

Abstract
Inflammatory bowel diseases are chronic relapsing immune-mediated diseases of the intestinal tract with multifaceted manifestations and treatment related morbidity. Faecal and blood tests, radiological, endoscopic and histologic investigations are now widely used for managing both ulcerative colitis and Crohn’s disease. Over the years, a number of new investigations have been proposed but not widely adopted yet. Patients with Crohn’s disease may have multiple causes of diarrhoea, not always attributable to disease exacerbation, but sometimes linked to bile acid malabsorption; we have a reliable serum test, C4, that allows us to recognize and treat this cause of diarrhoea efficaciously and not empirically, but it is not available or used widely. There is genetic inter-individual variability in drug responses, in terms of both efficacy and toxicity, leading to high rates of therapeutic failure. Patients treated with thiopurine or, more rarely, 5-aminosalicylic acid may suffer from unpredictable and serious adverse events, some of these with pathogenesis related to genetic variants: myelosuppression, acute pancreatitis and nephrotoxicity. The identification of pre-treatment genetic tests can optimize therapeutic choice and avoid adverse events. With regard to biological drugs, patients can experience primary non-response or loss of response due to induction of immune responses to the drugs affecting drug efficacy and determining hypersensitivity reactions. We have specifically reviewed a number of investigations, whose use is currently limited, and highlighted four tests that deserve to be more widely incorporated in clinical practice as these could improve medical decision-making and patient outcomes.
Keywords: bile acid malabsorption, C4 test, HLA and anti-drug antibody, immunogenicity, inflammatory bowel disease, Nudix hydrolase 15, thiopurine S-methyltransferase, TPMT testing
Introduction
Inflammatory bowel diseases (IBDs) are chronic relapsing–remitting diseases of the gastrointestinal tract whose management is changing with close monitoring of disease control, but there is less adoption of prediction of adverse events due to drugs or tests to explore symptoms not directly related to inflammation.1,2 The aetiology is complex, involving the interaction of genetic predisposition, environmental triggers, microbial dysbiosis and immunological disorders.3 Immune dysregulation in IBD depends on the overproduction of several pro-inflammatory cytokines, which are responsible for intestinal inflammation and constitute targets for current and future therapeutic development.4 Investigations that are now widely used include therapeutic drug levels and anti-drug antibodies for biologics, faecal calprotectin for monitoring tight control of disease, endoscopic investigations, transabdominal ultrasonography and magnetic resonance enterography for assessment of intestinal healing, small intestinal and pan-enteric video capsule endoscopy as well as other blood tests such as C-reactive protein, genotypes and metabolites for thiopurine monitoring – these are now part of comprehensive investigation and staging of IBD.
Over the years, a number of new investigations have been proposed but these have not been adopted widely at all, despite strong supporting evidence. On the other hand, tests such as faecal calprotectin have been well known since the nineties but required changing disease management strategies and strong commercial support from diagnostic assay industries and pharmaceutical companies to become widely available. While the identification of stratification markers for disease progress and drug response could improve medical decision-making, patient outcomes and costs,5 robust validation of these biomarkers is still necessary and some, such as anti-Saccharomyces cerevisiae antibodies (ASCA), perinuclear anti-neutrophil cytoplasmic antibodies (pANCA), other microbial antibodies and mucosal healing panels, have not proved to be highly valuable despite commercial availability and therefore are infrequently used in clinical practice.
We have reviewed a small number of investigations that can stake a strong claim to be adopted in routine IBD clinical practice, but currently their usage is limited. In particular, the four tests highlighted below deserve serious consideration in most routine clinical practice involving IBD patients. We have reviewed the rationale for advocating these tests to be adopted in management of IBD patients.
Methodology
Combined automated and manual literature searches were performed on PubMed/Medline using the search terms: inflammatory bowel disease/Ulcerative Colitis/Crohn’s disease (5-ASA/mesalamine/adverse effects/adverse events/pancreatitis/nephrotoxicity) (thiopurine/pancreatitis/hepatotoxicity) [‘anti-TNF’/‘anti-TNFalpha (α)’/‘TNF inhibitor’/‘TNF-alpha (α) inhibitor’/‘anti-tumour necrosis factor’/‘TNF antagonist’ ‘bile acid malabsorption’ AND ‘anti-drug antibodies’ ‘ADA’(infliximab/adalimumab/golimumab/)] AND immunogenicity/diarrhoea.
No other limits were applied. The search results were manually searched; the number of articles identified at the beginning was 685, of which 101 were selected for their clinical relevance and alignment to the goal of this review by NB and SG. The bibliographies of relevant papers and reviews were also searched to identify suitable papers for inclusion.
Diagnosis of bile acid diarrhoea in Crohn’s disease – 7α-hydroxy-4-cholesten-3-one
Bile acids are predominantly absorbed in the ileum by an active transport process.6 Ileal resection causes bile acid malabsorption resulting from imbalances in the homoeostasis of bile acids in the enterohepatic circulation.7 Bile acid diarrhoea is common, and likely under-diagnosed, but it should be considered relatively early in the differential diagnosis of chronic diarrhoea.7 As early as 1969 Hoffman et al. demonstrated the effects of cholestyramine as symptomatic treatment, improving faecal consistency by abolishing bile acid-induced secretion of water and electrolytes in the colon.8
Patients with Crohn’s disease (CD) may have multiple causes of diarrhoea and it is common for these patients to receive cholestyramine or other bile acid sequestrant (BAS) drugs empirically without testing but this approach has limitations and is not precise or predictably effective.
Bile acid malabsorption (BAM) has been reported in up to 50% of adult patients with CD, especially those with ileal involvement and dysfunction or resection.9,10 Depending on the extent of disease or resection, this usually predisposes to diarrhoea, but may also cause steatorrhoea with malabsorption of fat soluble vitamins and formation of gallstones and kidney stones.11,12 Secretory diarrhoea (bile acid diarrhoea) is due to the effects of unabsorbed bile acids (BAs) on various mechanisms, such as adenylate cyclase affecting water and electrolyte absorption, in the colonic epithelium. This may be compounded by an increase in intestinal permeability and also motility, produced by actions of primary and secondary bile acids on the farnesoid X and G-protein-coupled bile acid receptors.13
There are several causes for the increase in BAs entering the colon in active ileal CD.14 Ileal dysfunction produces malabsorption of BAs, due to a decrease in BA absorptive transporters, particularly the apical sodium-linked BA transporter.10,15 Active inflammatory disease also reduces synthesis of the regulatory hormone, fibroblast growth factor 19 (FGF19), and this results in excess BA synthesis, with increased BA precursors.16 Similarly, ileal resection reduces the amount of specialized tissue for active BA absorption and FGF19 production. These changes in the enterohepatic circulation and synthesis of BAs can be measured to help the differential diagnosis of symptoms in people with CD.
The gold standard in diagnosing BAM is the 75seleno-homocholic-acid-taurine (SeHCAT) test,12 which is a relatively simple low-gamma radiation nuclear medicine test requiring two scans 7 days apart, which will detect increased loss of the tracer. Patients with CD and a previous ileal resection who have diarrhoea have a >90% likelihood of an abnormal SeHCAT, which means that the predictive value of this test is mostly redundant in them. In CD without resection, results are more variable.17 Forty-eight-hour stool collection to measure faecal BAs is hardly ever used and cumbersome. Patients with BAM or reduced levels of FGF19 also develop compensatory increases in the synthesis of BA precursors, specifically the intermediate in the classical synthetic pathway, 7α-hydroxy-4-cholesten-3-one (C4; also called 7αC4) (Figure 1). This can be measured in fasting serum or plasma. There is a good inverse correlation between C4 test and the SeHCAT-test18 and the blood test has many advantages: the C4 test is easier for patients to perform, less time consuming and burdensome, and less expensive, although it only provides a measure at a single time point.19 Whether the C4 test can provide useful supporting evidence to indicate ileal inflammation in CD will require further investigations.
Figure 1.

Bile acid metabolism pathway.
Two major pathways are involved in bile acid synthesis. The classic pathway is controlled by CYP7A1 in the endoplasmic reticulum. CYP8B1 is required to synthesize cholic acid and CYP27A1 is able to form chenodeoxycholic acid. Patients with loss of bile acids, as in ileal disease or resection, develop compensatory increases in the synthesis of bile acids precursors, specifically the intermediate in the classical synthetic pathway, 7α-hydroxy-4-cholesten-3-one (C4).
AKR1C4, aldo-keto reductase family 1 member C4; AKR1D1, aldo-keto reductase family 1 member D1; CYP7A1, cytochrome P450 family 7 subfamily A member 1; CYP8B1, cytochrome P450 family 8 subfamily B member 1; CYP27A1, cytochrome P450 family 27 subfamily A member 1; HSD3B7, hydroxy-delta-5-steroid dehydrogenase, 3 beta- and steroid delta-isomerase 7.
Several studies found increased C4 levels in 42–46% of CD patients with ileal disease and in 55% of those with ileal resections.9,10 Furthermore, elevated C4 levels were detected also in 14% of CD patients with only colonic involvement.9,20 A cut-off concentration of C4 of 48.3 ng/ml or greater identified patients with diarrhoea attributable to bile acid malabsorption with 90.9% sensitivity, 84.4% specificity.21 C4 testing is available to clinicians but not widely across the world and an abnormal value should be used to guide appropriate and effective treatment, in particular, the use of BAS drugs.
C4 is a relatively simple, straightforward test that should be available more widely, ideally performed by laboratories serving hospitals or regionally. While this may be helpful for investigation of diarrhoea in general, it can play an important role in investigating an important morbidity in CD patients. (Figure 2.)
Figure 2.

C4 test: when to use it and positive and negative aspects.
BAM, bile acid malabsorption; C4, 7α-hydroxy-4-cholesten-3-one; CD, Crohn’s disease
Prediction of thiopurine-induced myelosuppression – NUDT15 genotypes
Thiopurines, consisting of azathioprine (AZA) and its analogues 6-mercaptopurine and 6-thioguanine, are the most commonly prescribed immunosuppressive agents in IBD used to maintain corticosteroid-free remission, prevent postoperative recurrence22 and to avoid the development of antidrug antibodies in those receiving anti-tumour necrosis factor (TNF)-α.23 They are effective and cheap but their use is limited by several adverse events: 17% of Europeans using thiopurines develop adverse events24 and this percentage is higher in Asian populations, despite doses of thiopurines in Asian countries being lower than in Europe.25,26
Genetic polymorphisms have been identified as important determinants of adverse events and a detailed meta-analysis showed that thiopurine s-methyltransferase (TPMT) polymorphisms were significantly associated with AZA-induced overall adverse effects.27 TPMT catalyses the S-methylation of thiopurines; its activity is inversely proportional to the levels of thioguanine nucleotide metabolites (6-TGN), whose accumulation determines most of the adverse events.
However, TPMT polymorphisms cannot explain all episodes of AZA-related adverse events and, furthermore, a normal TPMT genotype cannot exclude the development of side effects. Weinshilboum et al.28 showed that TPMT activity has a bimodal distribution in the general population: 89% have high enzymatic activity, 11% intermediate activity and only 0.3% lack activity. Currently, 37 alleles responsible for TPMT deficiency (TPMT*2-38) are known29 but four allelic variants, TPMT*2, *3B, *3C and *3A, were found in more than 80% of Caucasian and the most frequent was *3A.30 The frequency of TPMT*3C, which is associated with low or intermediate TPMT activity, is between 1.1% and 2.9% in Japanese people.31
Among adverse events there is thiopurine-induced myelosuppression (TIM), which occurs in 4% of European individuals and in up to 15% of Asian individuals.24 Most patients are asymptomatic, but when serious opportunistic infections occur in IBD patients with TIM there is an estimated mortality of 1%.32 There is substantial evidence linking TPMT and Nudix hydrolase 15 (NUDT15) enzyme activity to TIM.33 TIM has been attributed to low TPMT activity;34 nevertheless in a multicentre study, thiopurine therapy was prescribed with classic therapeutic dosage and with dose adjusted according to the TPMT mutations: the overall proportion of TIM was the same in the two groups.35
Although the standard dose of thiopurines in Japan (AZA: 1–2 mg/kg per day) is half of that in Europe (AZA: 2–2.5 mg/kg per day), and approximately 10% of Europeans36 versus 3% of Asians carry TPMT genetic variants, the incidence of TIM in Asian populations is higher than that in Caucasians.25,37 These data suggest that in IBD patients bone-marrow suppression is not solely dependent on TPMT activity, but is also associated with other genetic and environmental factors.38,39
This difference is explained by widespread genetic variation in NUDT15 in East Asian populations, approximately 10%, which has now been identified as a determinant of TIM.40,41 Moreover a recent case–control study described NUDT15 variants also in patients of European ancestry: three NUDT15 coding variants in chromosome 13, including p.Gly17_Val18del, were associated with TIM independent of TPMT genotype and thiopurine dose in European patients.38 NUDT15 catalyses the conversion of cytotoxic thioguanine metabolites to non-toxic thioguanine metabolites. Genetic variants lead to low NUDT15 enzyme activity and high levels of cytotoxic thioguanine that may lead to myelosuppression40 (Figure 3). Japanese and Korean studies have revealed that severe leukopenia and complete hair loss are inevitable in patients with the homozygous variant of NUDT15 R139C (T/T genotype),42 and patients with heterozygous variant (C/T genotype) experience early leukopenia more frequently than those with wild-type genotype (C/C genotype).42–45 For alopecia, it is a well-recognized, dose-dependent adverse event in Asian populations, with an incidence of around 1.5%,25,37 and it is rare in Europeans.24 It is recommended that treatment with thiopurines should be avoided for patients with the T/T genotype and low-dose mercaptopurine (0.2–0.3mg/kg per day) may be used for C/T genotype.42
Figure 3.
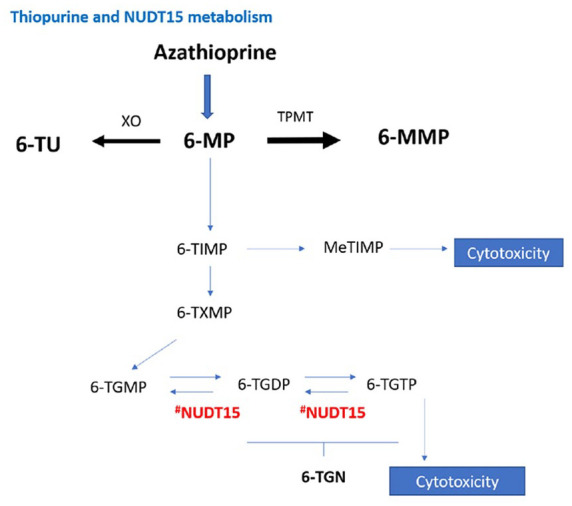
Thiopurine metabolism and role of NUDT15.
NUDT15 catalyses the conversion of cytotoxic thioguanine metabolites to non-toxic thioguanine metabolites. Genetic variants lead to low NUDT15 enzyme activity and high levels of cytotoxic thioguanine that may lead to myelosuppression.
6-MMP, 6-methylmercaptopurine; 6-MP, 6-mercaptopurine; 6-TGDP, 6-tioguanine diphosphate; 6-TGMP, 6-tioguanine mono-phosphate; 6-TGN, 6-tioguanine nucleotide; 6-TGTP, 6-tioguanine tri-phosphate; 6-TIMP, 6-thiosine 5 ¢ -monophosphate; 6-TU, 6-thiouric acid; 6-TXMP, 6-thioxanthosine 5 ¢-monophosphate; Me-TIMP, Me-thiosine 5 ¢ -monophosphate; NUDT15, Nudix hydrolase 15 enzyme; TPMT, thiopurine methyltransferase; XO, xanthine oxidase.
More recent studies have identified additional NUDT15 genetic variants predictive for TIM in Asian populations.43,46
Patients with both TPMT and NUDT15 genetic variants are at excessive risk of TIM if they receive standard thiopurine dosing. The Clinical Pharmacogenetics Implementation Consortium has published detailed dosing recommendations based on TPMT and NUDT15 genotypes: a reduction of starting doses (30–80% of target dose) should be considered for TPMT or NUDT15 intermediate metabolizers, while 10% of target dose or the use of an alternative agent should be used for TPMT or NUDT15 poor metabolizers.47
TPMT testing is cost-effective48 and widely available in routine service laboratories: in UK it was used by 67% of clinicians prior to AZA prescription49 whilst, worldwide, testing is used by 43% of gastroenterologists in the management of IBD.50 In the United States, the Food and Drug Administration has suggested the genotyping of TPMT before starting AZA or mercaptopurine treatment to prevent myelotoxicity.51 With a significant proportion of patients developing myelotoxicity despite TPMT testing, routine use of both TPMT and NUDT15 genotype should be considered as a routine prior to thiopurine therapy in IBD. Currently in Europe and North America, NUDT15 genotypes are not routinely checked despite significant presence of patients with relevant ethnicity. Prior to using thiopurine, it is recommended that clinicians should consider testing for NUDT15 not only in East Asian populations, but, as the study by Walker et al. suggests, also in European ancestry patients.38 Exome-wide association studies have shown that in NUDT15 variants may be associated with thiopurine associated myelosuppression in IBD patients of European ancestry (odds ratio 38.2, 95% confidence interval 5.1–286.1).46
If NUDT15 genotyping is not available, thiopurine needs to be started at a low dose and titrated up gradually to a minimal effective dose in order to minimize risk of bone marrow toxicity and therefore slows achieving optimum therapeutic doses.
Prediction of thiopurine-induced pancreatitis: HLA Class II haplotypes
Acute pancreatitis after thiopurine therapy (thiopurine-induced pancreatitis; TIP), which usually occurs within the first few weeks of therapy, is a well-recognized, idiosyncratic, unpredictable dose-independent adverse drug reaction with an incidence of approximately 4–7% in patients with IBD.24 The pathogenesis is likely related to genetic variants in HLA-DQA1*02:01-HLA-DRB1*07:01.52 The risk allele frequency in Europeans is 27% with a risk of approximately 17% of developing TIP in homozygous patients.53 Although these data show that the potential of pre-treatment HLA-DQA1-HLA-DRB1 genotyping would be useful to avoid administration of thiopurines to patients with IBD who are homozygous, it has not yet been incorporated in clinical treatment protocols. As mentioned above, HLA Class II panel could provide a relatively useful solution as the test is relatively inexpensive.
Prediction of immunogenicity to anti-TNF monoclonal antibodies: HLA Class II haplotypes
Biological therapy has transformed the management of IBD:54 since the introduction of infliximab for CD in 1998, TNF inhibitors have become widely used in moderate-to-severe IBD, in patients with extensive disease, in CD with stricturing and penetrating phenotypes or in patients who do not tolerate or do not respond to conventional therapies. Despite their established efficacy, up to one-third of patients with IBD will have no response to these agents (primary non-response) and another third will fail TNF-antagonist therapy after initial response (loss-of-response).55
Treatment failure in many cases is due to the formation of anti-drug antibodies (ADAs)56 that can also cause serious adverse events such as allergic infusion reactions and vasculitis.57,58 Immunogenicity is more common (65%) in patients treated with infliximab (a murine–human chimeric monoclonal antibody) than with adalimumab (38%), a fully human monoclonal antibody.56,59 Risk of immunogenicity can be reduced with combination immunomodulator therapy and for infliximab this strategy improves treatment outcomes.59 Despite these benefits, many patients are still treated with anti-TNF monotherapy because of concerns about the increased risk of adverse drug reactions, opportunistic infections and malignancies associated with combination therapy.60 As a result of clinical evidence, assessment of immunogenicity is now a mandatory requirement of the European Medicines Agency and the Food and Drug Administration prior to approval of all biological agents:61–63 the ability to identify patients at increased risk of immunogenicity may influence the choice of anti-TNF treatment and the use of preventive strategies, including combination with immunomodulator. Retrospective studies have suggested that variants in FCGR3A64 and HLA-DRB1*0365 increase susceptibility to immunogenicity to anti-TNF therapy. These associations did not achieve genome-wide significance and are yet to be independently replicated.66 Recently, the HLA-DQA1*05 haplotype, in particular the specific alleles HLA-DQA1*05:01 and HLA-DQA1*05:05, were identified as a genetic determinant of immunogenicity to TNF-antagonists: it is associated with a two-fold increased risk of immunogenicity.66 So pre-treatment HLA-DQA1*05 and HLA-DRB1*03 genetic testing thus has the potential to personalize TNF-antagonist therapy and should lead to preventive measures such as the use of concomitant immunomodulator to maximize response.66,67 This strategy will also spare patients combination therapy (thiopurines) if it is not required. Further replication studies from other geographical regions are necessary.
While combination therapy of anti-TNF monoclonal antibodies with thiopurines may reduce the rate of immunogenicity, it also increases the risk of infections, neoplastic lesions and myelotoxicity. For these reasons a stratified approach to determine the requirement of combination therapy to prevent immunogenicity by the use of HLA Class II genotyping is an elegant solution worthy of widespread adoption in clinical practice. HLA genotyping is inexpensive and relatively easy to introduce (Figure 4). However, the findings will require confirmation in cohorts from different geographical regions and ethnicities.
Figure 4.

Thiopurine-induced pancreatitis and immunogenicity predictors: positive and negative aspects.
TNF, tumour necrosis factor
Miscellaneous investigations that are promising but not ready for primetime
Predicting 5-aminosalicylic acid nephrotoxicity: HLA Class II haplotypes
5-Aminosalicylates (5-ASAs) are safe, cheap and effective drugs prescribed to induce and maintain steroid-free remission in patients with mild to moderately active ulcerative colitis (UC). The use of these agents for most patients is lifelong, so the long-term toxicity should not be underestimated. Common side effects associated with 5-ASAs include flatulence, abdominal pain, nausea, diarrhoea, headache, dyspepsia and nasopharyngitis, which may occur in up to 10% of patients. There are other two adverse events, rare but more serious: pancreatitis (0.3%) and nephrotoxicity (0.2%).68 Regarding pancreatitis in a randomized controlled trial comparing mesalamine 2.4 g versus 4.8 g daily, it was found that in both groups, only one patient in each developed pancreatitis. Both clinical episodes resolved upon discontinuation of the drug and pancreatitis was postulated to be secondary to mesalamine hypersensitivity.69
Nephrotoxicity has been reported for both sulfasalazine and the newer 5-ASA agents: there is an annual risk of 0.26% and an incidence of one case per 4000 patient-years.70 A review of the UK General Practice Research Database calculated the incidence at 0.17 cases per 100 patients per year but the authors noted that only 13% of these patients had a histological diagnosis of interstitial nephritis.71 The 5-ASA-induced nephrotoxicity is not associated with duration of therapy and has a probable genetic basis: HLA-DRB1*03:01 has been identified as one of its determinants.72 Carriership of the risk allele is associated with a three-fold increased risk of renal injury after 5-ASA administration. However, the high frequency of this risk allele in the general population and the low frequency of the adverse event limits its clinical utility.72 Currently only yearly monitoring of renal function is recommended: if there is an increase in serum creatinine, it is important to check urine electrolytes and proteinuria. Discontinuation of mesalamine is suggested if fractional excretion of sodium is >2% or in the presence of proteinuria.68,73
Predicting myelotoxicity and haematological cancers after thiopurine: ITPase enzymes
Adverse events of thiopurines can be divided into: dose-independent, such as pancreatitis and flu-like illness, and dose-dependent, such as myelosuppression and hepatotoxicity. As mentioned above TPMT polymorphisms explains many but not all of thiopurine-related adverse events.
Another significant enzyme, involved in the biotransformation of thiopurine drugs is inosine triphosphatase (ITPA). It catalyses the pyrophosphohydrolysis of inosine triphosphate (ITP) to inosine monophosphate, preventing the accumulation of potentially toxic ITPs.74 Two mutations reduce activity of the ITPA causing the most effect: IVS2 21AC (rs7270101) and p.P32T (c.94C/A, rs1127354). Deficiency in the ITPase activity occurs in approximately 1 in 1000 Caucasians, while in Asian populations the frequency is of 14–19%.75 Furthermore, it was observed that the ITPA c.94C/A genotype makes a contribution to the concentration of 6-methylmercaptopurine in red blood cells and the occurrence of hepatotoxicity and acute lymphoblastic leukaemia in paediatric patients.76
Predicting response to treatment with anti-TNF antibodies: TREM1, OSM, gene expression profiling of CD8+ T lymphocytes
Arijs et al. demonstrated that various genes involved in the inflammatory cascade account for resistance to anti-TNFa therapy and predicted the response to infliximab therapy with 89% accuracy.77 Among these, IL13RA2 was the highest ranked common gene for both CD and UC analyses. Recently, expansion of apoptosis-resistant intestinal TNFR2+ IL-23R+ T-cells has been associated with resistance to anti-TNF therapy in CD.78
Triggering receptor expressed on myeloid cells 1 (TREM1)
TREM1 looks a promising predictive biomarker for anti-TNF therapy in CD, although conflicting results are currently reported.79,80 It is a receptor expressed on innate immune cells, which amplify inflammatory signals triggered by Toll-like receptors contributing to the pathophysiology of many acute and chronic inflammatory conditions.81 Pre-test probabilities for primary (non)-response to anti-TNF therapy could be optimized using mucosal TREM1 expression or blood TREM1 levels.79,82 However, more evidence is required and blood TREM1 may be less accurate.
Oncostatin M (OSM)
An important and highly expressed cytokine in IBD patients is OSM. It has been proved that a single-nucleotide polymorphism in the OSM locus is strongly associated with risk of developing IBD,83 while mucosal OSM correlates closely with histopathological disease severity84 and furthermore it is associated with anti-TNF resistant disease. OSM is part of the IL-6 cytokine family85 that can induce signalling via the JAK-STAT pathway, the phosphatidylinositol-3-kinase (PI3K)-Akt pathway, and mitogen activated protein kinase cascades via heterodimeric receptors such as gp130, OSMR and leukaemia inhibitory factor receptor-β.86 However, OSM and OSMR expression was increased in patients with IBD who required surgery, suggesting an association with complicated disease, and high expression in pre-treatment biopsies was strongly associated with primary non-responsiveness to anti-TNF therapy.77,84,87
This association was confirmed in two additional prospective UC patient cohorts treated with infliximab and golimumab88,89 and also an analysis of five datasets demonstrates that high baseline OSM expression in the intestinal mucosa is reproducibly associated with decreased responsiveness to anti-TNF therapy.84 Similar results have also been shown for vedolizumab and corticosteroids,90,91 even though findings concerning high levels of mucosal OSM and response to vedolizumab are still preliminary.92 At present, mucosal OSM should probably be considered as a novel pharmacodynamic marker predicting disease severity and response to therapy.93
Gene expression profile of CD8+T
Lee et al. at first described a prognostic transcriptional signature in CD8 + T cells able to separate IBD patients into two phenotypically distinct subgroups.94 This can be explained because CD8 + T cell gene expression signature corresponds to differences in T cell exhaustion. T cell exhaustion is the phenomenon by which effector T cells progressively lose their ability to respond to target antigens. Patients with more T cell exhaustion had a better prognosis with longer time to disease relapse and fewer flares over time.95 This team developed a blood-based test, qPCR-based classifier, to identify the same subgroups without cell separation, thus creating a test more suitable for IBD clinical practice. The two subgroups were IBD1 (exhaustion low) or IBD2 (exhaustion high) and they experienced very different disease courses: IBD1 subgroup had consistently more aggressive disease, which was characterized by the need to escalate treatment earlier, with immunomodulators, biological therapies or surgery.96
This new biomarker could be a valuable tool for the clinician to stratify patients to receive personalized therapy, predicting the course of the disease, and a randomized trial is ongoing.
Conclusion
Patients with IBD, due to the chronic relapsing disease course, have to take medications for a lifetime to maintain disease remission, improve quality of life and prevent long-term complications such as uncontrolled bleeding, colorectal cancer and surgery.97 Patients who achieve endoscopic remission have improved long term outcomes compared with those who do not.98 Although the outcomes of medical therapies have greatly improved over the last decades, substantial individual variability remains in terms of both efficacy and toxicity: CD and UC are heterogeneous disorders and we cannot apply a ‘one-size-fits-all’ principle in terms of treatment strategy. Many patients with IBD do not achieve disease remission: they lose response after initial successful treatment or develop severe drug-induced adverse events.
The lack and/or loss of response, the concern of safety and the control on health budget are continuously driving IBD research: not only trying to find alternative therapeutic target, but also characterizing pre-treatment tests to incorporate into clinical daily IBD management.
It will be problematic to have precise guidance for clinical practice but several new markers have been identified as strong determinants of (adverse) response to drugs used in the management of IBD.
In this review we show that some of these can guide clinicians in choosing the right therapeutic strategy, predicting the risk of adverse events, and therefore they should be considered for use in clinical practice (Table 1): TPMT/NUDT15 genotype and HLA-DQA1*02:01/HLA-DRB1*07:01 predict myelosuppression and pancreatitis induced by thiopurine respectively; while as regards immunogenicity to TNF-antagonists, HLA-DQA1*05:01/HLA-DQA1*05:05/HLA-DRB1*03 are useful in predicting response or adverse events to therapies. The C4 test is a simple and inexpensive test that can help the clinician to understand the real causes of diarrhoea in CD patients pre- and even post-surgery. Other new investigations are promising but not ready for primetime as they still lack strong evidence (Table 2): HLA-DRB1*03:01 as predictor of 5-ASA-induced renal injury, ITPA for haematological cancer and TREM1/OSM/gene profile of CD8+T in non-response of TNF therapy. However, the uptake of routine pre-treatment testing to better stratify IBD patients is slow and extensive validation is required.
Table 1.
Tests strongly recommended in inflammatory bowel disease clinical practice.
| Pre-treatment test | Response | References |
|---|---|---|
| ○ C4 test | Diarrhoea in Crohn’s disease due to BAM | Vijayvargiya et al.19; Battat et al.21 |
| ○ TPMT genotype ○ NUDT15 genotype |
Myelosuppression, thiopurine induced | Coenen et al.35; Colombel et al.38; Moriyama et al.40,41; Kakuta et al.42 |
| ○ HLA-DQA1*02:01 ○ HLA-DRB1*07:01 |
Acute pancreatitis, thiopurine induced | Wilson et al.52 |
| ○ HLA-DQA1*02:01 ○ HLA-DQA1*05:05 ○ HLA-DRB1*03 |
Immunogenicity to TNF-antagonists | Garcês et al.61; Sazonovs et al.66 |
BAM, bile acid malabsorption; NUDT15, Nudix hydrolase 15; TPMT, thiopurine s-methyltransferase
Table 2.
Tests promising but not ready for primetime.
| Pre-treatment test | Response | References |
|---|---|---|
| ○ HLA-DRB1*03:01 | Renal injury, 5-ASA induced | Heap et al.72 |
| ○ ITPA1 | Lymphoblastic leukaemia in paediatric IBD patients | Marsh et al.75; Smid et al.76 |
| ○ TREM1 ○ OSM ○ CD8+T gene profile |
Non-response to anti-TNF therapy | Gaujoux et al.79; Verstockt et al.80,82; West et al.84; Lee et al.94; Biasci et al.96 |
5-ASA, 5-aminosalicylate; IBD, inflammatory bowel disease; ITPA, inosine triphosphatase. OSM, oncostatin M; TNF, tumour necrosis factor; TREM1, triggering receptor expressed on myeloid cells 1.
Recent efforts led by the United Kingdom IBD Genetics Consortium have successfully conducted prospective and retrospective studies about genetic pre-treatment tests in the context of IBD management.66,99 However, additional large international consortia are needed to facilitate the collection of rigorous cohorts of patients who develop (rare) adverse events and future studies should focus on the cost-effectiveness of these tests in the different ethnic populations.
Footnotes
Conflict of interest statement: The authors declare that there is no conflict of interest.
Funding: This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
ORCID iDs: Nunzia Labarile  https://orcid.org/0000-0002-8512-7726
https://orcid.org/0000-0002-8512-7726
Julian Walters
https://orcid.org/0000-0001-9720-5835
Contributor Information
Nunzia Labarile, Institute Translational of Medicine, Institute of Immunology and Immunotherapy and NIHR Birmingham Biomedical Research Centre, University Hospitals NHS Foundation Trust and University of Birmingham, Heritage Building, Mindelsohn Way, Birmingham B15 2TH, UK; Department of Emergency and Organ Transplantation, Section of Gastroenterology, University of Bari, Italy.
Subrata Ghosh, Institute Translational of Medicine, Institute of Immunology and Immunotherapy and NIHR Birmingham Biomedical Research Centre, University Hospitals NHS Foundation Trust and University of Birmingham, Birmingham, UK.
Siew C Ng, Department of Medicine and Therapeutics, Institute of Digestive Disease, State Key Laboratory of Digestive Diseases, Li Ka Shing Institute of Health Science, The Chinese University of Hong Kong, Hong Kong, China.
Julian Walters, Division of Digestive Diseases, Imperial College London, Imperial College Healthcare, London, UK.
Marietta Iacucci, Institute Translational of Medicine, Institute of Immunology and Immunotherapy and NIHR Birmingham Biomedical Research Centre, University Hospitals NHS Foundation Trust and University of Birmingham, Birmingham, UK.
References
- 1. Torres J, Mehandru S, Colombel JF, et al. Crohn’s disease. Lancet 2017; 389: 1741–1755. [DOI] [PubMed] [Google Scholar]
- 2. Ungaro R, Mehandru S, Allen PB, et al. Ulcerative colitis. Lancet 2017; 389: 1756–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Uniken Venema WT, Voskuil MD, Dijkstra G, et al. The genetic background of inflammatory bowel disease: from correlation to causality. J Pathol 2017; 241: 146–158. [DOI] [PubMed] [Google Scholar]
- 4. Chen ML, Sundrud MS. Cytokine networks and T-Cell subsets in inflammatory bowel diseases. Inflamm Bowel Dis 2016; 22: 1157–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Medrano LM, Taxonera C, Márquez A, et al. Role of TNFRSF1B polymorphisms in the response of Crohn’s disease patients to infliximab. Hum Immunol 2014; 75: 71–75. [DOI] [PubMed] [Google Scholar]
- 6. Borgström B, Lundh G, Hofmann A. The site of absorption of conjugated bile salts in man. Gastroenterology 1968; 54(Suppl): 781–783. [PubMed] [Google Scholar]
- 7. Mottacki N, Simrén M, Bajor A. Review article: bile acid diarrhoea - pathogenesis, diagnosis and management. Aliment Pharmacol Ther 2016; 43: 884–898. [DOI] [PubMed] [Google Scholar]
- 8. Hofmann AF, Poley JR. Cholestyramine treatment of diarrhea associated with ileal resection. N Engl J Med 1969; 281: 397–402. [DOI] [PubMed] [Google Scholar]
- 9. Lenicek M, Duricova D, Komarek V, et al. Bile acid malabsorption in inflammatory bowel disease: assessment by serum markers. Inflamm Bowel Dis 2011; 17: 1322–1327. [DOI] [PubMed] [Google Scholar]
- 10. Camilleri M, Nadeau A, Tremaine WJ, et al. Measurement of serum 7alpha-hydroxy-4-cholesten-3-one (or 7alphaC4), a surrogate test for bile acid malabsorption in health, ileal disease and irritable bowel syndrome using liquid chromatography-tandem mass spectrometry. Neurogastroenterol Motil 2009; 21: 734-e43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vítek L, Carey MC. Enterohepatic cycling of bilirubin as a cause of “black” pigment gallstones in adult life. Eur J Clin Invest 2003; 33: 799–810. [DOI] [PubMed] [Google Scholar]
- 12. Merrick MV, Eastwood MA, Anderson JR, et al. Enterohepatic circulation in man of a gamma-emitting bile-acid conjugate, 23-selena-25-homotaurocholic acid (SeHCAT). J Nucl Med 1982; 23: 126–130. [PubMed] [Google Scholar]
- 13. Lack L, Weiner IM. In vitro absorption of bile salts by small intestine of rats and guinea pigs. Am J Physiol 1961; 200: 313–317. [DOI] [PubMed] [Google Scholar]
- 14. Nolan JD, Johnston IM, Walters JRF. Altered enterohepatic circulation of bile acids in Crohn’s disease and their clinical significance: a new perspective. Expert Rev Gastroenterol Hepatol 2013; 7: 49–56. [DOI] [PubMed] [Google Scholar]
- 15. Jung D, Fantin AC, Scheurer U, et al. Human ileal bile acid transporter gene ASBT (SLC10A2) is transactivated by the glucocorticoid receptor. Gut 2004; 53: 78–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nolan JD, Johnston IM, Pattni SS, et al. Diarrhea in Crohn’s disease: investigating the role of the ileal hormone fibroblast growth factor 19. J Crohns Colitis 2015; 9: 125–131. [DOI] [PubMed] [Google Scholar]
- 17. Smith MJ, Cherian P, Raju GS, et al. Bile acid malabsorption in persistent diarrhoea. J R Coll Physicians Lond 2000; 34: 448–451. [PMC free article] [PubMed] [Google Scholar]
- 18. Bajor A, Kilander A, Fae A, et al. Normal or increased bile acid uptake in isolated mucosa from patients with bile acid malabsorption. Eur J Gastroenterol Hepatol 2006; 18: 397–403. [DOI] [PubMed] [Google Scholar]
- 19. Vijayvargiya P, Camilleri M, Shin A, et al. Methods for diagnosis of bile acid malabsorption in clinical practice. Clin Gastroenterol Hepatol 2013; 11: 1232–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pattni SS, Brydon WG, Dew T, et al. Fibroblast growth factor 19 and 7α-hydroxy-4-cholesten-3-one in the diagnosis of patients with possible bile acid diarrhea. Clin Transl Gastroenterol 2012; 3: e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Battat R, Duijvestein M, Vande Casteele N, et al. Serum concentrations of 7α-hydroxy-4-cholesten-3-one are associated with bile acid diarrhea in patients with Crohn’s disease. Clin Gastroenterol Hepatol 2019; 17: 2722–2730e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chande N, Patton PH, Tsoulis DJ, et al. Azathioprine or 6-mercaptopurine for maintenance of remission in Crohn’s disease. Cochrane Database Syst Rev 2015; 10: CD000067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nielsen OH, Vainer B, Rask-Madsen J. Review article: the treatment of inflammatory bowel disease with 6-mercaptopurine or azathioprine. Aliment Pharmacol Ther 2001; 15: 1699–1708. [DOI] [PubMed] [Google Scholar]
- 24. Chaparro M, Ordás I, Cabré E, et al. Safety of thiopurine therapy in inflammatory bowel disease: long-term follow-up study of 3931 patients. Inflamm Bowel Dis 2013; 19: 1404–1410. [DOI] [PubMed] [Google Scholar]
- 25. Qiu Y, Mao R, Zhang S, et al. Safety profile of thiopurines in Crohn disease: analysis of 893 patient-years follow-up in a Southern China Cohort. Medicine (Baltimore) 2015; 94: e1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kakuta Y, Kawai Y, Okamoto D, et al. NUDT15 codon 139 is the best pharmacogenetic marker for predicting thiopurine-induced severe adverse events in Japanese patients with inflammatory bowel disease: a multicenter study. J Gastroenterol 2018; 53: 1065–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liu YP, Xu HQ, Li M, et al. Association between thiopurine S-methyltransferase polymorphisms and azathioprine-induced adverse drug reactions in patients with autoimmune diseases: a meta-analysis. PLoS One 2015; 10: e0144234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Weinshilboum RM, Sladek SL. Mercaptopurine pharmacogenetics: monogenic inheritance of erythrocyte thiopurine methyltransferase activity. Am J Hum Genet 1980; 32: 651–662. [PMC free article] [PubMed] [Google Scholar]
- 29. Kim HY, Lee SH, Lee MN, et al. Complete sequence-based screening of TPMT variants in the Korean population. Pharmacogenet Genomics 2015; 25: 143–146. [DOI] [PubMed] [Google Scholar]
- 30. Ban H, Andoh A, Tanaka A, et al. Analysis of thiopurine S-methyltransferase genotypes in Japanese patients with inflammatory bowel disease. Intern Med 2008; 47: 1645–1648. [DOI] [PubMed] [Google Scholar]
- 31. Kubota T, Nishida A, Takeuchi K, et al. Frequency distribution of thiopurine S-methyltransferase activity in red blood cells of a healthy Japanese population. Ther Drug Monit 2004; 26: 319–321. [DOI] [PubMed] [Google Scholar]
- 32. Gisbert JP, Gomollón F. Thiopurine-induced myelotoxicity in patients with inflammatory bowel disease: a review. Am J Gastroenterol 2008; 103: 1783–1800. [DOI] [PubMed] [Google Scholar]
- 33. Liu Y, Meng Y, Wang L, et al. Associations between the NUDT15 R139C polymorphism and susceptibility to thiopurine-induced leukopenia in Asians: a meta-analysis. Onco Targets Ther 2018; 11: 8309–8317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Haglund S, Taipalensuu J, Peterson C, et al. IMPDH activity in thiopurine-treated patients with inflammatory bowel disease - relation to TPMT activity and metabolite concentrations. Br J Clin Pharmacol 2008; 65: 69–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Coenen MJH, de Jong DJ, van Marrewijk CJ, et al. Identification of patients with variants in TPMT and dose reduction reduces hematologic events during thiopurine treatment of inflammatory bowel disease. Gastroenterology 2015; 149: 907–917e7. [DOI] [PubMed] [Google Scholar]
- 36. Schaeffeler E, Fischer C, Brockmeier D, et al. Comprehensive analysis of thiopurine S-methyltransferase phenotype-genotype correlation in a large population of German-Caucasians and identification of novel TPMT variants. Pharmacogenetics 2004; 14: 407–417. [DOI] [PubMed] [Google Scholar]
- 37. Takatsu N, Matsui T, Murakami Y, et al. Adverse reactions to azathioprine cannot be predicted by thiopurine S-methyltransferase genotype in Japanese patients with inflammatory bowel disease. J Gastroenterol Hepatol 2009; 24: 1258–1264. [DOI] [PubMed] [Google Scholar]
- 38. Colombel JF, Ferrari N, Debuysere H, et al. Genotypic analysis of thiopurine S-methyltransferase in patients with Crohn’s disease and severe myelosuppression during azathioprine therapy. Gastroenterology 2000; 118: 1025–1030. [DOI] [PubMed] [Google Scholar]
- 39. Dewit O, Moreels T, Baert F, et al. Limitations of extensive TPMT genotyping in the management of azathioprine-induced myelosuppression in IBD patients. Clin Biochem 2011; 44: 1062–1066. [DOI] [PubMed] [Google Scholar]
- 40. Moriyama T, Nishii R, Perez-Andreu V, et al. NUDT15 polymorphisms alter thiopurine metabolism and hematopoietic toxicity. Nat Genet 2016; 48: 367–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Moriyama T, Yang YL, Nishii R, et al. Novel variants in NUDT15 and thiopurine intolerance in children with acute lymphoblastic leukemia from diverse ancestry. Blood 2017; 130: 1209–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kakuta Y, Naito T, Onodera M, et al. NUDT15 R139C causes thiopurine-induced early severe hair loss and leukopenia in Japanese patients with IBD. Pharmacogenomics J 2016; 16: 280–285. [DOI] [PubMed] [Google Scholar]
- 43. Yang SK, Hong M, Baek J, et al. A common missense variant in NUDT15 confers susceptibility to thiopurine-induced leukopenia. Nat Genet 2014; 46: 1017–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lee YJ, Hwang EH, Park JH, et al. NUDT15 variant is the most common variant associated with thiopurine-induced early leukopenia and alopecia in Korean pediatric patients with Crohn’s disease. Eur J Gastroenterol Hepatol 2016; 28: 475–478. [DOI] [PubMed] [Google Scholar]
- 45. Asada A, Nishida A, Shioya M, et al. NUDT15 R139C-related thiopurine leukocytopenia is mediated by 6-thioguanine nucleotide-independent mechanism in Japanese patients with inflammatory bowel disease. J Gastroenterol 2016; 51: 22–29. [DOI] [PubMed] [Google Scholar]
- 46. Walker GJ, Harrison JW, Heap GA, et al. Association of genetic variants in NUDT15 with thiopurine-induced myelosuppression in patients with inflammatory bowel disease. JAMA 2019; 321: 773–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Relling MV, Schwab M, Whirl-Carrillo M, et al. Clinical pharmacogenetics implementation consortium guideline for thiopurine dosing based on TPMT and NUDT15 genotypes: 2018 update. Clin Pharmacol Ther. 2019; 105: 1095–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gauba V, Saldanha M, Vize C, et al. Thiopurine methyltransferase screening before azathioprine therapy. Br J Ophthalmol 2006; 90: 923–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Fargher EA, Tricker K, Newman W, et al. Current use of pharmacogenetic testing: a national survey of thiopurine methyltransferase testing prior to azathioprine prescription. J Clin Pharm Ther. 2007; 32: 187–195. [DOI] [PubMed] [Google Scholar]
- 50. Roblin X, Oussalah A, Chevaux JB, et al. Use of thiopurine testing in the management of inflammatory bowel diseases in clinical practice: a worldwide survey of experts. Inflamm Bowel Dis 2011; 17: 2480–2487. [DOI] [PubMed] [Google Scholar]
- 51. U.S. Food & Drug Administration. Approved drug products with therapeutic equivalence evaluations (Orange Book), http://www.fda.gov/drugs/drug-approvals-and-databases/approved-drug-products-therapeutic-equivalence-evaluations-orange-book (2020, accessed 24 March 2020).
- 52. Wilson A, Jansen LE, Rose RV, et al. HLA-DQA1-HLA-DRB1 polymorphism is a major predictor of azathioprine-induced pancreatitis in patients with inflammatory bowel disease. Aliment Pharmacol Ther 2018; 47: 615–620. [DOI] [PubMed] [Google Scholar]
- 53. Heap GA, Weedon MN, Bewshea CM, et al. HLA-DQA1-HLA-DRB1 variants confer susceptibility to pancreatitis induced by thiopurine immunosuppressants. Nat Genet 2014; 46: 1131–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Van der Valk ME, Mangen MJJ, Leenders M, et al. Healthcare costs of inflammatory bowel disease have shifted from hospitalisation and surgery towards anti-TNFα therapy: results from the COIN study. Gut 2014; 63: 72–79. [DOI] [PubMed] [Google Scholar]
- 55. Allez M, Karmiris K, Louis E, et al. Report of the ECCO pathogenesis workshop on anti-TNF therapy failures in inflammatory bowel diseases: definitions, frequency and pharmacological aspects. J Crohns Colitis 2010; 4: 355–366. [DOI] [PubMed] [Google Scholar]
- 56. Vermeire S, Gils A, Accossato P, et al. Immunogenicity of biologics in inflammatory bowel disease. Ther Adv Gastroenterol. 2018; 11: 1756283X17750355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Matucci A, Pratesi S, Petroni G, et al. Allergological in vitro and in vivo evaluation of patients with hypersensitivity reactions to infliximab. Clin Exp Allergy 2013; 43: 659–664. [DOI] [PubMed] [Google Scholar]
- 58. Zidi I, Bouaziz A, Ben Amor N. Golimumab and immunogenicity? 2010 and beyond. Pharm 2011; 66: 233–243. [PubMed] [Google Scholar]
- 59. Kennedy NA, Heap GA, Green HD, et al. Predictors of anti-TNF treatment failure in anti-TNF-naive patients with active luminal Crohn’s disease: a prospective, multicentre, cohort study. Lancet Gastroenterol Hepatol 2019; 4: 341–353. [DOI] [PubMed] [Google Scholar]
- 60. Radstake TRDJ, Svenson M, Eijsbouts AM, et al. Formation of antibodies against infliximab and adalimumab strongly correlates with functional drug levels and clinical responses in rheumatoid arthritis. Ann Rheum Dis 2009; 68: 1739–1745. [DOI] [PubMed] [Google Scholar]
- 61. Garcês S, Demengeot J, Benito-Garcia E. The immunogenicity of anti-TNF therapy in immune-mediated inflammatory diseases: a systematic review of the literature with a meta-analysis. Ann Rheum Dis 2013; 72: 1947–1955. [DOI] [PubMed] [Google Scholar]
- 62. European Medicines Agency. Guideline on Immunogenicity assessment of therapeutic proteins, https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-immunogenicity-assessment-therapeutic-proteins-revision-1_en.pdf (2017).
- 63. Pedras-Vasconcelos JA. The immunogenicity of therapeutic proteins-what you don’t know cam hurt YOU and the patients. https://www.fda.gov/files/drugs/published/The-immunogenicity-of-therapeutic-proteins—what-you-don%E2%80%99t-know-can-hurt-YOU-and-the-patient—Fall-2014.pdf (2014).
- 64. Romero-Cara P, Torres-Moreno D, Pedregosa J, et al. A FCGR3A polymorphism predicts anti-drug antibodies in chronic inflammatory bowel disease patients treated with anti-TNF. Int J Med Sci 2018; 15: 10–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Billiet T, Vande Casteele N, Van Stappen T, et al. Immunogenicity to infliximab is associated with HLA-DRB1. Gut 2015; 64: 1344–1345. [DOI] [PubMed] [Google Scholar]
- 66. Sazonovs A, Kennedy NA, Moutsianas L, et al. HLA-DQA1*05 carriage associated with development of anti-drug antibodies to infliximab and adalimumab in patients with Crohn’s disease. Gastroenterology 2020; 158: 189–199. [DOI] [PubMed] [Google Scholar]
- 67. Liu M, Degner J, Davis JW, et al. Identification of HLA-DRB1 association to adalimumab immunogenicity. PLoS One 2018; 13: e0195325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lichtenstein GR, Barrett AC, Bortey E, et al. Long-term safety and tolerability of once-daily mesalamine granules in the maintenance of remission of ulcerative colitis. Inflamm Bowel Dis 2014; 20: 1399–1406. [DOI] [PubMed] [Google Scholar]
- 69. Lichtenstein GR, Kamm MA, Boddu P, et al. Effect of once- or twice-daily MMX mesalamine (SPD476) for the induction of remission of mild to moderately active ulcerative colitis. Clin Gastroenterol Hepatol 2007; 5: 95–102. [DOI] [PubMed] [Google Scholar]
- 70. Gisbert JP, Luna M, González-Lama Y, et al. Effect of 5-aminosalicylates on renal function in patients with inflammatory bowel disease: 4-year follow-up study. Gastroenterol Hepatol 2008; 31: 477–484. [DOI] [PubMed] [Google Scholar]
- 71. Van Staa TP, Travis S, Leufkens HGM, et al. 5-aminosalicylic acids and the risk of renal disease: a large British epidemiologic study. Gastroenterology 2004; 126: 1733–1739. [DOI] [PubMed] [Google Scholar]
- 72. Heap GA, So K, Weedon M, et al. Clinical features and HLA association of 5-aminosalicylate (5-ASA)-induced nephrotoxicity in inflammatory bowel disease. J Crohns Colitis. 2016; 10: 149–158. [DOI] [PubMed] [Google Scholar]
- 73. Sandborn WJ, Korzenik J, Lashner B, et al. Once-daily dosing of delayed-release oral mesalamine (400-mg tablet) is as effective as twice-daily dosing for maintenance of remission of ulcerative colitis. Gastroenterology 2010; 138: p1286–p1296E3. [DOI] [PubMed] [Google Scholar]
- 74. Sumi S, Marinaki AM, Arenas M, et al. Genetic basis of inosine triphosphate pyrophosphohydrolase deficiency. Hum Genet 2002; 111: 360–367. [DOI] [PubMed] [Google Scholar]
- 75. Marsh S, King CR, Ahluwalia R, et al. Distribution of ITPA P32T alleles in multiple world populations. J Hum Genet 2004; 49: 579–581. [DOI] [PubMed] [Google Scholar]
- 76. Smid A, Karas-Kuzelicki N, Milek M, et al. Association of ITPA genotype with event-free survival and relapse rates in children with acute lymphoblastic leukemia undergoing maintenance therapy. PLoS One 2014; 9: e109551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Arijs I, Li K, Toedter G, et al. Mucosal gene signatures to predict response to infliximab in patients with ulcerative colitis. Gut 2009; 58: 1612–1619. [DOI] [PubMed] [Google Scholar]
- 78. Schmitt H, Billmeier U, Dieterich W, et al. Expansion of IL-23 receptor bearing TNFR2+ T cells is associated with molecular resistance to anti-TNF therapy in Crohn’s disease. Gut 2019; 68: 814–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Gaujoux R, Starosvetsky E, Maimon N, et al. Cell-centred meta-analysis reveals baseline predictors of anti-TNFα non-response in biopsy and blood of patients with IBD. Gut 2019; 68: 604–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Verstockt B, Verstockt S, Blevi H, et al. TREM-1, the ideal predictive biomarker for endoscopic healing in anti-TNF-treated Crohn’s disease patients? Gut 2019; 68: 1531–1533. [DOI] [PubMed] [Google Scholar]
- 81. Carrasco K, Boufenzer A, Jolly L, et al. TREM-1 multimerization is essential for its activation on monocytes and neutrophils. Cell Mol Immunol 2019; 16: 460–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Verstockt B, Verstockt S, Dehairs J, et al. Low TREM1 expression in whole blood predicts anti-TNF response in inflammatory bowel disease. EBioMedicine 2019; 40: 733–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Jostins L, Ripke S, Weersma RK, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012; 491: 119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. West NR, Hegazy AN, Owens BMJ, et al. Oncostatin M drives intestinal inflammation and predicts response to tumor necrosis factor-neutralizing therapy in patients with inflammatory bowel disease. Nat Med 2017; 23: 579–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Garbers C, Hermanns HM, Schaper F, et al. Plasticity and cross-talk of interleukin 6-type cytokines. Cytokine Growth Factor Rev 2012; 23: 85–97. [DOI] [PubMed] [Google Scholar]
- 86. Hermanns HM. Oncostatin M and interleukin-31: cytokines, receptors, signal transduction and physiology. Cytokine Growth Factor Rev 2015; 26: 545–558. [DOI] [PubMed] [Google Scholar]
- 87. Arijs I, De Hertogh G, Lemaire K, et al. Mucosal gene expression of antimicrobial peptides in inflammatory bowel disease before and after first infliximab treatment. PLoS One 2009; 4: e7984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Sandborn WJ, Rutgeerts P, Feagan BG, et al. Colectomy rate comparison after treatment of ulcerative colitis with placebo or infliximab. Gastroenterology 2009; 137: 1250–1260; quiz 1520. [DOI] [PubMed] [Google Scholar]
- 89. Sandborn WJ, Feagan BG, Marano C, et al. Subcutaneous golimumab induces clinical response and remission in patients with moderate-to-severe ulcerative colitis. Gastroenterology 2014; 146: 85–95; quiz e14–e15. [DOI] [PubMed] [Google Scholar]
- 90. Verstockt S, Verstockt B, Vermeire S. Oncostatin M as a new diagnostic, prognostic and therapeutic target in inflammatory bowel disease (IBD). Expert Opin Ther Targets 2019; 23: 943–954. [DOI] [PubMed] [Google Scholar]
- 91. Haberman Y, Karns R, Dexheimer PJ, et al. Ulcerative colitis mucosal transcriptomes reveal mitochondriopathy and personalized mechanisms underlying disease severity and treatment response. Nat Commun 2019; 10: 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Zhou H, Xi L, Ziemek D, et al. Molecular profiling of ulcerative colitis subjects from the TURANDOT trial reveals novel pharmacodynamic/efficacy biomarkers. J Crohns Colitis 2019; 13: 702–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Dreesen E, Baert F, Laharie D, et al. Monitoring a combination of calprotectin and infliximab identifies patients with mucosal healing of Crohn’s disease. Clin Gastroenterol Hepatol 2020; 18: 637–646e11. [DOI] [PubMed] [Google Scholar]
- 94. Lee JC, Lyons PA, McKinney EF, et al. Gene expression profiling of CD8+ T cells predicts prognosis in patients with Crohn disease and ulcerative colitis. J Clin Invest 2011; 121: 4170–4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. McKinney EF, Lee JC, Jayne DRW, et al. T-cell exhaustion, co-stimulation and clinical outcome in autoimmunity and infection. Nature 2015; 523: 612–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Biasci D, Lee JC, Noor NM, et al. A blood-based prognostic biomarker in IBD. Gut 2019; 68: 1386–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Baumgart DC, Sandborn WJ. Crohn’s disease. Lancet 2012; 380: 1590–1605. [DOI] [PubMed] [Google Scholar]
- 98. Peyrin-Biroulet L, Ferrante M, Magro F, et al. Results from the 2nd scientific workshop of the ECCO. I: impact of mucosal healing on the course of inflammatory bowel disease. J Crohns Colitis 2011; 5: 477–483. [DOI] [PubMed] [Google Scholar]
- 99. Gearry RB, Barclay ML, Burt MJ, et al. Thiopurine S-methyltransferase (TPMT) genotype does not predict adverse drug reactions to thiopurine drugs in patients with inflammatory bowel disease. Aliment Pharmacol Ther 2003; 18: 395–400. [DOI] [PubMed] [Google Scholar]