

Abstract
目的
探讨中链酰基辅酶A脱氢酶缺乏症(MCADD)中国人群流行病学特征、表型、基因型及预后。
方法
回顾性分析2009年1月至2018年6月期间经高效液相色谱串联质谱(HPLC-MS/MS)筛查并结合基因检测诊断为MCADD的新生儿资料。
结果
2 674 835例接受筛查的新生儿中诊断MCADD的12例(1/222 902)。其中10例接受基因检测,发现ACADM基因16个突变位点的13种突变类型:7种为已报道突变(p.T150Rfs*4、p.M1V、p.R206C、p.R294T、p.G310R、p.M328V、p.G362E);5种新突变(p.N194D、p.A324P、p.N366S、c.118+3A > G、c.387+1del G)和1例11号外显子缺失,以p.T150Rfs*4最常见(4/16)。ACADM基因突变位点检出率80%。未见表型-基因型相关性。确诊后给予饮食指导及对症治疗,随访4~82个月期间未见急性代谢失衡发作,除1例合并脑发育不良外均预后良好。
结论
MCADD在中国南方人群相对罕见;p.T150Rfs*4为中国人群热点突变;筛查阳性的病例建议联合辛酰基肉碱检测及基因判断。
Keywords: 中链酰基辅酶A脱氢酶, 患病率, 基因型, 新生儿
Abstract
Objective
To investigate the epidemiological characteristics, phenotype, genotype, and prognosis of medium-chain acyl-CoA dehydrogenase deficiency (MCADD) in the Chinese population.
Methods
A retrospective analysis was performed for the clinical data of the neonates who underwent screening with high-performance liquid chromatography-tandem mass spectrometry from January 2009 to June 2018 and were diagnosed with MCADD by gene detection.
Results
A total of 2674835 neonates underwent neonatal screening, among whom 12 were diagnosed with MCADD. Gene detection was performed for 10 neonates with MCADD and found 13 mutation types at 16 mutation sites of the ACADM gene, among which there were 7 reported mutations (p.T150Rfs*4, p.M1V, p.R206C, p.R294T, p.G310R, p.M328V, and p.G362E), 5 novel mutations (p.N194D, p.A324P, p.N366S, c.118+3A > G, and c.387+1del G), and 1 exon 11 deletion; p.T150Rfs*4 was the most common mutation (4/16). The detection rate of mutation sites in the ACADM gene was 80%. No phenotype-genotype correlation was observed. Dietary guidance and symptomatic treatment were given after confirmed diagnosis. No acute metabolic imbalance was observed within 4-82 months of follow-up. All neonates had good prognosis except one who had brain dysplasia.
Conclusions
MCADD is relatively rare in southern China, and p.T150Rfs*4 is a common mutation in the Chinese population. Cases with positive screening results should be evaluated by octanoylcarnitine C8 value and gene detection.
Keywords: Medium-chain acyl-CoA dehydrogenase, Prevalence rate, Genotype, Neonate
中链酰基辅酶A脱氢酶缺乏症(medium chain acyl-CoA dehydrogenase deficiency, MCADD;OMIM 201450)是由于ACADM(OMIM 607008)基因异常导致中链酰基辅酶A脱氢酶(medium chain acyl-CoA dehydrogenase, MCAD)功能缺陷,因此能量生成减少和毒性代谢产物蓄积,属于常染色体隐性遗传病,1982年由Kølvraa首次报道[1]。该病起病隐匿,首次发病的病死率及神经系统后遗症发生率高,其患病率、基因型存在明显种族差异[2]。目前国内尚无MCADD流行病学、基因型及远期预后的报道。本研究基于浙江省新生儿疾病筛查中心2009年1月至2018年6月的高效液相色谱串联质谱(high-performance liquid chromatography tandem mass spectrometry, HPLC-MS/MS)筛查资料库,分析中国新生儿MCADD患病率,探讨其表型、基因型特征及预后,为MCADD防控提供循证依据。
1. 资料与方法
1.1. 研究对象
2009年1月至2018年6月2 674 835例新生儿接受HPLC-MS/MS筛查。样本的采集和检测均在监护人签署知情同意书的情况下完成。
1.2. HPLC-MS/MS检测酰基肉碱和尿有机酸
生后72~120 h足底采血,滴于Schleicher & Schuell 903滤纸,阴干。干血滤纸片经多种氨基酸、肉碱测定试剂盒(串联质谱法,PerKin Elmer)处理,经串联质谱仪检测酰基肉碱。
对于干血滤纸酰基肉碱筛查的疑诊新生儿取新鲜尿样进行尿有机酸检测,予以尿素酶、盐酸羟铵、氢氧化钠和盐酸处理,加入内标17烷酸,乙酸乙酯萃取2次,经甲基硅烷化衍生后上机(岛津GC/MS-QP2010)检测。
1.3. 基因突变分析
对于干血滤纸酰基肉碱筛查疑诊MCADD的病例,经家长知情同意,取患儿及父母外周血标本进行二代高通量测序(靶向测序包,包括ACADVL、ACADM、ACADS、SLC22A5、SLC25A20、CPT1A、CPT2、HADH、HADHA、HADHB、ETFA、ETFB、ETFDH、ETHE1、ACAD8、ACADSB、OXCT1、SUCLA2、SUCLG1、SUCLG2、TMEM70等基因)。检测ACADM基因突变并进行Sanger测序验证,检测到ACADM单个变异位点者加用q-PCR方法检测。
1.4. MCADD诊断及筛查可疑的判断依据
筛查可疑的判断参照文献[2-3],以辛酰基肉碱(C8)高于正常2倍,或C8合并C8/C10(辛酰基肉碱/葵酰基肉碱)增高者作为MCADD可疑病例。MCADD诊断根据基因检测发现ACADM基因突变或根据C8持续增高确诊[2]。
2. 结果
2.1. 筛查结果
共筛查2 674 835例新生儿,可疑阳性161例,其中12例持续3次以上增高,其余恢复正常,12例中10例进行基因检测(2例拒绝基因检测)。结合C8和基因检测结果,最终12例足月新生儿确诊为MCADD,患病率1/222 902。除外不合格血片及筛查阳性患者,C8、C8/C10正常范围分别为0.02~0.17 µmol/L和0.44~2.00。
2.2. 特殊生化结果
确诊为MCADD的病例初筛时C8为0.58~11.39 µmol/L,C8/C10为0.91~21.08;随访中C8、C8/C10波动明显,除1例C8/C10有1次正常,其余均高于正常范围;12例新生儿均检测了尿有机酸,仅1例合并戊二酸及辛二酸增高。见表 1。
1.
MCADD患儿临床特征汇总
| 病例号 | 性别 | 出生胎龄 (周) |
出生体重 (kg) |
初筛C8 (μmol/L) |
初筛C8/C10 | C8波动范围 (μmol/L) |
随访时间 (月) |
其它异常 |
| 注:C0正常范围:10.28~54.24 μmol/L;SGPT正常范围:0~40 U/L。 | ||||||||
| 1 | 女 | 40 | 3.3 | 2.53 | 21.08 | 2.53~3.47 | 82 | C0 10.0 μmol/L, SGPT 114 U/L |
| 2 | 男 | 40 | 3.6 | 5.14 | 11.96 | 1.83~5.14 | 64 | C0 6.52 μmol/L |
| 3 | 男 | 38 | 3.0 | 5.95 | 4.19 | 0.27~5.95 | 57 | 3羟基戊二酸、 辛二酸增高 |
| 4 | 男 | 39 | 3.6 | 1.77 | 3.34 | 0.3~1.77 | 26 | C0 9.43 μmol/L |
| 5 | 男 | 38 | 2.9 | 0.76 | 3.62 | 0.57~1.16 | 23 | 脑白质发育不良 |
| 6 | 男 | 37 | 4.6 | 11.32 | 17.15 | 1.31~11.32 | 19 | C0 9.8 μmol/L |
| 7 | 男 | 39 | 3.5 | 1.52 | 12.67 | 1.03~6.17 | 16 | C0 7.67 μmol/L, SGPT 114 U/L |
| 8 | 女 | 39 | 3.0 | 5.93 | 9.72 | 0.76~0.93 | 11 | 卵圆孔未闭 |
| 9 | 女 | 40 | 3.2 | 10.35 | 15.68 | 2.29~10.35 | 10 | |
| 10 | 男 | 37 | 2.6 | 8.00 | 13.11 | 1.01~8.00 | 8 | 房间隔缺损 |
| 11 | 男 | 38 | 3.5 | 11.17 | 15.73 | 4.20~11.73 | 5 | |
| 12 | 女 | 39 | 3.2 | 11.39 | 14.24 | 2.67~11.39 | 4 | |
2.3. 基因检测结果
10例进行基因检测的患儿均检测到ACADM基因突变,共发现13种突变类型(未发现其它可致C8增高的相关基因变异):包括7种已报道的突变:p.T150Rfs*4、p.M1V、p.R206C、p.R294T、p.G310R、p.M328V、p.G362E和5种新突变:p.N194D、p.A324P、p.N366S、c.118+3A>G、c.387+1del G,其中p.T150Rfs*4为框移突变,c.118+3A>G、c.387+1del G为剪接突变,其余均为错义突变,所有突变均遗传自父亲或者母亲;1例11号外显子缺失(发现单个突变位点者加做q-PCR)为父源性,其母亲为11号外显子重复(HPLC-MS/MS检测未见异常)。见表 2及图 1~2。以p.T150Rfs*4的发生率最高,占25%,其余均为散发。3种新的错义突变用PolyPhen-2(http://genetics.bwh.harvard.edu/pph2/)、SIFT(http://sift.jcvi.org/)、MutationTaster(http://www.mutationtaster.org/)、FATHMM(http://fathmm.biocompute.org.uk/)等进行功能预测提示有害,见表 3。另2种新剪接突变对蛋白功能影响大,据ACMG评判标准归为致病性。患儿基因突变位点分散,未能发现生化表型与基因型的相关性。
2.
MCADD患儿变异位点汇总(NM_000016.4)
| 病例号 | 突变1 (母源) | 属性 | 突变2 (父源) | 属性 |
| 注:ACADM基因变异分类评估通过检索ExAC、GnomAD、1000 Genomes、ClinVar、PubMed等数据库综合评估。 | ||||
| 1 | p.R294T, c.881G>C | 致病/可能致病 | p.A324P, c.970G>C | 新位点 |
| 2 | p.N194D, c.580A>G | 新位点 | del exon 11 | |
| 3 | c.387+1 delG, splicing | 新位点 | p.M328V, c.982A>G | 致病 |
| 5 | p.G310R, c.928G>A | 致病 | c.118+3A>G, splicing | 新位点 |
| 6 | p.N366S, c.1097A>G | 新位点 | ||
| 7 | c.449_452 del CTGA, p.T150Rfs*4 | 致病 | ||
| 8 | p.M1V, c.1A>G | 可能致病 | p.R206C, c.616C>T | 致病 |
| 9 | c.449-452 del CTGA, p.T150Rfs*4 | 致病 | c.449-452del CTGA, p.T150Rfs*4 | 致病 |
| 10 | c.449-452 del CTGA, p.T150Rfs*4 | 致病 | ||
| 11 | p.G362E, c.1085G>A | 致病 | ||
1.
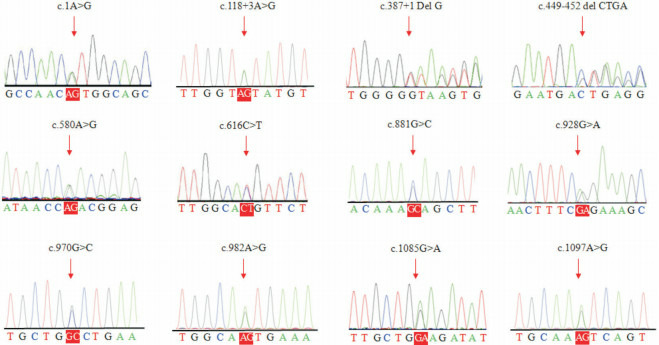
13种突变的Sanger测序图
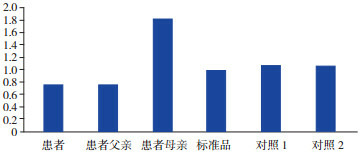
2.
病例2 ACADM基因11号外显子改变
病例2及其父亲的11号外显子缺失。
3.
3种新的错义突变的致病性预测
| Amino acid change | Func.ref | SIFT | Polyphen2 | MutationTaster | FATHMM | PROVEAN | MetaLR | M-CAP | ||
| 注:[D] Deleterious or Damaging;[T] Tolerated | ||||||||||
| exon7 | c.580A>G | p.N194D | nonsynonymous | D | D | D | D | D | D | D |
| exon11 | c.970G>C | p.A324P | nonsynonymous | D | D | D | D | D | D | D |
| exon11 | c.1097A>G | p.N366T | nonsynonymous | T | D | D | D | D | D | D |
2.4. 临床表型及治疗结局
诊断后均给予饮食指导及健康教育。随访至4~82月龄,随访中未见急性代谢失衡发作,5例患儿无症状状态下各出现1次C0降低,发生在5月龄至4岁,经左卡尼汀(每日50~100 mg/kg)口服,1个月后C0均恢复正常,停药后未发现C0改变;随访中除1例C8/C10有1次正常,其余C8、C8/C10检测均高于正常。出生时合并肝功能损害的2例,经复方甘草酸苷、肌苷治疗3个月后恢复正常;血糖、肝肾功能、肌酸激酶、心肌酶随访均正常。心脏超声检查提示合并房间隔缺损、卵圆孔未闭各1例,6个月时自行恢复;所有患儿心电图未见异常,心超未见心肌病表现或心功能异常。合并脑发育不良1例(MRI提示脑白质髓鞘发育落后)。除合并脑发育不良的1例,所有患儿随访体格发育正常,Bayley发育量表检测均正常。
3. 讨论
MCADD患病率存在明显种族差异,新生儿患病率以德国较高,为1 : 4 900~1 : 8 500;美国1 : 13 000~1 : 19 000;亚洲人群患病率相对较低,日本新生儿患病率为1 : 51 000;中国台湾地区1 : 263 500 [2, 4]。本研究总结10年新生儿串联质谱筛查数据,获得中国南方人群MCADD患病率为1 : 222 902,与我国台湾地区接近。
MCAD是酰基辅酶A脱氢酶家族成员之一,位于线粒体基质,特异性催化中链脂肪酸β氧化的第一步。MCAD功能缺陷时己酰基肉碱(C6)以及C8、C10增高,C8升高更明显,是其特征性变化,因此将C8作为MCADD新生儿筛查指标。但继发肉碱缺乏时,C0低,C6~C10升高不明显,结合C8/C10比值可提高敏感性及准确性[2, 5-6]。本研究基于260余万新生儿HPLC-MS/MS筛查数据,得出C8、C8/C10正常范围为0.02~0.17 µmol/L和0.44~2.00,为MCADD新生儿筛查切值的确立及诊断提供了一定的循证依据,但中国MCADD新生儿C8、C8/C10筛查切值的建立有待不同地区更多样本数据的积累及综合分析软件的研发。本研究所有病例初筛时C8、C8/C10均高于正常范围2倍以上,随访中除1例C8/C10有1次检测正常,其余均高于正常范围。但需注意以C8筛查新生儿群体MCADD,在无代谢压力如发热、饥饿、手术、预防接种等诱发分解代谢的情况下,可出现假阴性;C8在其他遗传代谢病、接受丙戊酸钠治疗或喂食富含中链甘油三酯饮食的新生儿也会升高[2]。国外不同筛查中心以酰基肉碱分析筛查MCADD的阳性预测值差异显著,约8%~78%[7]。诊断后分析工具R4S(https://www.nbstrn.org/research-tools/lab-performance-database)及CLIR(https://www.clir-r4s.org/)可以对被认为异常的酰基肉碱结果进行综合分析和鉴别以提高筛查、诊断效率[8]。
MCADD患者的尿二羧酸(己二酸、辛二酸、葵二酸等)可增高,但病情稳定时正常。因此尿有机酸分析适用于MCADD急性发作的诊断,不适于新生儿筛查[2]。本研究仅1例合并尿戊二酸及辛二酸增高。文献[9]报道急性发作期5-羟基己酸、己基甘氨酸、苯丙基甘氨酸、环庚甘氨酸特别是亚乙基甘氨酸是MCADD的额外生化标志,可作为诊断的首选检测。
ACADM基因是目前发现的MCADD唯一致病基因,位于染色体1p31.1,包含12个外显子,迄今已至少报道160余种突变位点,以错义突变为主(http://www.hgmd.cf.ac.uk/ac/index.php)[2]。MCADD在欧裔尤其是北欧人群很常见,最常见的突变是位于11号外显子的c.985A>G(p.K304E),第二常见突变是c.199T>C(p.Y42H)[2],本研究均未发现这些突变。阿拉伯人的奠基者突变可能是c.362C>T [10]。日本、韩国人群有449-452del4(p.T150Rfs*4)突变位点报道[11-12]。本研究10例患儿共发现13种变异,以错义突变为主,其中p.T150Rfs*4为热点突变,占25%,提示该突变可能为亚洲人群的热点突变。
MCADD可通过基因检测发现ACADM等位基因致病性突变确诊,或通过MCAD酶活性检测确诊,目前多通过基因检测诊断[2]。本研究10例患儿均检测到ACADM基因突变,共发现13种突变包括新突变和已报道突变,因检测到等位基因的16个突变位点,突变位点检出率80%,低于国外文献报道[2]。由于某些突变位点位于内含子剪接部位、调控区或存在片段缺失,通过常用高通量测序不能发现,故一部分常染色体隐性遗传代谢病患者仅能检测到1个突变位点甚至不能检测到突变位点,这部分患者除了通过全基因测序、q-PCR寻找突变位点,还可依据其特殊生化表型进行鉴别诊断。如本组患儿中4例经高通量测序仅检测到单个变异位点,其中1例经q-PCR发现父源性ACADM 11号外显子缺失得以确诊;另外3例的家长拒绝进一步遗传学检测,随访至5~19月龄,在无任何应激、疾病情况下C8及C8/C10比值持续增高,且高通量测序未发现其他致C8增高的遗传代谢病基因(如ETFA、ETFB、ETFDH基因)变异,最终诊断为MCADD。Janzen等[13]报道1种快速酶检测方法诊断30例MCADD。但MCAD酶活性检测方法较复杂,皮肤、肝脏等组织标本获取困难,临床难以推广。因此,对于筛查阳性病例建议联合C8及基因检测进行确诊。
MCADD典型临床表现是在饥饿、疾病或应激状态下的低血糖、呕吐、昏睡、癫痫发作,多在3~24月龄起病,也可在成年期起病,间歇期往往正常或终身无症状;MCADD患者死亡率高,发病的患者中约25%死亡,成人期急性起病的患者死亡率可达50%。约1/3的患者于急性发病后出现后遗症。轻症、成人型患者仍有猝死风险[2]。因此对MCADD的早期诊治是改善预后的关键。MCADD患者应注意避免禁食、保证热量供应,避免高脂饮食及富含中链脂肪酸奶粉[14]。Derks等[15]研究了MCADD无症状患儿禁食耐受时间,6~12个月婴儿的最长禁食时间应小于8小时;1~2岁幼儿小于10小时;>2岁者小于12小时。急性期患者最重要的治疗是通过口服碳水化合物逆转分解代谢,促进持续的合成代谢。如果患者无法通过口服维持正常代谢,应立即静脉注射葡萄糖:25%葡萄糖2 mL/kg稀释后静脉推注或10%葡萄糖以每分钟10~12 mg/kg的速度输注以维持高于5 mmol/L的血糖水平[2]。本组病例诊断后均给予饮食指导及健康教育,随访至4~82月龄未见急性代谢失衡发作,随访中5例出现C0减低,经护肝、口服左卡尼汀等对症治疗,预后良好。
MCADD基因型与临床表型的关系尚不明确,由于影响因素较复杂,目前研究表明通过基因型不能准确预测患者的临床表型或疾病严重程度[2]。同一家族MCADD患者具有不同临床表型很常见;轻症生化表型的个体仍有可能出现危及生命的症状[16]。本研究发现同一患儿在无急性代谢失衡状态下C8及其比值波动明显,亦提示其表型-基因型无明确相关性。
综上所述,MCADD在中国南方人群相对罕见,ACADM突变以错义突变为主,多数散发,p.T150Rfs*4为中国人群热点突变,筛查阳性的病例建议联合C8及基因检测进行诊断。
Biography
童凡, 女, 硕士研究生, 主任医师
Funding Statement
国家重点研发计划(2017YFC1001704,2017YFC1001702);国家自然科学基金(81741090)
References
- 1.Kølvraa S, Gregersen N, Christensen E, et al. In vitro fibroblast studies in a patient with C6-C10-dicarboxylic aciduria:evidence for a defect in general acyl-CoA dehydrogenase. Clin Chim Acta. 1982;126(1):53–67. doi: 10.1016/0009-8981(82)90361-8. [DOI] [PubMed] [Google Scholar]
- 2.Matern D, Rinaldo P. Medium-chain acyl-coenzyme A dehydrogenase deficiency. Seattle: University of Washington; 2015. [PubMed] [Google Scholar]
- 3.Oerton J, Khalid JM, Besley G, et al. Newborn screening for medium chain acyl-CoA dehydrogenase deficiency in England:prevalence, predictive value and test validity based on 1.5 million screened babies. J Med Screen. 2011;18(4):173–181. doi: 10.1258/jms.2011.011086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chien YH, Lee NC, Chao MC, et al. Fatty acid oxidation disorders in a chinese population in Taiwan. JIMD Rep. 2013;11:165–172. doi: 10.1007/978-3-642-37328-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Matern D. Acylcarnitines[M]//Blau N, Duran M, Gibson KM, et al. Physician's Guide to the Diagnosis, Treatment, and Followup of Inherited Metabolic Diseases. Heidelberg: SpringerVerlag, 2014: 775-784.
- 6.Hall PL, Wittenauer A, Hagar A. Newborn screening for medium chain acyl-CoA dehydrogenase deficiency:performance improvement by monitoring a new ratio. Mol Genet Metab. 2014;113(4):274–277. doi: 10.1016/j.ymgme.2014.10.007. [DOI] [PubMed] [Google Scholar]
- 7.Lindner M, Hoffmann GF, Matern D. Newborn screening for disorders of fatty-acid oxidation:experience and recommendations from an expert meeting. J Inherit Metab Dis. 2010;33(5):521–526. doi: 10.1007/s10545-010-9076-8. [DOI] [PubMed] [Google Scholar]
- 8.Rinaldo P. Retrospective and prospective date mining to develop continuous, covariate-adjusted reference and disease percentiles for biomarkers of metabolic disease. Clin Chem Lab Med. 2017;55:1200–1201. [Google Scholar]
- 9.Gregersen N, Kølvraa S, Rasmussen K, et al. General (mediumchain) acyl-CoA dehydrogenase deficiency (non-ketotic dicarboxylic aciduria):quantitative urinary excretion pattern of 23 biologically significant organic acids in three cases. Clin Chim Acta. 1983;132(2):181–191. doi: 10.1016/0009-8981(83)90246-2. [DOI] [PubMed] [Google Scholar]
- 10.Al-Hassnan ZN, Imtiaz F, Al-Amoudi M, et al. Medium-chain acyl-CoA dehydrogenase deficiency in Saudi Arabia:incidence, genotype, and preventive implications. http://cn.bing.com/academic/profile?id=01542c33c2c42167e0e4719aaf7171a0&encoded=0&v=paper_preview&mkt=zh-cn. J Inherit Metab Dis. 2010;33(Suppl 3):S263–S267. doi: 10.1007/s10545-010-9143-1. [DOI] [PubMed] [Google Scholar]
- 11.Shigematsu Y, Hirano S, Hata I, et al. Newborn mass screening and selective screening using electrospray tandem mass spectrometry in Japan. J Chromatogr B Analyt Technol Biomed Life Sci. 2002;776(1):39–48. doi: 10.1016/S1570-0232(02)00077-6. [DOI] [PubMed] [Google Scholar]
- 12.Ensenauer R, Winters JL, Parton PA, et al. Genotypic differences of MCAD deficiency in the Asian population:novel genotype and clinical symptoms preceding newborn screening notification. Genet Med. 2005;7(5):339–343. doi: 10.1097/01.GIM.0000164548.54482.9D. [DOI] [PubMed] [Google Scholar]
- 13.Janzen N, Hofmann AD, Schmidt G, et al. Non-invasive test using palmitate in patients with suspected fatty acid oxidation defects:disease-specific acylcarnitine patterns can help to establish the diagnosis. Orphanet J Rare Dis. 2017;12(1):187. doi: 10.1186/s13023-017-0737-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Piercy H, Machaczek K, Ali P, et al. Parental experiences of raising a child with medium chain acyl-CoA dehydrogenase deficiency. http://cn.bing.com/academic/profile?id=46ad8e4f79cf9cf86ffbb138473ffd85&encoded=0&v=paper_preview&mkt=zh-cn. Glob Qual Nurs Res. 2017;4:2333393617707080. doi: 10.1177/2333393617707080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Derks TG, Van Spronsen FJ, Rake JP, et al. Safe and unsafe duration of fasting for children with MCAD deficiency. Eur J Pediatr. 2007;166(1):5–11. doi: 10.1007/s00431-006-0186-0. [DOI] [PubMed] [Google Scholar]
- 16.Dessein AF, Fontaine M, Andresen BS, et al. A novel mutation of the ACADM gene (c.145C>G) associated with the common c.985A>G mutation on the other ACADM allele causes mild MCAD deficiency:a case report. Orphanet J Rare Dis. 2010;5:26. doi: 10.1186/1750-1172-5-26. [DOI] [PMC free article] [PubMed] [Google Scholar]