Severo Ochoa. LREC laboratory was supported by projects PI16/01477 from Plan Estatal de I + D + I 2013–2016, Instituto de Salud Carlos III (ISCIII), Subdirección General de Evaluación y Fomento de la Investigación, Ministerio de Economía y Competitividad (Gobierno de España) and Fondo Europeo de Desarrollo Regional (FEDER); and ED431C2017/57 from the Consellería de Cultura, Educación e Ordenación Universitaria (Xunta de Galicia) and FEDER. S.-C. F.-S. was holder of a PhD fellowship (FPU15/02644) from the Secretaría General de Universidades, Spanish Ministerio de Educación, Cultura y Deporte.
Author contributions
Conceptualization (MRR), Formal analysis (SCFS, MdT, LC, MRR), Funding Acquisition (JB, MRR), Investigation (SCFS, LC, MdT, MB, JMG, MRR), Resources (JB, MdT), Supervision (MdT, MS, JB, MRR), Writing—Original Draft Preparation (MRR), Writing—Review & Edit (SCFS, MdT, LC, JB, MRR).
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Saskia-Camille Flament-Simon, María de Toro, and Liubov Chuprikova.
Margarita Salas is deceased.
Supplementary information
is available for this paper at 10.1038/s41598-020-69356-6.
References
- 1.Kaas RS, Friis C, Ussery DW, Aarestrup FM. Estimating variation within the genes and inferring the phylogeny of 186 sequenced diverse Escherichia coli genomes. BMC Genomics. 2012;13:577. doi: 10.1186/1471-2164-13-577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.von Wintersdorff CJ, Penders J, van Niekerk JM, Mills ND, Majumder S, van Alphen LB, et al. Dissemination of antimicrobial resistance in microbial ecosystems through horizontal gene transfer. Front. Microbiol. 2016;7:173. doi: 10.3389/fmicb.2016.00173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Redrejo-Rodríguez M, Ordóñez CD, Berjón-Otero M, Moreno-González J, Aparicio-Maldonado C, Forterre P, et al. Primer-independent DNA synthesis by a family B DNA polymerase from self-replicating mobile genetic elements. Cell Rep. 2017;21:1574–1587. doi: 10.1016/j.celrep.2017.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Krupovic M, Koonin EV. Self-synthesizing transposons: unexpected key players in the evolution of viruses and defense systems. Curr. Opin. Microbiol. 2016;31:25–33. doi: 10.1016/j.mib.2016.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Krupovic M, Bamford DH, Koonin EV. Conservation of major and minor jelly-roll capsid proteins in Polinton (Maverick) transposons suggests that they are bona fide viruses. Biol. Direct. 2014;9:6. doi: 10.1186/1745-6150-9-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kapitonov VV, Jurka J. Self-synthesizing DNA transposons in eukaryotes. Proc. Natl. Acad. Sci. USA. 2006;103:4540–4545. doi: 10.1073/pnas.0600833103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pritham EJ, Putliwala T, Feschotte C. Mavericks, a novel class of giant transposable elements widespread in eukaryotes and related to DNA viruses. Gene. 2007;390:3–17. doi: 10.1016/j.gene.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 8.Krupovic M, Makarova KS, Forterre P, Prangishvili D, Koonin EV. Casposons: a new superfamily of self-synthesizing DNA transposons at the origin of prokaryotic CRISPR-Cas immunity. BMC Biol. 2014;12:36. doi: 10.1186/1741-7007-12-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kohler CD, Dobrindt U. What defines extraintestinal pathogenic Escherichia coli? Int. J. Med. Microbiol. 2011;301:642–647. doi: 10.1016/j.ijmm.2011.09.006. [DOI] [PubMed] [Google Scholar]
- 10.Lee E, Lee Y. Prevalence of Escherichia coli carrying pks Islands in bacteremia patients. Ann. Lab. Med. 2018;38:271–273. doi: 10.3343/alm.2018.38.3.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sarowska J, Futoma-Koloch B, Jama-Kmiecik A, Frej-Madrzak M, Ksiazczyk M, Bugla-Ploskonska G, et al. Virulence factors, prevalence and potential transmission of extraintestinal pathogenic Escherichia coli isolated from different sources: recent reports. Gut Pathog. 2019;11:10. doi: 10.1186/s13099-019-0290-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krupovic M, Shmakov S, Makarova KS, Forterre P, Koonin EV. recent mobility of casposons, self-synthesizing transposons at the origin of the CRISPR-cas immunity. Genome Biol. Evol. 2016;8:375–386. doi: 10.1093/gbe/evw006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mamani R, Flament-Simon SC, Garcia V, Mora A, Alonso MP, Lopez C, et al. Sequence types, clonotypes, serotypes, and virotypes of extended-spectrum beta-lactamase-producing Escherichia coli causing bacteraemia in a Spanish hospital over a 12-year period (2000 to 2011) Front. Microbiol. 2019;10:1530. doi: 10.3389/fmicb.2019.01530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garcia-Menino I, Garcia V, Mora A, Diaz-Jimenez D, Flament-Simon SC, Alonso MP, et al. Swine enteric colibacillosis in Spain: pathogenic potential of mcr-1 ST10 and ST131 E. coli isolates. Front. Microbiol. 2018;9:2659. doi: 10.3389/fmicb.2018.02659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Flament-Simon SC, Nicolas-Chanoine MH, Garcia V, Duprilot M, Mayer N, Alonso MP, et al. Clonal structure, virulence factor-encoding genes and antibiotic resistance of Escherichia coli, causing urinary tract infections and other extraintestinal infections in humans in Spain and France during 2016. Antibiotics (Basel) 2020 doi: 10.3390/antibiotics9040161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Flament-Simon SC, Garcia V, Duprilot M, Mayer N, Alonso MP, Garcia-Menino I, et al. High prevalence of ST131 subclades C2–H30Rx and C1–M27 among extended-spectrum beta-lactamase-producing Escherichia coli causing human extraintestinal infections in patients from two hospitals of Spain and France during 2015. Front. Cell. Infect. Microbiol. 2020;10:125. doi: 10.3389/fcimb.2020.00125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Flament-Simon SC, Duprilot M, Mayer N, Garcia V, Alonso MP, Blanco J, et al. Association between kinetics of early biofilm formation and clonal lineage in Escherichia coli. Front. Microbiol. 2019;10:1183. doi: 10.3389/fmicb.2019.01183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Richter TKS, Hazen TH, Lam D, Coles CL, Seidman JC, You Y, et al. Temporal variability of Escherichia coli diversity in the gastrointestinal tracts of Tanzanian children with and without exposure to antibiotics. mSphere. 2018 doi: 10.1128/mSphere.00558-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Clermont O, Christenson JK, Daubie AS, Gordon DM, Denamur E. Development of an allele-specific PCR for Escherichia coli B2 sub-typing, a rapid and easy to perform substitute of multilocus sequence typing. J. Microbiol. Methods. 2014;101:24–27. doi: 10.1016/j.mimet.2014.03.008. [DOI] [PubMed] [Google Scholar]
- 20.Stoppe NC, Silva JS, Carlos C, Sato MIZ, Saraiva AM, Ottoboni LMM, et al. Worldwide phylogenetic group patterns of Escherichia coli from commensal human and wastewater treatment plant isolates. Front. Microbiol. 2017;8:2512. doi: 10.3389/fmicb.2017.02512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Massot M, Daubie AS, Clermont O, Jaureguy F, Couffignal C, Dahbi G, et al. Phylogenetic, virulence and antibiotic resistance characteristics of commensal strain populations of Escherichia coli from community subjects in the Paris area in 2010 and evolution over 30 years. Microbiology. 2016;162:642–650. doi: 10.1099/mic.0.000242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fais T, Delmas J, Barnich N, Bonnet R, Dalmasso G. Colibactin: more than a new bacterial toxin. Toxins (Basel) 2018 doi: 10.3390/toxins10040151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bossuet-Greif N, Dubois D, Petit C, Tronnet S, Martin P, Bonnet R, et al. Escherichia coli ClbS is a colibactin resistance protein. Mol. Microbiol. 2016;99:897–908. doi: 10.1111/mmi.13272. [DOI] [PubMed] [Google Scholar]
- 24.Gonzalez-Alba JM, Baquero F, Canton R, Galan JC. Stratified reconstruction of ancestral Escherichia coli diversification. BMC Genomics. 2019;20:936. doi: 10.1186/s12864-019-6346-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wirth T, Falush D, Lan R, Colles F, Mensa P, Wieler LH, et al. Sex and virulence in Escherichia coli: an evolutionary perspective. Mol. Microbiol. 2006;60:1136–1151. doi: 10.1111/j.1365-2958.2006.05172.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou Z, Alikhan NF, Mohamed K, Fan Y, Agama Study G, Achtman M The EnteroBase user's guide, with case studies on Salmonella transmissions, Yersinia pestis phylogeny, and Escherichia core genomic diversity. Genome Res. 2020;30:138–152. doi: 10.1101/gr.251678.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Page AJ, Cummins CA, Hunt M, Wong VK, Reuter S, Holden MT, et al. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics. 2015;31:3691–3693. doi: 10.1093/bioinformatics/btv421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vielva L, de Toro M, Lanza VF, de la Cruz F. PLACNETw: a web-based tool for plasmid reconstruction from bacterial genomes. Bioinformatics. 2017;33:3796–3798. doi: 10.1093/bioinformatics/btx462. [DOI] [PubMed] [Google Scholar]
- 29.Rozwandowicz M, Brouwer MSM, Fischer J, Wagenaar JA, Gonzalez-Zorn B, Guerra B, et al. Plasmids carrying antimicrobial resistance genes in Enterobacteriaceae. J. Antimicrob. Chemother. 2018;73:1121–1137. doi: 10.1093/jac/dkx488. [DOI] [PubMed] [Google Scholar]
- 30.Partridge SR, Kwong SM, Firth N, Jensen SO. Mobile genetic elements associated with antimicrobial resistance. Clin. Microbiol. Rev. 2018 doi: 10.1128/CMR.00088-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Adelowo OO, Helbig T, Knecht C, Reincke F, Mausezahl I, Muller JA. High abundances of class 1 integrase and sulfonamide resistance genes, and characterisation of class 1 integron gene cassettes in four urban wetlands in Nigeria. PLoS ONE. 2018;13:e0208269. doi: 10.1371/journal.pone.0208269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perez-Etayo L, Berzosa M, Gonzalez D, Vitas AI. Prevalence of integrons and insertion sequences in ESBL-producing E. coli isolated from different sources in Navarra, Spain. Int. J. Environ. Res. Public Health. 2018 doi: 10.3390/ijerph15102308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Odetoyin BW, Labar AS, Lamikanra A, Aboderin AO, Okeke IN. Classes 1 and 2 integrons in faecal Escherichia coli strains isolated from mother-child pairs in Nigeria. PLoS ONE. 2017;12:e0183383. doi: 10.1371/journal.pone.0183383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deng Y, Bao X, Ji L, Chen L, Liu J, Miao J, et al. Resistance integrons: class 1, 2 and 3 integrons. Ann. Clin. Microbiol. Antimicrob. 2015;14:45. doi: 10.1186/s12941-015-0100-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 36.Cardona G, Mir A, Rossello F, Rotger L, Sanchez D. Cophenetic metrics for phylogenetic trees, after Sokal and Rohlf. BMC Bioinform. 2013;14:3. doi: 10.1186/1471-2105-14-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Garcillan-Barcia MP, Francia MV, de la Cruz F. The diversity of conjugative relaxases and its application in plasmid classification. FEMS Microbiol. Rev. 2009;33:657–687. doi: 10.1111/j.1574-6976.2009.00168.x. [DOI] [PubMed] [Google Scholar]
- 38.Cury J, Touchon M, Rocha EPC. Integrative and conjugative elements and their hosts: composition, distribution and organization. Nucleic Acids Res. 2017;45:8943–8956. doi: 10.1093/nar/gkx607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Balbuena JA, Miguez-Lozano R, Blasco-Costa I. PACo: a novel procrustes application to cophylogenetic analysis. PLoS ONE. 2013;8:e61048. doi: 10.1371/journal.pone.0061048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hutchinson MC, Cagua EF, Balbuena JA, Stouffer DB, Poisot T. paco: implementing procrustean approach to cophylogeny in R. Methods Ecol. Evol. 2017;8:932–940. doi: 10.1111/2041-210x.12736. [DOI] [Google Scholar]
- 41.Pratama AA, Chaib De Mares M, van Elsas JD. Evolutionary history of bacteriophages in the genus Paraburkholderia. Front. Microbiol. 2018;9:835. doi: 10.3389/fmicb.2018.00835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang IN, Yeh WB, Lin NS. Phylogeography and coevolution of bamboo mosaic virus and its associated satellite RNA. Front. Microbiol. 2017;8:886. doi: 10.3389/fmicb.2017.00886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guinée, P. A. M., Jansen, W. H., Wadström, T., Sellwood, R. Escherichia Coli associated with Neonatal Diarrhoea in piglets and calves. In de Leeuw, P. W., Guinée, P. A. M., editors. Laboratory Diagnosis in Neonatal Calf and Pig Diarrhoea: Proceedings of a Workshop on Diagnostic Techniques for Enteropathogenic Agents Associated with Neonatal Diarrhoea in Calves and Pigs, held at the Central Veterinary Institute, Department of Virology, Lelystad, The Netherlands, June 3–5, 1980. Dordrecht: Springer Netherlands; 1981. pp. 126–162.
- 44.Clermont O, Christenson JK, Denamur E, Gordon DM. The Clermont Escherichia coli phylo-typing method revisited: improvement of specificity and detection of new phylo-groups. Environ. Microbiol. Rep. 2013;5:58–65. doi: 10.1111/1758-2229.12019. [DOI] [PubMed] [Google Scholar]
- 45.Weissman SJ, Johnson JR, Tchesnokova V, Billig M, Dykhuizen D, Riddell K, et al. High-resolution two-locus clonal typing of extraintestinal pathogenic Escherichia coli. Appl. Environ. Microbiol. 2012;78:1353–1360. doi: 10.1128/AEM.06663-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Blanco M, Blanco JE, Dahbi G, Alonso MP, Mora A, Coira MA, et al. Identification of two new intimin types in atypical enteropathogenic Escherichia coli. Int. Microbiol. 2006;9:103–110. [PubMed] [Google Scholar]
- 47.Johnson JR, Porter S, Johnston B, Kuskowski MA, Spurbeck RR, Mobley HL, et al. Host characteristics and bacterial traits predict experimental virulence for Escherichia coli bloodstream isolates from patients with urosepsis. Open Forum Infect. Dis. 2015;2:o fv083. doi: 10.1093/ofid/ofv083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Spurbeck RR, Dinh PC, Jr, Walk ST, Stapleton AE, Hooton TM, Nolan LK, et al. Escherichia coli isolates that carry vat, fyuA, chuA, and yfcV efficiently colonize the urinary tract. Infect. Immun. 2012;80:4115–4122. doi: 10.1128/IAI.00752-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Johnson TJ, Wannemuehler Y, Doetkott C, Johnson SJ, Rosenberger SC, Nolan LK. Identification of minimal predictors of avian pathogenic Escherichia coli virulence for use as a rapid diagnostic tool. J. Clin. Microbiol. 2008;46:3987–3996. doi: 10.1128/JCM.00816-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Orden JA, Dominguez-Bernal G, Martinez-Pulgarin S, Blanco M, Blanco JE, Mora A, et al. Necrotoxigenic Escherichia coli from sheep and goats produce a new type of cytotoxic necrotizing factor (CNF3) associated with the eae and ehxA genes. Int. Microbiol. 2007;10:47–55. [PubMed] [Google Scholar]
- 51.CLSI. Performance standards for antimicrobial susceptibility testing, 27th Ed. CLSI Supplement M100S: Clinical Laboratory Standard Institute (CLSI); 2017.
- 52.Magiorakos AP, Srinivasan A, Carey RB, Carmeli Y, Falagas ME, Giske CG, et al. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: an international expert proposal for interim standard definitions for acquired resistance. Clin. Microbiol. Infect. 2012;18:268–281. doi: 10.1111/j.1469-0691.2011.03570.x. [DOI] [PubMed] [Google Scholar]
- 53.Seemann T. ABRicate: mass screening of contigs for antimicrobial resistance or virulence genes. 2019.
- 54.Zankari E, Hasman H, Cosentino S, Vestergaard M, Rasmussen S, Lund O, et al. Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 2012;67:2640–2644. doi: 10.1093/jac/dks261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Beghain J, Bridier-Nahmias A, Le Nagard H, Denamur E, Clermont O. ClermonTyping: an easy-to-use and accurate in silico method for Escherichia genus strain phylotyping. Microb. Genom. 2018 doi: 10.1099/mgen.0.000192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Starikova, E. V., Tikhonova, P. O., Prianichnikov, N. A., Rands, C. M., Zdobnov, E. M., Govorun, V.M. Phigaro: high throughput prophage sequence annotation. bioRxiv, 598243, doi:10.1101/598243 (2019). [DOI] [PubMed]
- 57.Arndt D, Grant JR, Marcu A, Sajed T, Pon A, Liang Y, et al. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016;44:W16–21. doi: 10.1093/nar/gkw387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Couvin D, Bernheim A, Toffano-Nioche C, Touchon M, Michalik J, Neron B, et al. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 2018;46:W246–W251. doi: 10.1093/nar/gky425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cury J, Jove T, Touchon M, Neron B, Rocha EP. Identification and analysis of integrons and cassette arrays in bacterial genomes. Nucleic Acids Res. 2016;44:4539–4550. doi: 10.1093/nar/gkw319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Moura A, Soares M, Pereira C, Leitao N, Henriques I, Correia A. INTEGRALL: a database and search engine for integrons, integrases and gene cassettes. Bioinformatics. 2009;25:1096–1098. doi: 10.1093/bioinformatics/btp105. [DOI] [PubMed] [Google Scholar]
- 61.Loytynoja A. Phylogeny-aware alignment with PRANK. Methods Mol. Biol. 2014;1079:155–170. doi: 10.1007/978-1-62703-646-7_10. [DOI] [PubMed] [Google Scholar]
- 62.Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods. 2017;14:587–589. doi: 10.1038/nmeth.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, et al. BLAST+: architecture and applications. BMC Bioinform. 2009;10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sullivan MJ, Petty NK, Beatson SA. Easyfig: a genome comparison visualizer. Bioinformatics. 2011;27:1009–1010. doi: 10.1093/bioinformatics/btr039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Soding J, Biegert A, Lupas AN. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005;33:W244–W248. doi: 10.1093/nar/gki408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Huerta-Cepas J, Szklarczyk D, Heller D, Hernandez-Plaza A, Forslund SK, Cook H, et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019;47:D309–D314. doi: 10.1093/nar/gky1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kanehisa M, Sato Y, Morishima K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 2016;428:726–731. doi: 10.1016/j.jmb.2015.11.006. [DOI] [PubMed] [Google Scholar]
- 68.Paradis E, Claude J, Strimmer K. APE: analyses of phylogenetics and evolution in R language. Bioinformatics. 2004;20:289–290. doi: 10.1093/bioinformatics/btg412. [DOI] [PubMed] [Google Scholar]
- 69.Revell LJ. phytools: an R package for phylogenetic comparative biology (and other things) Methods Ecol. Evol. 2012;3:217–223. doi: 10.1111/j.2041-210X.2011.00169.x. [DOI] [Google Scholar]
- 70.Galili T. dendextend: an R package for visualizing, adjusting and comparing trees of hierarchical clustering. Bioinformatics. 2015;31:3718–3720. doi: 10.1093/bioinformatics/btv428. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.