
Fig. 1: Design of a selective, radical C–H amination for the synthesis of β-amino alcohols.
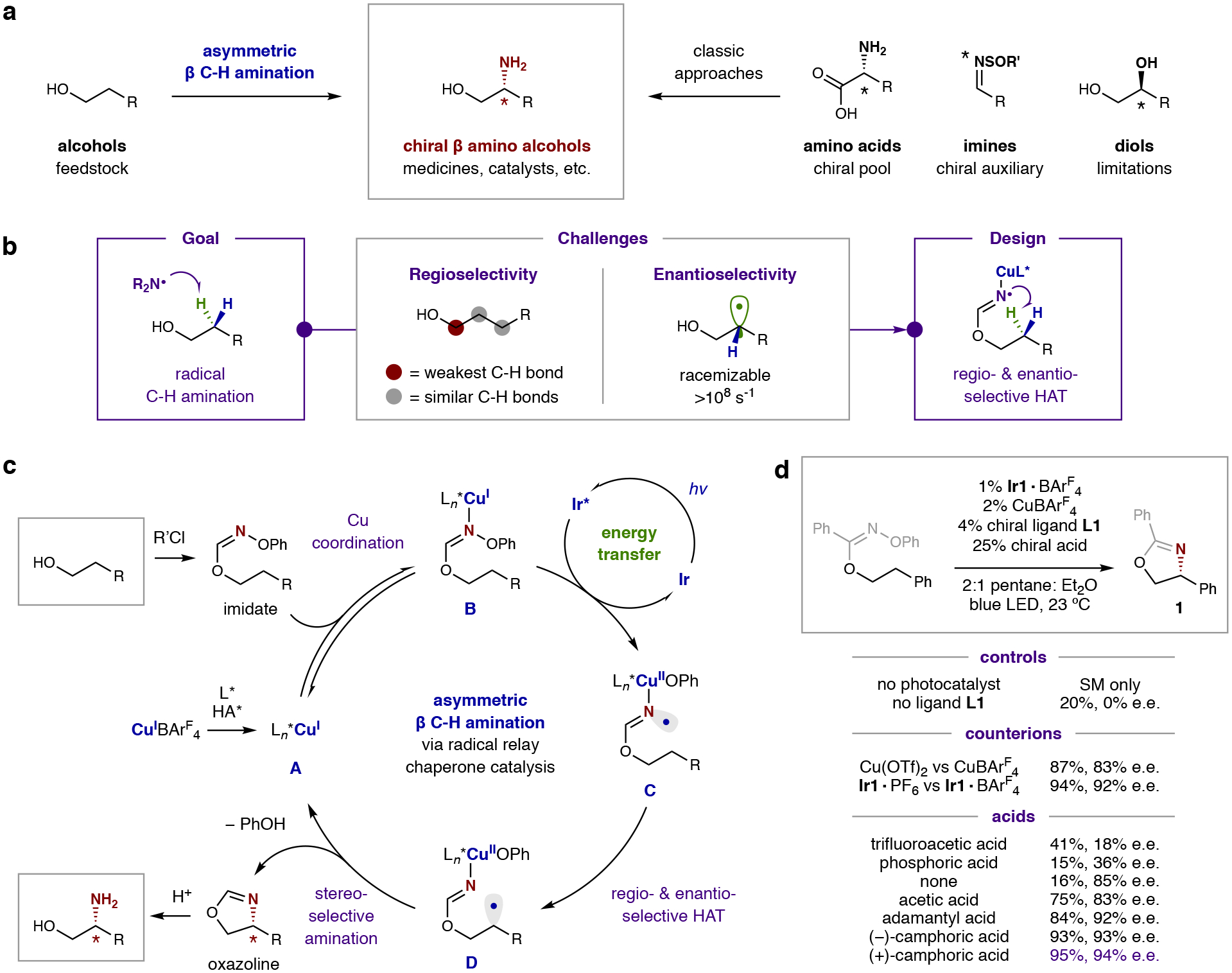
a, Radical C–H amination of cheap and abundant alcohols enables access to the privileged β-amino alcohol architecture – complementing classic approaches that are limited by availability of chiral pool precursors (e.g. amino acids), stoichiometric chiral auxiliaries (e.g. imines), or multi-step manipulation of chiral reagents (e.g. diols). b, Dual challenges for a radical approach include selective formation and trapping of a transient radical intermediate. Specifically, β-regioselectivity is disfavored compared to abstraction of the weaker α C–H bond by an N-centered radical. Moreover, enantioselectivity is both challenging to control and retain in this process, as it may rapidly erode by radical epimerization. c, Proposed mechanism: Alcohol addition to an imidoyl chloride chaperone furnishes an oxime imidate, which binds to a chiral Cu catalyst. This complex then forms an N-centered radical via selective, triplet sensitization by an excited Ir photocatalyst. Regio- and enantio-selective HAT, followed by stereoselective amination, affords a chiral oxazoline. Hydrolysis yields the enantioenriched β-amino alcohol. d, Development of this asymmetric β-C–H amination required a multi-catalytic strategy, wherein a photocatalyst selectively excites a chiral Cu catalyst complex when it is bound to an imidate-activated alcohol. Mechanistic probes illustrate the necessity of each component in ensuring the efficiency and selectivity of this radical C–H amination.