
Figure 1.
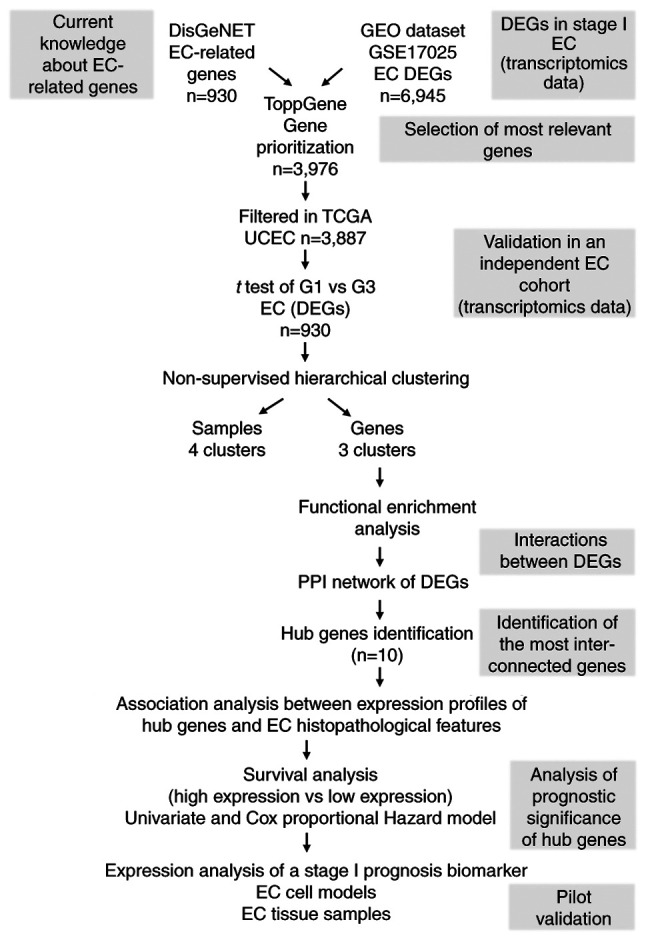
Workflow diagram of text and data mining tools used for the identification of prognostic EC biomarkers. Genes associated with the entire repertoire of EC-related disease terms were gathered from DisGeNET in order to cover a higher spectrum of genes with potential prognostic value in EC. These genes were used as a reference for further selection and prioritization of EC-DEGs retrieved from the GEO microarray dataset GSE17025. The prioritized genes were then subjected to hierarchical clustering analysis using TGCA UCEC RNAseq dataset, which led to the identification of a set of DEGs able to generate sample and gene clusters. Since sample cluster segregation has a lack of prognostic significance, gene clusters were further analyzed. To determine if DEGs belonged to the same pathways, enrichment analysis and PPI network analysis were carried out. A set of ‘hub genes’ were identified, and statistical association analysis with EC clinicopathological parameters and survival analysis was performed. Finally, pilot validation studies were performed using EC cell models and tissue specimens. EC, endometrial cancer; DEGs, differentially expressed genes; GEO, Gene Expression Omnibus; TCGA, The Cancer Genome Atlas; UCEC, Uterine Corpus Endometrioid Cancer; PPI, protein-protein interaction.