
Fig. 2: Mechanistic experiments.
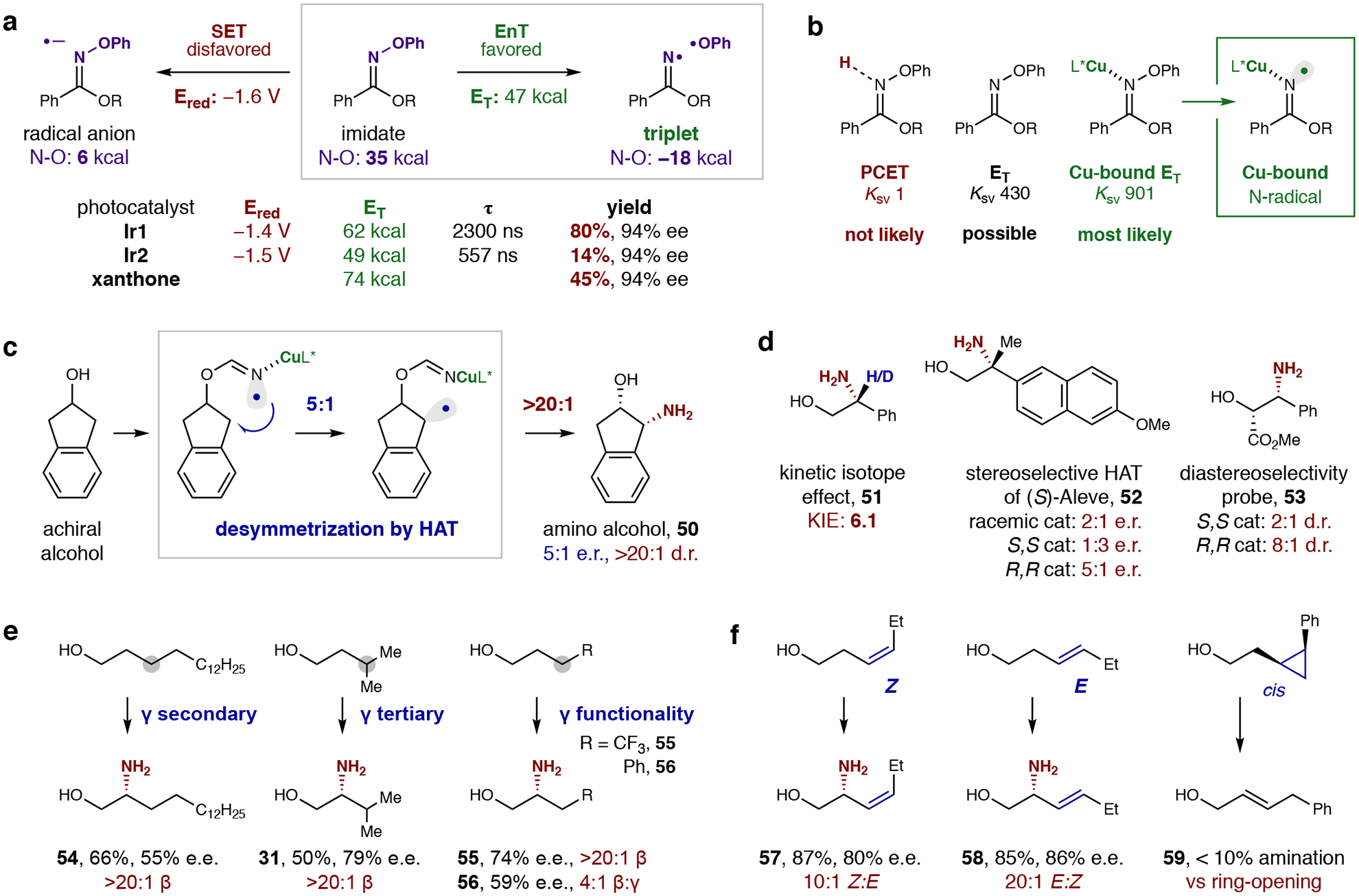
a, An energy transfer (EnT) pathway (vs single-electron transfer: SET) is supported by CV and DFT data. Specifically, a less reducing photocatalyst (Ir1 vs Ir2) provides greater efficiency due to a higher triplet energy (ET) and longer-lived excited state (τ). Triplet sensitizer, xanthone, also affords reactivity – indicating the imidate triplet, which is computed to have a significantly weakened N–O bond, is the source of N-centered radical. b, Photo-quenching experiments illustrate the favorability of Cu catalyst coordination during the triplet sensitization and subsequent N-radical generation. c, Desymmetrization of a secondary alcohol demonstrates the ability of this Cu-bound N-radical to discriminate among H-atoms by enantioselective HAT (5:1 e.r.). High diastereoselectivity (>20:1 d.r.) illuminates the role of Cu radical trapping in further upgrading the high enantioselectivity observed for primary alcohols. d, A significant KIE indicates HAT is the product-determining step. Stereoselectivity of the HAT was further probed with two chiral alcohols. Selectivity and efficiency are highly catalyst-dependent for both (S)-Aleve, which contains a β-stereocenter, as well as an enantioenriched secondary alcohol. e, A highly regioselective 1,5-HAT (>20:1 β) outcompetes 1,6-HAT even when much weaker γ-C–H bonds are present. f, Radical clocks indicate radical trapping by Cu is more rapid than allyl radical isomerization, but slower than cyclopropyl ring-opening.