

Abstract
Introduction:
Cutaneous squamous cell carcinoma (cSCC) is the second most frequent cancer; it can be locally invasive and metastatic. cSCC is an immense clinical and economic problem given its sheer incidence and potential morbidity and mortality, particularly in the elderly and immunocompromised. Epigenetics has emerged as one of the most exciting areas of human biology, impacting virtually all areas of cellular physiology. Inhibition of an epigenetic enzyme is a potential treatment of cSCC.
Areas Covered:
We provide an overview of the development of inhibitors targeting the lysine demethylase, LSD1 (KDM1A), the first histone demethylase discovered. We summarize current treatment modalities for cSCC and provide rationale for why epigenome-targeting therapies, and particularly LSD1 inhibitors, may be a novel and effective approach for treating pre-malignant and malignant cSCCs. A search was conducted in PubMed utilizing the combination of ‘LSD1’ with keywords such as ‘epidermis’, ‘squamous cell carcinoma’ or ‘skin’. Relevant papers from 2000–2020 were reviewed.
Expert Opinion:
Given the ability of LSD1 inhibitors to promote epidermal differentiation and enhance anti-tumor immune responses, LSD1 inhibitors may offer a highly effective therapeutic approach for the prevention and treatment of these ubiquitous cancers.
Keywords: Epigenetics, Histone Demethylase, LSD1, Squamous Cell Carcinoma, Skin Cancer
1. INTRODUCTION
1.1. Histone Modifiers Regulate Cellular Fate, Identity, and Disease
A person is born with genetically identical somatic cells, each containing the same DNA sequence.
Despite this, the cell types across the body have remarkable differences in form and function due to the unique activation or silencing of genes in the DNA code. These differential gene expression programs are heavily regulated through epigenetic mechanisms. Used here, epigenetics refers to those processes that regulate gene expression and cellular phenotypes independent of changes to the DNA sequence. Epigenetic regulation of gene expression is primarily mediated through alterations of chromatin structure and organization. For example, epigenetic mechanisms can make a genomic region more open and accessible tending to increase its gene expression, while compacting chromatin will tend to repress its transcription [1]. A primary level of chromatin structure is its organization into nucleosomes, the bundles of histone proteins around which DNA wraps. The post-translational modification of histones has profound impacts on chromatin structure and gene expression, making histones one of the main carriers of epigenetic information [2]. These covalent modifications are added, removed, or identified with extreme specificity by a host of well-characterized histone modifier enzymes. In line with this, histone modifiers have essential roles in regulating gene expression during organismal development, cellular differentiation, and in establishing and maintaining cellular fate and identity [3]. Disruption of these processes can result in disease, and epigenetic dysregulation is increasingly being appreciated as a driver of developmental disorders and cancer [4]. In fact, mutations in chromatin modifiers occur in approximately 50% of all human cancers and are commonly associated with poor disease prognosis [5]. Given this, and the inherently reversible nature of epigenetic modifications, there has been significant interest in targeting epigenetic disruption in disease. This has resulted in the development of numerous epigenetic inhibitors, several of which have already reached the clinic for the treatment of cancer [6].
1.2. Cutaneous Squamous Cell Carcinoma: Current Treatments and Unmet Clinical Needs
Epigenetic regulation plays a particularly crucial role in self-renewing somatic epithelia which constantly rely on a balance between proliferation and differentiation in order to maintain tissues homeostasis [7]. An example of this is the epidermis in the skin which turnovers approximately every 4 weeks in humans [8]. Here, basal layer stem cells give rise to the upper layers of the epidermis through precise coordination of multiple, stepwise differentiation programs. The product is a stratified epithelial barrier that guards the body from external insult and water loss. However, these protective capacities are not limitless, and chronic environmental damage can contribute to the pathogenesis of skin disease including cancer [9]. Indeed, increased recreational sun exposure, combined with the aging of the U.S. population, is leading to an epidemic of cutaneous neoplasia [10], [11]. Keratinocyte cancers (KCs), consisting of cutaneous squamous cell carcinoma (cSCC) and basal cell carcinoma (BCC), are the two most common of all human malignancies and collectively outnumber all other cancers combined [12]. While BCCs are typically thought to outnumber cSCCs, recent Medicare claims data in the U.S. suggests that they are equally common [11]. In line with this, the incidence of cSCC, the focus of this Review, has increased over 200 percent in the last three decades [13]. Furthermore, the premalignant neoplasms that precede the development of cSCC, known as actinic keratoses (AKs), account for over 10% of dermatology visits, have a prevalence of almost 40 million, and annual costs of over $1 billion (U.S.D.) [14]. Collectively, these data highlight the enormity of the public health and economic problem that cSCCs are, and the need to develop better strategies for prevention and treatment.
Current therapies for primary cSCC consist of standard local excision and Mohs micrographic surgery (MMS) for regions where tissue conservation is important (such as the head or neck), all with 4–6-mm margins. MMS is performed in cases of high-risk cSCC and in regions of limited tissue [15]. Other destructive methods are employed such as electrodessication and curettage for low risk cSCC. Scarring and disfigurement can therefore be quite common, underscoring the potential benefit of effective topical therapies. While topical therapies are also commonly utilized in the treatment of AKs and early cSCC limited to the epidermal layer of the skin, frequently referred to as squamous cell carcinoma in-situ (SCCis), they are limited by either a lack of efficacy or deleterious side effects that frequently hinder use. These include the antimetabolite 5-fluoruracil (5-FU), topical retinoids, immunomodulators such as imiquimod, and ingenol mebutate [16]. While 5-FU demonstrates the best efficacy [17], its use is particularly restricted by common and frequently severe localized skin reactions. Indeed, given these limitations, there remains a need for better topical therapies that may exert both a direct and local “field effect” phenomenon to treat both visible and subclinical lesions [18] (Figure 1).
Figure 1:
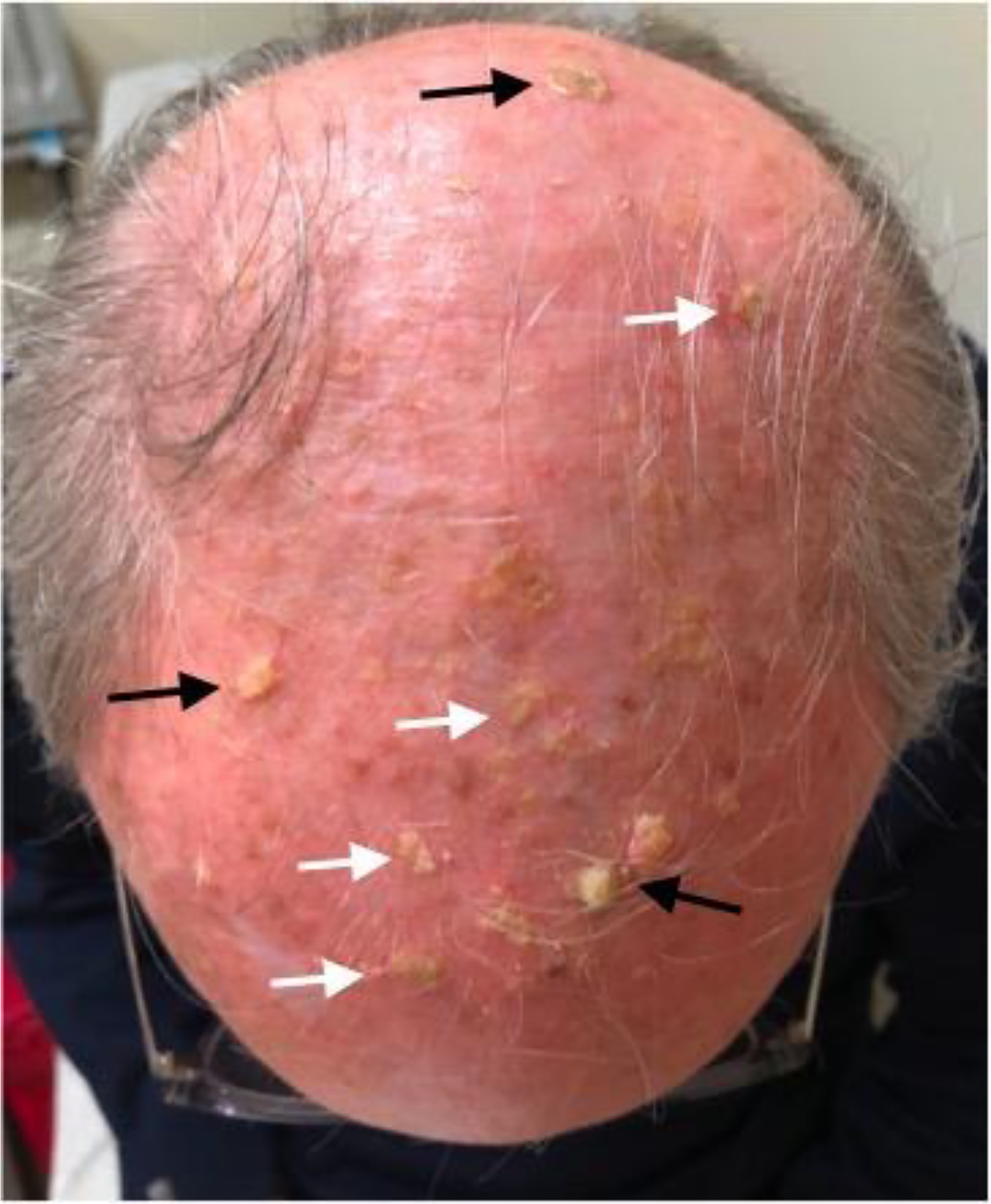
cSCC frequently arises in sun-exposed areas, particularly in the elderly and immunocompromised. Patients with numerous clinically visible and even subclinical cancerous field would particularly benefit from improved topical therapeutic options. For example, the scalp of this patient displays multiple cSCCs (black arrows) and AKs (white arrows), many of which display unclear clinical borders and thus presenting a surgical challenge. A topical therapy for the treatment of this “field” would be highly beneficial. Image published with patient consent.
More aggressive treatment is employed for advanced and metastatic cSCC. For localized lymph node metastases, surgery followed by adjuvant radiation and/or systemic therapy is recommended [15]. Patients with distant metastases typically have received cisplatin and/or systemic 5-FU chemotherapies, though epidermal growth factor receptor inhibitors such as cetuximab and panitumumab have also demonstrated some efficacy. Following the approval of pembrolizumab for patients with advanced head and neck SCC, emerging studies have looked at immunotherapy approaches for cSCC. Notably, the PD-1 inhibitor, cepilumab, was recently discovered to demonstrate some response for metastatic cSCC [19]. In all of these cases, treatments are effective in less than half of patients, and are associated with numerous side effects, highlighting the potential space and continued need for new and novel therapies.
Interestingly, chromatin and histone modifiers are among some of the most commonly mutated genes in cSCC, particularly in metastatic tumors [20], [21]. Understanding the specific transcriptional and epigenetic networks required for proper epidermal development and homeostasis, and how these may become dysregulated to promote cancer, may provide unique opportunity for the use of epigenetic therapy in cSCC. The epigenetic enzyme lysine specific demethylase 1 (LSD1 or KDM1A) is a histone demethylase and is overexpressed in numerous human malignancies which often associates with poor disease prognosis [22]–[30]. In the past decade there has been a rapid development of small molecule inhibitors to target LSD1 activity several of which are currently being tested in clinical trial for the treatment of cancer [31]. Given this, there is significant interest in determining if LSD1 inhibitors may be used therapeutically in other cancers. Here, we discuss the potential of LSD1 as a viable therapeutic target in cSCC, reviewing the historical features and functions of this protein as well as advances in LSD1 inhibitors that make it an attractive therapeutic target and how this may be particularly amenable as a therapy to treat cSCC.
2. Physiological and Oncogenic Functions of LSD1
Since its discovery just over 15 years ago, LSD1 has made landmark changes to our understanding of histone methylation during basic biological processes as well as cancer. Indeed, histone methylation was originally considered to be an irreversible process until the identification of LSD1. This provided the first irrefutable evidence that histone methylation is dynamic and catalyzed the discovery of the over ~20 known histone demethylases today. Consequently, LSD1 is an extremely well-investigated protein among epigenetic regulators. Hence, we will simply highlight some of the most fundamental of these findings, referring the reader to the following reviews for more in depth reading on other topics [23], [32]–[34]
LSD1 plays key roles during numerous biological processes across many tissues during mammalian development, including stem cell maintenance, differentiation, metabolism, and epithelial-to-mesenchymal transition (EMT) [35]–[39]. These diverse functions are frequently context-specific and primarily mediated through LSD1 association with unique binding partners that alter LSD1 substrate specificity, protein complex incorporation, and genomic localization. Consequently, LSD1 functions in various ways acting via 1) transcriptional repression by removing mono- and dimethyl group from histone 3 lysine 4 (H3K4) [40]; 2) transcriptional activation by removing repressive methylation marks on H3 lysine 9 (H3K9) in a small number of contexts [41]–[44]; 3) non-histone demethylation to regulate proteins such as p53, DNMT1, E2F1, STAT3, HIF1α and MYPT1[38], [45]–[48]; and, more recently, 4) non-catalytic functions, for example, altering the stabilization of interacting partners including the protein FBXW7 [49]. Given all of these functions, it is perhaps unsurprising that knockout of LSD1 in mice is lethal at embryonic day 6 suggesting this protein plays a key role during development [38], [39]. A central function of LSD1 across many tissues is the maintenance of stem cell or progenitor-like states [50]. In undifferentiated embryonic stem cells, LSD1 is highly expressed and represses the expression of lineage specific developmental programs [51]. LSD1 plays a similar role and is also required for proper differentiation in myocytes, neurons, adipocytes, and hematopoietic progenitors.
In addition to directing normal cellular functions, LSD1 is crucial to many oncogenic processes in cancer. In acute myeloid leukemia (AML), LSD1 is frequently overexpressed and associated with poor prognosis in patients. Mouse models of AML have revealed that LSD1 is required for disease development and functions to maintain the stem cell progenitor state of leukemia cells. Inhibiting LSD1 in AML mouse models and cell lines through genetic deletion or use of pharmacological inhibitors impairs proliferation and increases differentiation and apoptosis[52], [53]. These findings have advanced to clinical trials where multiple LSD1 inhibitors are currently being tested for treating AML. LSD1 has also been studied in the context of solid tumors, for example in breast and prostate cancers. Similar to hematological malignancies, LSD1 overexpression promotes cellular proliferation and is associated with aggressive phenotypes in these cancers. However, these studies also highlight an additional oncogenic function of LSD1 showing that it plays a role in mediating EMT and cancer cell migration and invasion. [24], [30], [44], [54], [55] Consistently, studies show that LSD1 binds with the Snail/Slug family transcription factors which promote cancer cell metastasis and LSD1 inhibitors can disrupt these interactions to block cancer cell invasion [56], [57]. It has also been recently discovered that LSD1 is key for suppressing endogenous double stranded RNA (dsRNA) levels and interferon responses in tumor cells. Inhibiting LSD1 leads to improved anti-tumor immune responses and causes refractory mouse melanoma tumors to respond to anti-PD-1 therapy [58]. This opens up a wide range of new potential therapeutic applications for LSD1 inhibitors given the recent successes of immunotherapies. Collectively, these data demonstrate the multifaceted oncogenic functions of LSD1 in cancer and highlight its status as an attractive therapeutic target.
3. Pharmacologically Targeting LSD1
The demethylase activity of LSD1 lies within its enzymatic FAD-dependent amine oxidase-like domain which shares structural homology with other flavin-containing monoamine oxidase (MAO) enzymes. Because of this, the first attempts to inhibit LSD1 activity utilized existing MAO inhibitors such as tranylcypromine (TCP) which is used clinically for treating depression[32], [59]. TCP sufficiently inhibits LSD1 activity by forming a covalent adduct with FAD in the active site of LSD1 to irreversibly inhibit enzyme function [60]. However, TCP has less than desirable specificity and potency for use in targeting LSD1 therapeutically, which has resulted in the development of numerous, highly specific LSD1 inhibitors. These can be broadly classified into two categories including inhibitors that, similarly to TCP, work via an irreversible mechanism of action or those that reversibly inhibit LSD1. Several LSD1 inhibitors are currently undergoing clinical trial either for applications in cancer or other diseases including 1) TCP; 2) ORY-1001 [61]; 3) IMG-7289; 4) INCB059872; 5) ORY-2001; and 6) CC-90011. A very recent, detailed description of each of these drugs and their advances in the clinic has been extensively reviewed [31]. Most of these inhibitors (2–5) are TCP derivatives and contain a shared primary molecular scaffold that irreversibly inhibits LSD1 by forming covalent FAD-adducts. However, as mentioned, relative to TCP they have better specificity and potency attributed to unique chemical modifications added to the TCP-scaffold phenyl ring and amine group [31]. CC-90011 (6, above) is the only reversible LSD1 inhibitor to reach the clinic, but highlights the potential of this broad category of compounds which attempt to curtail any enduring off-target effects that irreversible inhibitors can suffer from [62]. Generally reversible inhibitors, including CC-90011, have distinct molecular structures and mechanisms of action from TCP. For example, the reversible LSD1 inhibitor SP2509 is thought to function by reducing LSD1 binding with the Co-REST complex to hinder its demethylase functions. [63]
Among the LSD1 inhibitors being clinically tested, ORY-1001 is perhaps best described and has shown to be both highly selective and potent, inhibiting LSD1 activity with an IC50 of less than 20 nM [61]. Phase IIa clinical trials for ORY-1001 are currently underway for its use in AML. Despite the exciting potential of these clinical developments, three phase I clinical trials using the LSD1 inhibitor GSK-2879552, a TCP-analog that works irreversibly, have recently been terminated because of unfavorable risk benefit tied to drug side effects. This brings attention to the drawbacks of the systemic use of broad-based epigenetic inhibitors and the need for advances in the delivery strategies of these drugs to their target sites [64].
Although potentially effective as monotherapies, LSD1 inhibitors likely hold the most promising potential through their use in combination therapy. Indeed, it is well known that epigenetic drugs often produce synergistic effects when used in combination with other anti-cancer therapies [65]. As mentioned, one exciting combination that likely holds tremendous therapeutic potential is that of LSD1 inhibitors with PD-L1 blockade to improve anti-tumor immune responses [58]. In addition to this, others have observed synergistic effects of LSD1 inhibitors when combined with other known epigenetic inhibitors such as azacitidine, a clinically approved DNMT1 inhibitor, or histone deacetylase (HDAC) inhibitors[61]. Interestingly, others have taken this strategy even further by developing single molecule, dual action LSD1/HDAC inhibitors to comprehensively target these epigenetic enzyme complexes [66]. The promising anti-tumor properties of such compounds, and added benefits of a single molecule approach clinically, speak to the potential of this strategy and is an important consideration when considering LSD1 as a therapeutic target. Finally, LSD1 inhibitors are able to sensitize non-acute promyelocytic leukemias to treatment with all-trans-retinoic-acid, a strategy that is actively being tested in clinical trials [67].
Many existing LSD1 inhibitors, particularly TCP-analogs, were primarily designed to target the catalytic activity of this protein. However, when considering LSD1 as a pharmacological target it is also necessary to acknowledge its emerging noncatalytic roles [49], [68]. Indeed, an abundance of recent findings have demonstrated the array of critical, noncatalytic functions of histone modifiers during development and disease [69]. Interestingly, there have been several reports that existing LSD1 inhibitors both inhibit LSD1 catalytic activity and prevent its binding to genomic targets [68], [70]. This raises the question of whether the effects of LSD1 inhibitors are completely tied to inhibiting LSD1 catalytic activity or if disruption of noncatalytic functions of LSD1 may play a role in their anti-tumorigenic effects. Future work to more directly characterize the noncatalytic mechanisms of LSD1 and more directly inhibit these functions using pharmacological approaches will likely expand the already enormous therapeutic potential of targeting this protein.
4. CONCLUSION
This body of literature demonstrates the multifaceted biological functions of LSD1 during development, differentiation, and disease, particularly cancer. Together, the diverse range of mechanisms by which LSD1 promotes tumorigenesis and its frequent overexpression in cancer contributes significantly to its emergence as a promising and innovative clinical target. This interest has been reflected in the multitude of LSD1 inhibitors developed in the past few years many of which are being tested in clinical trials. Given all this, it is of significant interest to continue to expand our knowledge of this protein across a broad range of tissues and disease contexts in order to leverage its attractive qualities as a drug target. To this end, below we provide our expert opinion on the potential of LSD1 as a therapeutic target for cSCC.
5. EXPERT OPINION
LSD1 as a therapeutic target in cSCC
One of the first indications that LSD1 inhibitors may be used to treat cSCC came from data using human cSCC cell lines. Significantly, this study showed that two independent, human cSCC cell lines were sensitive to LSD1 inhibitors alone or in combination with HDAC inhibitors as measured using proliferation assays. Notably, cSCC cell lines were most sensitive to a single molecule, dual action LSD1/HDAC inhibitor. When tested in primary human keratinocytes this dual action inhibitor was also better tolerated than single agent LSD1 inhibitors [66]. This highlights the sensitivity of human cSCC cell lines to LSD1 inhibition and is an effect that may be exacerbated by comprehensively targeting epigenetic complexes containing LSD1 and HDACs. These data point to the need to understand how LSD1 may promote cSCC tumorigenesis as well as the basic biological roles of this protein in the epidermis.
Indeed, how LSD1 function in normal epithelial tissues or other squamous cell carcinomas (SCCs) remains a major gap in knowledge. This is particularly surprising given that LSD1 is commonly overexpressed and known to associate with poor prognosis in SCCs of the esophagus [27], [28], oral mucosa [26], [29], and lung [25] (Figure 2). Further, the opposing H3K4 histone methyltransferases, KMT2C (MLL3) and KMT2D (MLL4), display exceptionally high rates of loss-of-function mutations in SCCs with the highest rate of mutation occurring in cSCC (>50%) [20], [21]. Importantly, cSCC shares common mutational and transcriptional underpinnings with all other forms of SCC which together form a “pan-squamous” group due to their unified histological and genetic features [71], [72]. These genetic and clinical data support the idea that LSD1 may be a promising therapeutic target in cSCC and warrant further study of how KMT2C/D-LSD1 epigenetic dysregulation promotes cSCC. Given the incredible accessibility of the skin this knowledge may also provide critical insight into these other, often difficult to study SCCs.
Figure 2:
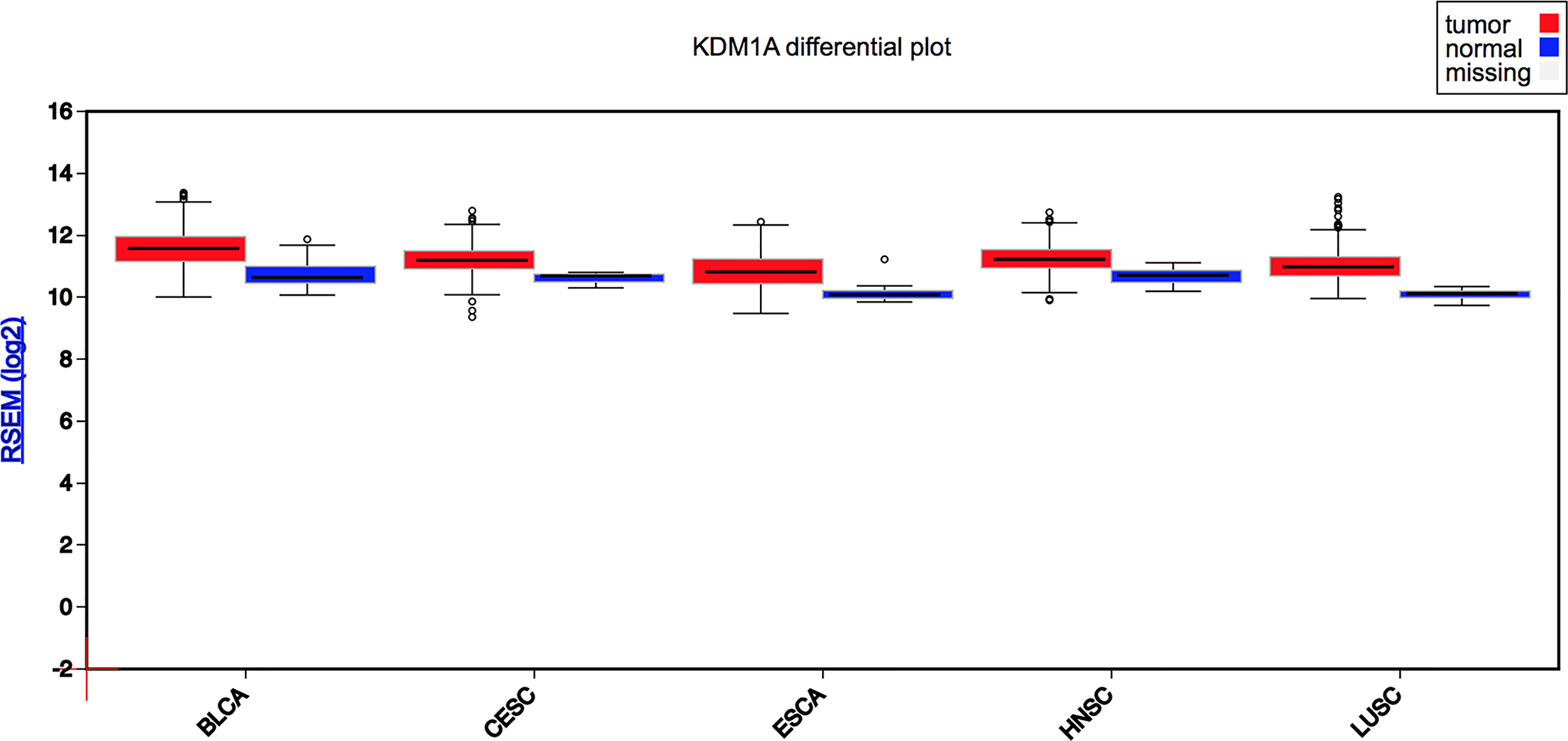
LSD1 (KDM1A) is typically overexpressed in human cancers, and particularly in all forms of SCC as measure by RNA-seq (BLCA = Bladder Cancer; CESC = Cervical SCC; ESCA = Esophageal SCC; HNSC = Head and Neck SCC; LUSC = Lung SCC; Source: FireBrowse from The Cancer Genome Atlas, TCGA). It should be noted that there is no gene expression data for cSCC in the TCGA at this time.
To this end, recent work done in epidermal progenitors (EPs), has demonstrated that pharmacological inhibition or genetic knockdown of LSD1 significantly upregulates gene expression programs involved in epidermal cell differentiation, development of the epidermis, and establishment of the skin barrier [73]. Interestingly, transcription factors that play key roles in altering cellular fate in the epidermis by promoting differentiation such GRHL1, GRHL3, NOTCH3, and KLF4 were among those genes upregulated by LSD1 inhibition. These data suggest LSD1 plays a key role in maintaining the stem-cell-like epidermal progenitor state by inhibiting the expression of key, fate-determining pro-differentiation transcription factors in the epidermis. Mechanistically, this is likely mediated through a cooperative interaction between LSD1 and SNAI2 given that LSD1 inhibition in EPs specifically reduces LSD1 occupation, increases H3K4 methylation, and increases expression at known SNAI2-repressed epidermal differentiation genes. LSD1 inhibition also had profound effects in a 3D-invasive organotypic (OTC) model of human cutaneous squamous cell carcinoma. Specifically, inhibiting LSD1 in this model significantly reduces cSCC growth as well as invasiveness into the dermis [73] (Figure 3). Like normal epidermal progenitors, cSCC organotypics treated with LSD1 inhibitor have increased levels of pro-differentiation transcription factors. However, given the multi-faceted oncogenic roles of LSD1 in cancer and the well-established roles of SNAI2 during EMT, it is likely that LSD1 likely works through diverse mechanisms to inhibit cSCC growth and invasion.
Figure 3:
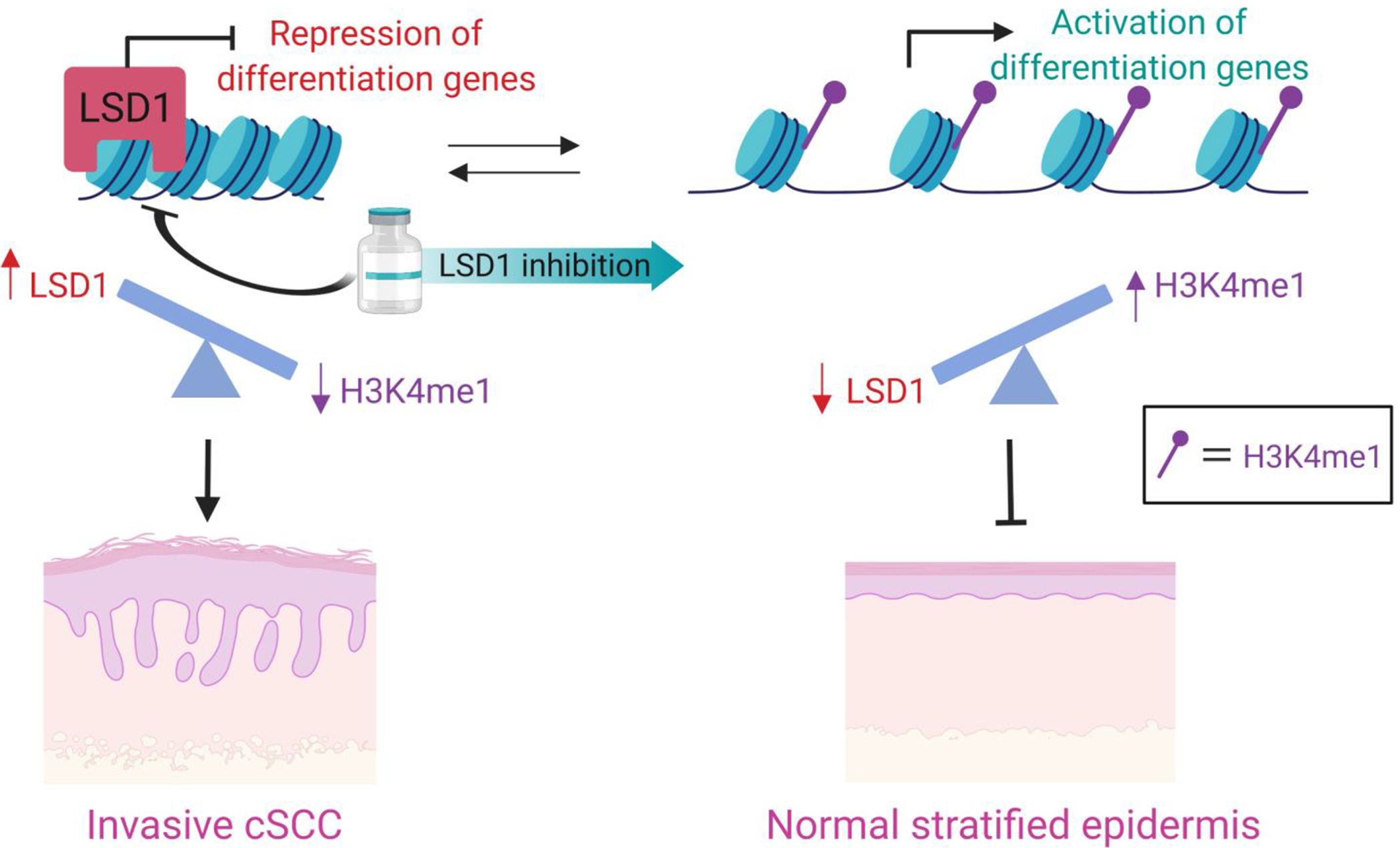
In the epidermis, LSD1 typically demethylates H3K4me1/2 to close chromatin at gene enhancers and promoters. This drives the repression of the expression of key pro-differentiation transcription factors. Consistent with this, overexpression of LSD1 in cancers and/or the frequent loss of function mutations in the major H3K4me1 methyltransferases, KMT2C and KMT2D (observed in more than 50% of cSCCs), can similarly tip the balance to promote a loss of H3K4me1 and, in turn, the expression of these differentiation genes. Loss of epidermal differentiation is a key driver of stemness and squamous carcnogenesis. By utilizing LSD1 inhibitors to reduce LSD1 function and activity, H3K4me1 can be increased and the expression of differentiation genes restored. This approach has been demonstrated to repress cSCC invasion in human organoid models and lead to a more normal epidermis.
The ability to repress invasiveness in cSCC is important given that the vast majority of disease-associated lethality occurs in patients with metastatic cSCC tumors[13]. These results provide significant rationale for testing the potential of LSD1 inhibitors to treat cSCC in vivo. Given the ability of LSD1 inhibition to activate anti-tumor immune responses, in vivo studies may also reveal how LSD1 inhibitors may be used to leverage the immune system and immune based therapies in cSCC. This approach may be particularly amenable to cSCC given the acknowledged importance of immune dysfunction in driving cSCC formation and relative success of immune-based therapies in this disease. The effect of LSD1 inhibitors in cSCC may be further enhanced by coupling LSD1 inhibitors with PD-1 blockade given the synergistic effects observed previously between these drugs in a mouse model of melanoma[58]. This could greatly potentiate the therapeutic potential of PD-1 blockade in cSCC patients given the limited efficacy of this therapy alone. LSD1 inhibition may also be especially well-suited as a treatment in cSCC given the direct accessibility of the skin and ability to deliver drugs topically which may avoid the negative side effects associated with systemic delivery of LSD1 inhibitors. Finally, given the huge clinical and economic burden presented by AKs, the ability of LSD1 inhibitors to promote epidermal differentiation and potentiate local immunosurveillance could represent a highly effective topical strategy to treat AKs and early SCCis, exerting both a local and field effect on both neoplasms and even subclinical clonal proliferations.
Article Highlights.
Cutaneous squamous cell carcinoma (cSCC) is the second most common human malignancy and has significant unmet clinical needs given the sheer enormity of the clinical incidence and economic burden of the disease.
Epigenetic therapy through inhibition of the histone demethylase LSD1 may offer a novel strategy for cSCC given its ability to promote epidermal differentiation and potentiate antitumor immunity.
Numerous LSD1 inhibitors have been developed and are at various stages of development, and these compounds offer a level of specificity not observed with many other epigenetic drugs.
Preclinical data demonstrates that LSD1 inhibitors can activate differentiation gene expression programs and repress cSCC invasion in human organoid models.
The ability to target LSD1 inhibitors directly onto the skin for the treatment of both premalignant (actinic keratoses) and malignant cSCCs may avoid some of the toxicities observed with systemic deliveries of these compounds.
Funding
The work of the authors was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) (K08AR070289), the Damon Runyon Cancer Foundation, the Dermatology Foundation, the Penn Head and Neck Cancer Center, the Penn Center for Excellence in Environmental Toxicology, and the Penn Epigenetics Institute, all to B.C.C., as well as the Penn Skin Biology and Diseases Resource-based Center (NIAMS 1P30AR069589–01). S.E. is supported by NIAMS (T32AR007465).
Footnotes
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
References
Papers of special note have been highlighted as either of interest (*) or of considerable interest (**) to readers
- [1].Allis CD and Jenuwein T, “The molecular hallmarks of epigenetic control,” Nat. Rev. Genet, vol. 17, no. 8, pp. 487–500, 2016, doi: 10.1038/nrg.2016.59. [DOI] [PubMed] [Google Scholar]
- [2].Lawrence M, Daujat S, and Schneider R, “Lateral Thinking: How Histone Modifications Regulate Gene Expression,” Trends Genet, vol. 32, no. 1, pp. 42–56, 2016, doi: 10.1016/j.tig.2015.10.007. [DOI] [PubMed] [Google Scholar]
- [3].Atlasi Y and Stunnenberg HG, “The interplay of epigenetic marks during stem cell differentiation and development,” Nat. Rev. Genet, vol. 18, no. 11, pp. 643–658, 2017, doi: 10.1038/nrg.2017.57. [DOI] [PubMed] [Google Scholar]
- [4].Rickels R and Shilatifard A, “Enhancer Logic and Mechanics in Development and Disease,” Trends Cell Biol, vol. 28, no. 8, pp. 608–630, 2018, doi: 10.1016/j.tcb.2018.04.003. [DOI] [PubMed] [Google Scholar]
- [5].Flavahan WA, Gaskell E, and Bernstein BE, “Epigenetic plasticity and the hallmarks of cancer,” Science (80-.)., vol. 357, no. 6348, 2017, doi: 10.1126/science.aal2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Biswas S and Rao CM, “Epigenetic tools (The Writers, The Readers and The Erasers) and their implications in cancer therapy,” Eur. J. Pharmacol, vol. 837, no. June, pp. 8–24, 2018, doi: 10.1016/j.ejphar.2018.08.021. [DOI] [PubMed] [Google Scholar]
- [7].Avgustinova A and Benitah SA, “Epigenetic control of adult stem cell function,” Nat. Rev. Mol. Cell Biol, vol. 17, no. 10, pp. 643–658, 2016, doi: 10.1038/nrm.2016.76. [DOI] [PubMed] [Google Scholar]
- [8].Gonzales K and Fuchs E, “Skin and its regenerative powers: an alliance between stem cells and their niches,” Dev. Cell, vol. 43, no. 4, pp. 387–401, 2017, doi: 10.1016/j.devcel.2017.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Valacchi G, Sticozzi C, Pecorelli A, Cervellati F, Cervellati C, and Maioli E, “Cutaneous responses to environmental stressors,” Ann. N. Y. Acad. Sci, vol. 1271, no. 1, pp. 75–81, 2012, doi: 10.1111/j.1749-6632.2012.06724.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Guy G, Machlin SR, Ekwueme DU, and Yabroff R, “Prevalence and Costs of Skin Cancer Treatment in the U.S., 2002–2006 and 2007–2011 Gery,” Am J Prev, vol. 48, no. 2, pp. 183–187, 2015, doi: 10.1016/j.amepre.2014.08.036.Prevalence. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Rogers HW, Weinstock MA, Feldman SR, and Coldiron BM, “Incidence estimate of nonmelanoma skin cancer (keratinocyte carcinomas) in the us population, 2012,” JAMA Dermatology, vol. 151, no. 10, pp. 1081–1086, 2015, doi: 10.1001/jamadermatol.2015.1187. [DOI] [PubMed] [Google Scholar]
- [12].Nehal KS and Bichakjian CK, “Update on keratinocyte carcinomas,” N. Engl. J. Med, vol. 379, no. 4, pp. 363–374, 2018, doi: 10.1056/NEJMra1708701. [DOI] [PubMed] [Google Scholar]; *Authoritative review describing our current understanding of keratinocyte cancers such as cSCC.
- [13].Karia P, Han J, and Schmults C, “Cutaneous squamous cell carcinoma: incidence of disease, nodal metastasis, and deaths from disease in the United States, 2012,” J Am Acad Dermatol, vol. 68, pp. 957–966, 2013. [DOI] [PubMed] [Google Scholar]
- [14].Siegel JA, Korgavkar K, and Weinstock MA, “Current perspective on actinic keratosis: a review,” Br. J. Dermatol, vol. 177, no. 2, pp. 350–358, 2017, doi: 10.1111/bjd.14852. [DOI] [PubMed] [Google Scholar]
- [15].Work G et al. , “Guidelines of care for the management of cutaneous squamous cell carcinoma,” J Am Acad Dermatol, vol. 78, no. 3, pp. 560–578, 2019, doi: 10.1016/j.jaad.2017.10.007.Guidelines. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Micali G, Lacarrubba F, Nasca M, and Schwartz R, “Topical pharmacotherapy for skin cancer: part I. Pharmacology,” J Am Acad Dermatol, vol. 70, no. 6, pp. e961–912; quiz 977–968, 2014. [DOI] [PubMed] [Google Scholar]
- [17].Jansen MHE et al. , “Randomized trial of four treatment approaches for actinic keratosis,” N. Engl. J. Med, vol. 380, no. 10, pp. 935–946, 2019, doi: 10.1056/NEJMoa1811850. [DOI] [PubMed] [Google Scholar]
- [18].Curtius K, Wright NA, and Graham TA, “An evolutionary perspective on field cancerization,” Nat. Rev. Cancer, vol. 18, no. 1, pp. 19–32, 2017, doi: 10.1038/nrc.2017.102. [DOI] [PubMed] [Google Scholar]
- [19].Migden MR et al. , “PD-1 blockade with cemiplimab in advanced cutaneous squamous-cell carcinoma,” N. Engl. J. Med, vol. 379, no. 4, pp. 341–351, 2018, doi: 10.1056/NEJMoa1805131. [DOI] [PubMed] [Google Scholar]
- [20].Yilmaz AS et al. , “Differential mutation frequencies in metastatic cutaneous squamous cell carcinomas versus primary tumors,” Cancer, vol. 123, no. 7, pp. 1184–1193, 2017, doi: 10.1002/cncr.30459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Li Y, Hanna G, Laga A, Haddad R, Lorch J, and Hammerman P, “Genomic analysis of metastatic cutaneous squamous cell carcinoma,” Clin Cancer Res, vol. 21, no. 6, pp. 1447–1456, 2015, doi: 10.1158/1078-0432.CCR-14-1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ding J et al. , “LSD1-mediated epigenetic modification contributes to proliferation and metastasis of colon cancer,” Br. J. Cancer, vol. 109, no. 4, pp. 994–1003, 2013, doi: 10.1038/bjc.2013.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Hosseini A and Minucci S, “A comprehensive review of lysine-specific demethylase 1 and its roles in cancer,” Epigenomics, vol. 9, no. 8, pp. 1123–1142, 2017, doi: 10.2217/epi-2017-0022. [DOI] [PubMed] [Google Scholar]
- [24].Li X et al. , “Overexpression of lysine-specific demethylase 1 promotes androgen-independent transition of human prostate cancer LNCaP cells through activation of the AR signaling pathway and suppression of the p53 signaling pathway,” Oncol. Rep, vol. 35, no. 1, pp. 584–592, 2016, doi: 10.3892/or.2015.4362. [DOI] [PubMed] [Google Scholar]
- [25].Lv T et al. , “Over-expression of LSD1 promotes proliferation, migration and invasion in non-small cell lung cancer,” PLoS One, vol. 7, no. 4, pp. 1–8, 2012, doi: 10.1371/journal.pone.0035065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Yuan C, Li Z, Qi B, Zhang W, Cheng J, and Wang Y, “High expression of the histone demethylase LSD1 associates with cancer cell proliferation and unfavorable prognosis in tongue cancer,” J. Oral Pathol. Med, vol. 44, no. 2, pp. 159–165, 2015, doi: 10.1111/jop.12220. [DOI] [PubMed] [Google Scholar]
- [27].Hoshino I et al. , “Histone Demethylase LSD1 Inhibitors Prevent Cell Growth by Regulating Gene Expression in Esophageal Squamous Cell Carcinoma Cells,” Ann. Surg. Oncol, vol. 23, no. 1, pp. 312–320, 2016, doi: 10.1245/s10434-015-4488-1. [DOI] [PubMed] [Google Scholar]
- [28].Hou G et al. , “LSD1 regulates Notch and PI3K/Akt/mTOR pathways through binding the promoter regions of notch target genes in esophageal squamous cell carcinoma,” Onco. Targets. Ther, vol. 12, pp. 5215–5225, 2019, doi: 10.2147/OTT.S207238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Alsaqer SF et al. , “Inhibition of LSD1 epigenetically attenuates oral cancer growth and metastasis,” Oncotarget, vol. 8, no. 43, pp. 73372–73386, 2017, doi: 10.18632/oncotarget.19637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lim S et al. , “Lysine-specific demethylase 1 (LSD1) is highly expressed in ER-negative breast cancers and a biomarker predicting aggressive biology.,” Carcinogenesis, vol. 31, pp. 512–520, 2010. [DOI] [PubMed] [Google Scholar]
- [31].Fang Y, Liao G, and Yu B, “LSD1/KDM1A inhibitors in clinical trials: Advances and prospects,” J. Hematol. Oncol, vol. 12, no. 1, pp. 1–14, 2019, doi: 10.1186/s13045-019-0811-9. [DOI] [PMC free article] [PubMed] [Google Scholar]; *Detailed review describing the LSD1 inhibitors currently in clincal trials.
- [32].Zheng Y-C et al. , “A systematic review of histone lysine-specific demethylase 1 and its inhibitors,” Med. Res. Rev, vol. 35, no. 5, pp. 1032–1071, 2015, doi: 10.1002/med. [DOI] [PubMed] [Google Scholar]
- [33].Maiques-Diaz A and Somervaille TCP, “LSD1: Biologic roles and therapeutic targeting,” Epigenomics, vol. 8, no. 8, pp. 1103–1116, 2016, doi: 10.2217/epi-2016-0009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Yang GJ, Lei PM, Wong SY, Ma DL, and Leung CH, “Pharmacological inhibition of LSD1 for cancer treatment,” Molecules, vol. 23, no. 12, pp. 1–20, 2018, doi: 10.3390/molecules23123194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Whyte WA et al. , “Enhancer decommissioning by LSD1 during embryonic stem cell differentiation,” Nature, vol. 482, no. 7384, pp. 221–225, 2014, doi: 10.1038/nature10805. [DOI] [PMC free article] [PubMed] [Google Scholar]; *First description of how LSD1 can repress differentiation through enhancer decommissioning.
- [36].Ambrosio S, Saccà CD, and Majello B, “Epigenetic regulation of epithelial to mesenchymal transition by the Lysine-specific demethylase LSD1/KDM1A,” Biochim. Biophys. Acta - Gene Regul. Mech, vol. 1860, no. 9, pp. 905–910, 2017, doi: 10.1016/j.bbagrm.2017.07.001. [DOI] [PubMed] [Google Scholar]
- [37].Sakamoto A et al. , “Lysine demethylase LSD1 coordinates glycolytic and mitochondrial metabolism in hepatocellular carcinoma cells,” Cancer Res, vol. 75, no. 7, pp. 1445–1456, 2015, doi: 10.1158/0008-5472.CAN-14-1560. [DOI] [PubMed] [Google Scholar]
- [38].Wang J et al. , “The lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylation,” Nat. Genet, vol. 41, no. 1, pp. 125–129, 2009, doi: 10.1038/ng.268. [DOI] [PubMed] [Google Scholar]
- [39].Wang J et al. , “Opposing LSD1 complexes function in developmental gene activation and repression programmes,” Nature, vol. 446, no. 7138, pp. 882–887, 2007, doi: 10.1038/nature05671. [DOI] [PubMed] [Google Scholar]
- [40].Shi Y et al. , “Histone demethylation mediated by the nuclear amine oxidase homolog LSD1,” Cell, vol. 119, no. 7, pp. 941–953, 2004, doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]; **Landmark paper that published the discovery of LSD1.
- [41].Metzger E et al. , “LSD1 demethylates repressive histone marks to promote androgen-receptor- dependent transcription,” Nature, vol. 437, no. 7057, pp. 436–439, 2005, doi: 10.1038/nature04020. [DOI] [PubMed] [Google Scholar]
- [42].Laurent B et al. , “A specific LSD1/KDM1A isoform regulates neuronal differentiation through H3K9 demethylation,” Mol. Cell, vol. 57, no. 6, pp. 957–970, 2015, doi: 10.1016/j.physbeh.2017.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].He Y et al. , “LSD1 promotes S-phase entry and tumorigenesis via chromatin co-occupation with E2F1 and selective H3K9 demethylation,” Oncogene, vol. 37, no. 4, pp. 534–543, 2018, doi: 10.1038/onc.2017.353. [DOI] [PubMed] [Google Scholar]
- [44].Carnesecchi J et al. , “ERRα induces H3K9 demethylation by LSD1 to promote cell invasion,” Proc. Natl. Acad. Sci. U. S. A, vol. 114, no. 15, pp. 3909–3914, 2017, doi: 10.1073/pnas.1614664114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Huang J et al. , “p53 is regulated by the lysine demethylase LSD1,” Nature, vol. 449, no. 7158, pp. 105–108, 2007, doi: 10.1038/nature06092. [DOI] [PubMed] [Google Scholar]
- [46].Kontaki H and Talianidis I, “Lysine Methylation Regulates E2F1-Induced Cell Death,” Mol. Cell, vol. 39, no. 1, pp. 152–160, 2010, doi: 10.1016/j.molcel.2010.06.006. [DOI] [PubMed] [Google Scholar]
- [47].Yang J et al. , “Reversible methylation of promoter-bound STAT3 by histone-modifying enzymes,” Proc. Natl. Acad. Sci. U. S. A, vol. 107, no. 50, pp. 21499–21504, 2010, doi: 10.1073/pnas.1016147107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Cho HS et al. , “Demethylation of RB regulator MYPT1 by histone demethylase LSD1 promotes cell cycle progression in cancer cells,” Cancer Res, vol. 71, no. 3, pp. 655–660, 2011, doi: 10.1158/0008-5472.CAN-10-2446. [DOI] [PubMed] [Google Scholar]
- [49].Lan H et al. , “LSD1 destabilizes FBXW7 and abrogates FBXW7 functions independent of its demethylase activity,” Proc. Natl. Acad. Sci. U. S. A, vol. 116, no. 25, pp. 12311–12320, 2019, doi: 10.1073/pnas.1902012116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Karakaidos P, Verigos J, and Magklara A, “Lsd1/kdm1a, a gate-keeper of cancer stemness and a promising therapeutic target,” Cancers (Basel), vol. 11, no. 12, pp. 1–21, 2019, doi: 10.3390/cancers11121821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Adamo A et al. , “LSD1 regulates the balance between self-renewal and differentiation in human embryonic stem cells,” Nat. Cell Biol, vol. 13, no. 6, pp. 652–661, 2011, doi: 10.1038/ncb2246. [DOI] [PubMed] [Google Scholar]
- [52].Somervaille TCP and Cleary ML, “Identification and characterization of leukemia stem cells in murine MLL-AF9 acute myeloid leukemia,” Cancer Cell, vol. 10, no. 4, pp. 257–268, 2006, doi: 10.1016/j.ccr.2006.08.020. [DOI] [PubMed] [Google Scholar]
- [53].Harris WJ et al. , “The Histone Demethylase KDM1A Sustains the Oncogenic Potential of MLL-AF9 Leukemia Stem Cells,” Cancer Cell, vol. 21, no. 4, pp. 473–487, 2012, doi: 10.1016/j.ccr.2012.03.014. [DOI] [PubMed] [Google Scholar]
- [54].Ketscher A et al. , “LSD1 controls metastasis of androgen-independent prostate cancer cells through PXN and LPAR6,” Oncogenesis, vol. 3, no. May, 2014, doi: 10.1038/oncsis.2014.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Serce N, Gnatzy A, Steiner S, Lorenzen H, Kirfel J, and Buettner R, “Elevated expression of LSD1 (Lysine-specific demethylase 1) during tumour progression from pre-invasive to invasive ductal carcinoma of the breast,” BMC Clin. Pathol, vol. 12, no. 1, p. 1, 2012, doi: 10.1186/1472-6890-12-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Ferrari-Amorotti G et al. , “Suppression of Invasion and Metastasis of Triple-Negative Breast Cancer Lines by Pharmacological or Genetic Inhibition of Slug Activity,” Neoplasia, vol. 16, no. 12, pp. 1047–1058, 2014, doi: 10.1016/j.neo.2014.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Ferrari-Amorotti G et al. , “Inhibiting interactions of lysine demethylase LSD1 with Snail/Slug blocks cancer cell invasion,” vol. 73, no. 1, pp. 235–245, 2013, doi: 10.1158/0008-5472.CAN-12-1739.Inhibiting. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Sheng W et al. , “LSD1 ablation stimulates anti-tumor immunity and enables checkpoint blockade,” Cell, vol. 174, no. 3, pp. 549–563, 2018, doi: 10.1016/j.cell.2018.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]; **Key study showing synergistic effects between LSD1 inhibitors and PD-1 blockade.
- [59].Yang M et al. , “Structural basis for the inhibition of the LSD1 histone demethylase by the antidepressant trans-2-phenylcyclopropylamine,” Biochemistry, vol. 46, no. 27, pp. 8058–8065, 2007, doi: 10.1021/bi700664y. [DOI] [PubMed] [Google Scholar]
- [60].Binda C et al. , “Biochemical, Structural, and Biological Evaluation of Trancypromine Derivatives as Inhibitors of Histone Demethylases LSD1 and LSD1,” J. Am. Chem. Soc, vol. 132, no. 19, pp. 6827–6833, 2010. [DOI] [PubMed] [Google Scholar]
- [61].Maes T et al. , “ORY-1001, a Potent and Selective Covalent KDM1A Inhibitor, for the Treatment of Acute Leukemia,” Cancer Cell, vol. 33, no. 3, pp. 495–511.e12, 2018, doi: 10.1016/j.ccell.2018.02.002. [DOI] [PubMed] [Google Scholar]
- [62].Mould DP, McGonagle AE, Wiseman DH, Williams EL, and Jordan AM, “Reversible Inhibitors of LSD1 as Therapeutic Agents in Acute Myeloid Leukemia: Clinical Significance and Progress to Date,” Med. Res. Rev, vol. 35, no. 3, pp. 586–618, May 2015, doi: 10.1002/med.21334. [DOI] [PubMed] [Google Scholar]
- [63].Ji Y-Y et al. , “Tying up tranylcypromine: Novel selective histone lysine specific demethylase 1 (LSD1) inhibitors,” Eur. J. Med. Chem, vol. 141, pp. 101–112, December 2017, doi: 10.1016/j.ejmech.2017.09.073. [DOI] [PubMed] [Google Scholar]
- [64].Cramer SA, Adjei IM, and Labhasetwar V, “Advancements in the delivery of epigenetic drugs,” Expert Opin. Drug Deliv, vol. 12, no. 9, pp. 1501–1512, 2015, doi: 10.1517/17425247.2015.1021678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Raynal NJM et al. , “Repositioning FDA-approved drugs in combination with epigenetic drugs to reprogram colon cancer epigenome,” Mol. Cancer Ther, vol. 16, no. 2, pp. 397–407, 2017, doi: 10.1158/1535-7163.MCT-16-0588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Kalin JH et al. , “Targeting the CoREST complex with dual histone deacetylase and demethylase inhibitors,” Nat. Commun, vol. 9, no. 1, p. 53, December 2018, doi: 10.1038/s41467-017-02242-4. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** Paper demonstrating LSD1 inhibitors and a dual action LSD1/HDAC inhibitor inhibit proliferation of human cSCC cell lines.
- [67].Schenk T et al. , “Inhibition of the LSD1 (KDM1A) demethylase reactivates the all- trans-retinoic acid differentiation pathway in acute myeloid leukemia,” Nat Med, vol. 18, no. 4, pp. 605–611, 2013, doi: 10.1038/nm.2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Maiques-Diaz A et al. , “Enhancer Activation by Pharmacologic Displacement of LSD1 from GFI1 Induces Differentiation in Acute Myeloid Leukemia,” Cell Rep, vol. 22, no. 13, pp. 3641–3659, 2018, doi: 10.1016/j.celrep.2018.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Aubert Y, Egolf S, and Capell BC, “The Unexpected Noncatalytic Roles of Histone Modifiers in Development and Disease,” Trends Genet, vol. 35, no. 9, pp. 645–657, 2019, doi: 10.1016/j.tig.2019.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].McGrath JP et al. , “Pharmacological inhibition of the histone lysine demethylase KDM1A suppresses the growth of multiple acute myeloid leukemia subtypes,” Cancer Res, vol. 76, no. 7, pp. 1975–1988, 2016, doi: 10.1158/0008-5472.CAN-15-2333. [DOI] [PubMed] [Google Scholar]
- [71].Campbell JD et al. , “Genomic, Pathway Network, and Immunologic Features Distinguishing Squamous Carcinomas,” Cell Rep, vol. 23, no. 1, pp. 194–212.e6, 2018, doi: 10.1016/j.celrep.2018.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Rustgi et al. , “SCC: Unified Perspective,” vol. 318, no. 24, pp. 2446–2456, 2018, doi: 10.1001/jama.2017.17923.Association. [DOI] [Google Scholar]
- [73].Egolf S et al. , “LSD1 Inhibition Promotes Epithelial Differentiation through Derepression of Fate-Determining Transcription Factors,” Cell Rep, vol. 28, no. 8, pp. 1981–1992.e7, 2019, doi: 10.1016/j.celrep.2019.07.058. [DOI] [PMC free article] [PubMed] [Google Scholar]; **Paper providing key insight into the funciton of LSD1 in normal kerationcytes and how LSD1 inhibitors reduce invasion in a model of human cSCC.