

Abstract
Nemaline myopathy is among the most common non-dystrophic congenital myopathies, and is characterized by the presence of nemaline rods in skeletal muscles fibers, general muscle weakness, and hypotonia. Although respiratory failure is the main cause of death in nemaline myopathy, only little is known regarding the contractile strength of the diaphragm, the main muscle of inspiration. To investigate diaphragm contractility, in the present study we took advantage of a mouse model for nebulin-based nemaline myopathy that we recently developed. In this mouse model, exon 55 of Neb is deleted (NebΔExon55), a mutation frequently found in patients. Diaphragm contractility was determined in permeabilized muscle fibers and was compared to the contractility of permeabilized fibers from three peripheral skeletal muscles: soleus, extensor digitorum longus, and gastrocnemius. The force generating capacity of diaphragm muscle fibers of NebΔExon55 mice was reduced to 25% of wildtype levels, indicating severe contractile weakness. The contractile weakness of diaphragm fibers was more pronounced than that observed in soleus muscle, but not more pronounced than that observed in extensor digitorum longus and gastrocnemius muscles. The reduced muscle contractility was at least partly caused by changes in cross-bridge cycling kinetics which reduced the number of bound cross-bridges. The severe diaphragm weakness likely contributes to the development of respiratory failure in NebΔExon55 mice and might explain their early, postnatal death.
Keywords: Diaphragm, Nebulin, Respiratory failure, Nemaline myopathy
1. Introduction
Nemaline myopathy (NM) is a non-dystrophic congenital myopathy described for the first time in the 1960s [1]. Eleven genes have been implicated in NM. Six of these genes code for components of the skeletal muscle thin filament: alpha actin 1 (ACTA1) [2], alpha- and beta-tropomyosin (TPM3 and TPM2) [3,4], nebulin (NEB) [5], leiomodin-3 (LMOD3) [6] and troponin T (TNNT1) [7]. Four genes code for proteins which play a role in the regulation and the stability of the thin filament: cofilin 2 (CFL2) [8] regulates actin filament dynamics; kelch family member 40 (KLHL40) is implicated in myogenesis [9] and in the stabilization of thin filament proteins [9,10] and kelch family member 41 (KLHL41) modulates ubiquitination of thin filament proteins [10,11]. The role of kelch repeat and BTB (POZ) domain containing 13 (KBTBD13) is unclear, but might involve a role in the ubiquitin-proteasome pathway [12]. The most recent implicated gene, MYO18B, codes for an unconventional myosin heavy chain expressed in cardiac and skeletal muscles. Its function is incompletely understood [13].
NM affects skeletal muscles, including the muscles of respiration. Consequently, patients with NM have reduced spirometric values such as forced vital capacity, peak expiratory flow, forced expiratory volume in 1 second [14–16], and they may suffer from the sensation of dyspnea [15–19]. Respiratory failure is the main cause of death in NM [20], and occurs even in ambulant patients who otherwise appear to be only mildly affected; respiratory failure may even be the presenting feature. This suggests that weakness of respiratory muscles may be much greater than that of peripheral skeletal muscles. The pathophysiology of respiratory muscle weakness is not completely understood, but might involve a dysfunctional diaphragm.The diaphragm is the main muscle of inspiration, and weakness of the diaphragm impairs respiration and causes dyspnea [21]. Thus, insights in diaphragm contractility in NM are important for developing treatment strategies.
The little information available regarding diaphragm contractility in NM is a consequence of technical and ethical limitations in assessing diaphragm contractility in vivo and in obtaining diaphragm muscle biopsies from patients. Therefore, in the present study we took advantage of a mouse model for nebulin-based NM that we recently developed [22]. Mutations in NEB account for more than 50% of the gene mutations reported in NM patients [23], and in this mouse model exon 55 of Neb is deleted, a mutation frequently found in patients [22]. To assess diaphragm contractility, we isolated permeabilized muscle fibers from the diaphragm and activated the fibers with calcium. Muscle fiber contractility was determined and compared to the contractility of muscle fibers of three peripheral skeletal muscles: soleus, extensor digitorum longus and gastrocnemius. Our results indicate that diaphragm muscle fibers of NebΔExon55 mice exhibit severe contractile weakness, a weakness that was more pronounced than that observed in soleus muscle, but not more pronounced than that in extensor digitorum longus (EDL) and gastrocnemius muscles.
2. Materials and methods
2.1. Generation of NebΔExon55 mice
The procedure for the generation of the NebΔExon55 mice has been previously described [22].
2.2. Muscle preparations
For contractility experiments we used diaphragm, soleus (slow-twitch), EDL (fast-twitch) and gastrocnemius (fast-twitch) muscles from wild-type (Wt) and knock-out littermate NebΔExon55 mice. The number of mice used is summarized in Table 1. Muscles were harvested at day 6, as NebΔExon55 mice die one week after birth [22].
Table 1.
Number of animals used, for each muscle-type, in Wt and NebΔExon55 mice.
| Diaphragm | Soleus | EDL | Gastrocnemius | |
|---|---|---|---|---|
| Wt | 6 | 6 | 7 | 6 |
| NebΔExon55 | 9 | 6 | 9 | 5 |
2.3. Permeabilized muscle fiber contractility
The procedures for permeabilized muscle fiber contractility experiments were as described previously [24], with small modifications [22]. Small fiber bundles (diameter ~70 μm) were dissected from the triton-X permeabilized muscles, and attached to aluminum foil clips. The clips were attached to a force transducer (model 403A, Aurora Scientific, Ontario, Canada) and length controller (model 315-CI, Aurora Scientific, Ontario, Canada). We used a setup (model 802D, Aurora Scientific) that was mounted on top of an inverted microscope (Zeiss Axio Observer A1, Zeiss, Thornwood, NY, USA). Sarcomere length was determined using a fast Fourier transformation on a region of interest on the real-time camera image using ASI 900B software (Aurora Scientific Inc., Ontario, Canada).
Experiments were performed at sarcomere length 2.3 μm. The width and depth of the muscle bundle were measured with a 40× objective. The cross-sectional area (CSA) was calculated from the average of three consecutive measurements made along the length of the muscle bundle. The temperature was kept constant at 20 °C using a TEC controller (ASI 825A, Aurora Scientific Inc. Ontario, Canada).
Various bathing solutions were used during the experimental protocols: a relaxing solution (pCa 9.0), a pre-activating solution with low EGTA concentration and several incremental Ca2+ solutions (pCa 7.0–4.5). Composition of these solutions has been described previously [24].
The preparations were activated at pCa 4.5 to obtain maximal Ca2+-activated force. Maximal active tension was obtained by divided the force generated at pCa 4.5 by CSA.
2.3.1. Cross-bridge cycling kinetics (Ktr)
Muscle bundles were first isometrically activated at pCa 4.5, and when a steady tension was reached, cross-bridges were disengaged by performing a quick release of 30% of the initial length, which reduced tension to zero. This was followed by unloaded shortening lasting 30 ms. The remaining bound cross-bridges were mechanically detached by rapidly (1 ms) re-stretching the muscle fiber bundles to its original length, after which tension redevelops. The Ktr was determined by fitting a double exponential through the force redevelopment curve (note that only the fast rate constant is reported as this is considered to reflect cross-bridge cycling kinetics).
2.3.2. Active stiffness
Following the Ktr protocol, active stiffness was determined by length variations of 0.3, 0.6 and 0.9% on the fiber, resulting in a force response. Stiffness is represented as the slope of the linear regression of the relationship between tension and length. A typical force trace, including the force response during the Ktr and active stiffness protocol, is shown in Fig. 1.
Fig. 1.
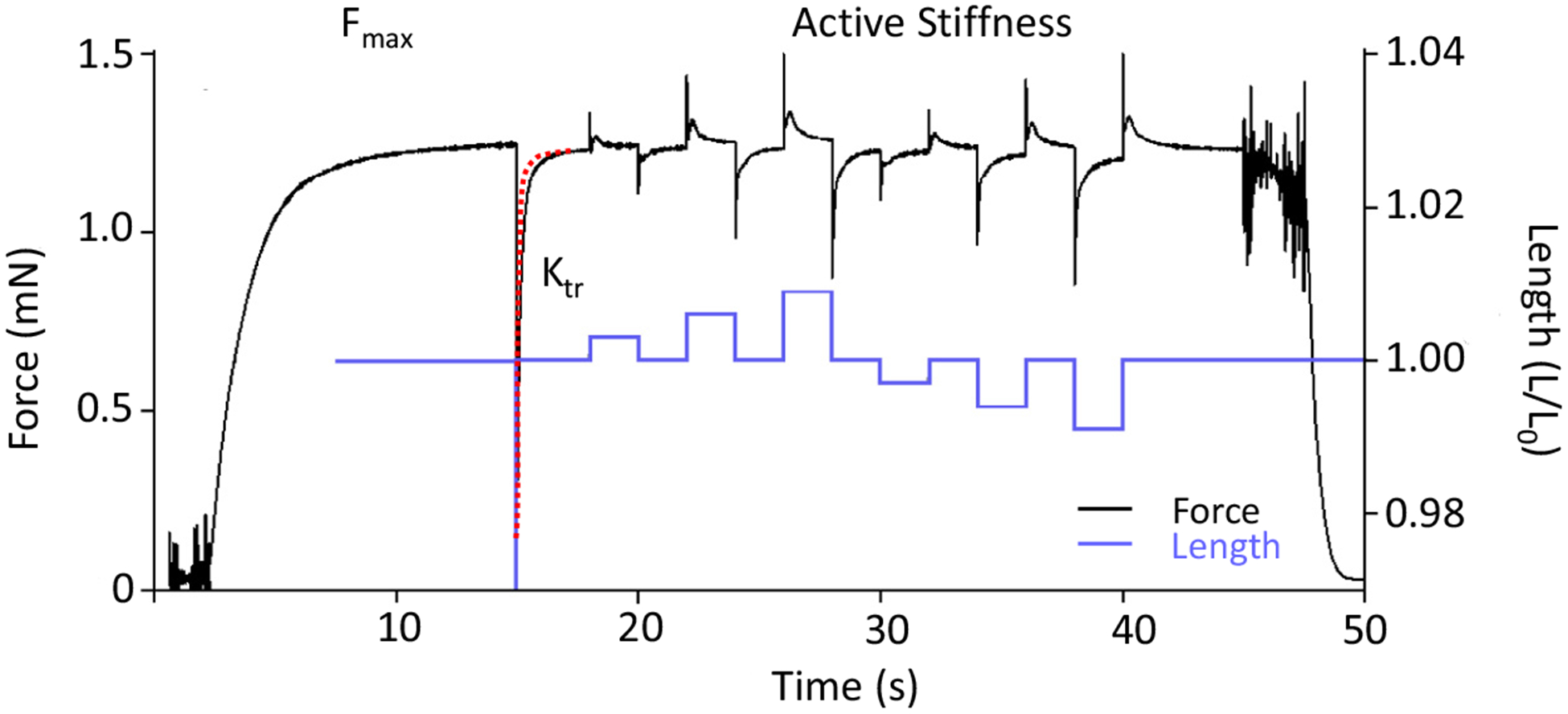
Example of a force and length trace of a permeabilized fiber bundle activated with exogenous calcium. The fiber bundle is exposed to saturating calcium concentration to determine the maximal force (Fmax). Second, the fiber is shortened by 30% of the initial length (Lo) to determine the rate constant of force redevelopment (Ktr, red dashed line shows the 2nd order exponential fit from which Ktr is determined). Finally, the fiber bundle is exposed to length variations of0.3, 0.6 and 0.9% (blue line) to determine the number of attached cross-bridges.
2.3.3. Calcium sensitivity of force generation
Preparations were in relaxing solution and exposed to pre-activating solution (pCa 9.0). Then, they were activated with solutions with a pCa ranging from 7.0 to 4.5 (Fig. 2A). The obtained force–pCa relation was fitted with a Hill equation, providing pCa50 (pCa giving 50% of maximal active tension) and the Hill coefficient, nH, an index of myofilament cooperativity (Fig. 2B).
Fig. 2.
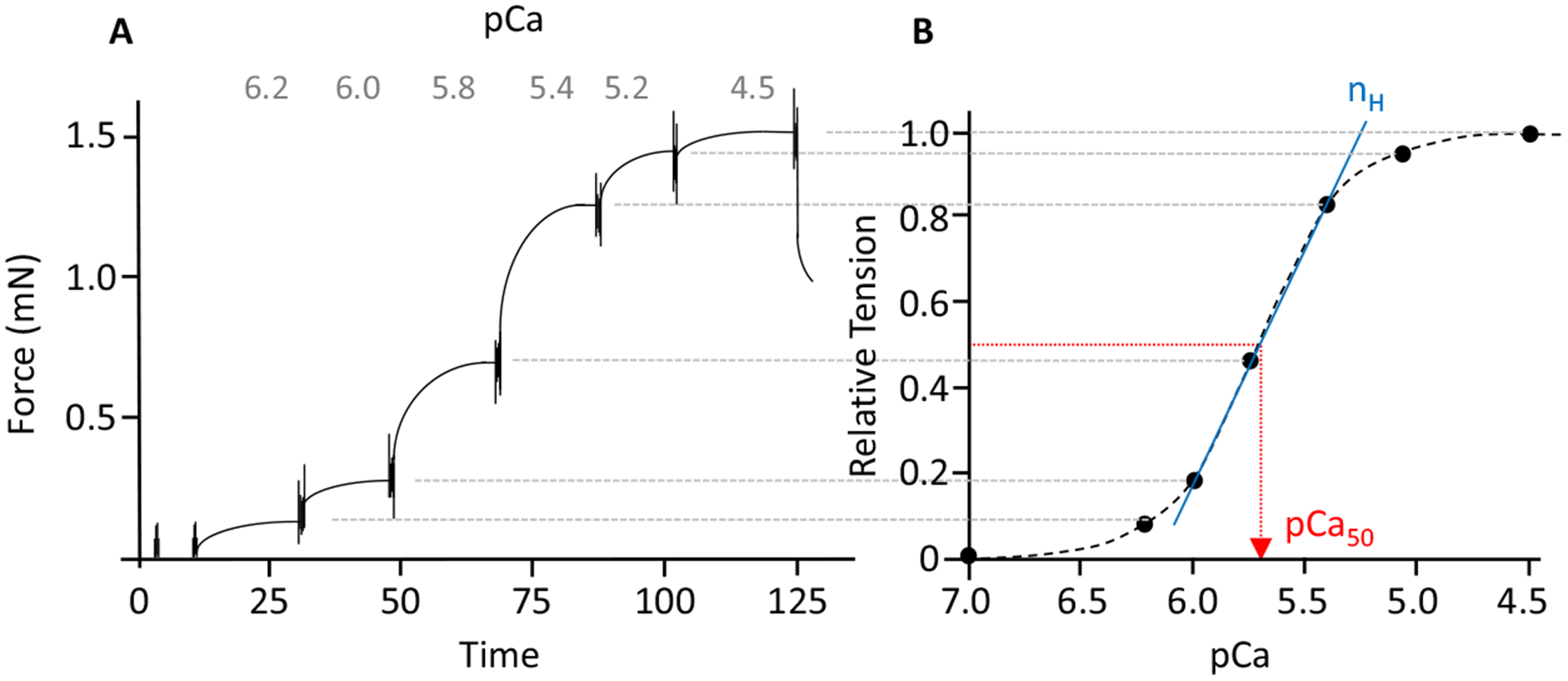
(A) Typical force trace representing the force response to incremental calcium concentrations, and (B) the corresponding force–pCa relation. The calcium concentration required for half-maximal activation (pCa50) was determined by fitting a modified Hill equation through the data point (the diagonal straight line shows the fit from which Hill coefficient (nH) is determined).
2.4. Fiber typing
As contractility is dependent on the myosin heavy chain (MyHC) isoforms expressed in the muscle fibers, a specialized sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) was used to determine the (MyHC) isoform composition of the muscle fiber preparations that we used in our contractility experiments. Muscles fibers were de-natured by boiling at 80 °C for 2 min in SDS sample buffer. The stacking gel contained a 4% acrylamide concentration (pH 6.7), and the separating gel contained 7% acryl-amide (pH 8.7) with 30% glycerol (v/v). The gels were run for 24 h at 15 °C and a constant voltage of 275 V. Finally, the gels were silver-stained, scanned [25], and analyzed with AIDA Image Analyzer software (Raytest Isotopenmessgeräte GmbH, Straubenhardt, Gemany).
2.5. Statistical analyses
Data are presented as mean ± SEM and % of the Wt. GraphPad Prism 6 was used to generate statistics. For statistical analyses t-test (Wt vs. NebΔexon55 for maximal active tension, Ktr, active stiffness, nH and pCa50) and one-way ANOVA (% of Wt between NebΔexon55) were performed. P < 0.05 was considered to be statistically significant.
3. Results
The results are shown in Table 2. Maximal active tension (force produced normalized to the cross sectional area) of diaphragm fibers was reduced by 76% in NebΔExon55 compared to Wt mice (22.7 ± 2.9 vs. 84.3 ± 2.87 mN/mm2, p < 0.0001, respectively). This reduction was significantly larger (p < 0.05) than the one observed in NebΔExon55 soleus muscle (reduction of 60%, 21.8 ± 1.4 vs. 53.4 ± 3.16 mN/mm2, p < 0.0001, respectively) but not significantly larger than that in NebΔExon55 EDL (reduction of 66%, 19.8 ± 1.2 vs. 60.6 ± 3.8 mN/mm2, p < 0.0001, respectively) and gastrocnemius muscles (reduction of 70%, 10.1 ± 1.3 vs. 33.2 ± 6.0 mN/mm2, p = 0.0074, respectively).
Table 2.
Contractility of permeabilized muscle bundles from Wt and NebΔExon55 mice.
| Diaphragm | Soleus | EDL | Gastrocnemius | |||||
|---|---|---|---|---|---|---|---|---|
| Wt (n = 30) | NebΔExon55 (n = 46) | Wt | NebΔExon55 | Wt | NebΔExon55 | Wt | NebΔExon55 | |
| Maximal active tension (mN/mm2) (% of Wt) | 1.00 ± 0.03 | 0.27 ± 0.03† | 1.00 ± 0.06 | 0.41 ± 0.02 | 1.00 ± 0.06 | 0.33 ± 0.02 | 1.00 ± 0.18 | 0.32 ± 0.04 |
| Ktr (s−1) (% of Wt) | 1.00 ± 0.09 | 0.47 ± 0.08†,‡ | 1.00 ± 0.04 | 0.62 ± 0.05 | 1.00 ± 0.02 | 0.63 ± 0.02 | 1.00 ± 0.04 | 0.50 ± 0.06 |
| Active stiffness (mN/mm2) (% of Wt) | 1.00 ± 0.06 | 0.25 ± 0.06 | 1.00 ± 0.13 | 0.35 ± 0.14 | 1.00 ± 0.04 | 0.28 ± 0.03 | 1.00 ± 0.03 | 0.27 ± 0.03 |
| Tension/stiffness ratio | 3.6 ± 0.25 | 3.1 ± 0.21 | 3.6 ± 0.3 | 3.0 ± 0.08 | 3.3 ± 0.26 | 2.8 ± 0.2 | 3.2 ± 0.32 | 2.9 ± 0.31 |
| pCa50 | 5.60 ± 0.01 | 5.50 ± 0.02* | 5.86 ± 0.05 | 5.71 ± 0.05* | 5.66 ± 0.04 | 5.50 ± 0.05* | 6.07 ± 0.05 | 5.91 ± 0.02* |
| nH | 4.62 ± 0.42 | 3.66 ± 0.32 | 1.65 ± 0.17 | 1.20 ± 0.10 | 2.22 ± 0.17 | 1.71 ± 0.17 | 3.11 ± 0.44 | 3.08 ± 0.63 |
n: fiber number;
p < 0.05 Wt vs. NebΔExon55;
p < 0.05 DIA NebΔExon55 vs. SOL NebΔExon55;
p < 0.05 DIA NebΔExon55 vs. EDL NebΔExon55.
Since the maximal tension of diaphragm fibers is reduced, and because nebulin affects cross-bridge cycling kinetics, we determined the rate constant of force redevelopment (Ktr). Ktr was significantly reduced in diaphragm (8.3 ± 0.4 s−1, p < 0.0001), soleus (8.0 ± 0.3 s−1, p = 0.0005), EDL(11.0 ± 0.7 s−1, p < 0.0001) and gastrocnemius (7.0 ± 0.7 s−1, p < 0.0001) NebΔExon55 muscles compared to the Wt muscles (17.6 ± 1.6, 12.9 ± 1.0, 17.4 ± 0.8, 14.5 ± 0.7, s−1, respectively), with the reduction most pronounced in the diaphragm (Table 2, p < 0.05).
To investigate whether the changes in cross-bridge cycling kinetics reduced the number of attached cross-bridges, we measured the active stiffness of the fibers by imposing rapid changes in muscles length. We observed a significant decrease of the active stiffness in NebΔExon55 diaphragm (7.2 ± 1.2 mN/mm2, p < 0.0001), soleus (6.3 ± 0.5 mN/mm2, p < 0.0001), EDL (6.4 ± 0.6 mN/mm2, p < 0.0001) and gastrocnemius (3.0 ± 0.3 mN/mm2, p = 0.0014) compared to Wt muscles (28.0 ± 1.7, 17.9 ± 1.1, 22.8 ± 3.0, 11.6 ± 1.7, mN/mm2, respectively), with no differences among muscle types (Table 2).
We estimated the force generated by cross-bridge by calculating the tension/stiffness ratio. This ratio was not significantly different in NebΔExon55 muscles and Wt and among muscles types: diaphragm (3.1 ± 0.21 vs. 3.6 ± 0.25, p = 0.2248, respectively), soleus (3.0 ± 0.08 vs. 3.6 ± 0.3, p = 0.1078, respectively), EDL (2.8 ± 0.2 vs. 3.3 ± 0.26, p = 0.1571, respectively) and gastrocnemius (2.9 ± 0.31 vs.3.2 ± 0.32, p = 0.4670, respectively).
To determine the calcium sensitivity of force generation, we measured force at a range of calcium concentrations from pCa 9.0 to 4.5. A rightward shift of the pCa curve, reflected by a decrease of the pCa50, was observed in NebΔExon55 muscles compare to Wt (Table 2): diaphragm (5.50 ± 0.02 vs. 5.60 ± 0.01, p = 0.0094), soleus (5.71 ± 0.05 vs. 5.86 ± 0.05, p = 0.022), EDL (5.50 ± 0.05 vs. 5.66 ± 0.04, p = 0.038) and gastrocnemius (5.90 ± 0.03 vs 6.07 ± 0.05, p = 0.032) (Table 2), with no difference among muscle types. This shift indicates that the calcium sensitivity of force is significantly reduced in the NebΔExon55muscles compare to Wt.
The Hill coefficient, representing the cooperativity of activation, is not affected in NebΔExon55 diaphragm (3.66 ± 0.32, p = 0.092), soleus (1.20 ± 0.05, p = 0.050), EDL (1.71 ± 0.17, p = 0.051) and gastrocnemius (3.01 ± 0.67, p = 0.7934) compared to Wt muscles (4.60 ± 0.42, 1.65 ± 0.17, 2.22 ± 0.17, 3.11 ± 0.44, respectively), with no differences among muscle types.
As contractility depends on the MyHC isoforms expressed in the muscle fibers, we analyzed the composition of MyHC isoforms in Wt muscle and NebΔExon55 muscles.
Mouse muscle is composed of several MyHC isoforms: type 1 (expressed in slow-twitch muscles) and three fast isoforms: type 2A, 2X and 2B (expressed in fast-twitch muscles). The results are shown in Table 3. MyHC type 1 and 2B are well separated on gels, whereas the top band is likely to be majority IIA that overlaps with a small amount of IIX that exists in mouse skeletal muscle [22]. We attempted to separate IIA and IIX bands, but were not successful and instead refer to the top band as IIA(X) (Table 3A). We observed that Wt muscles are composed of a majority of type 2B, followed by type 2A(X) and type 1 (Table 3B). In the NebΔExon55 diaphragm we observed a decrease of 18% of type 2B while type 2A(X) increased by 20% compared to Wt. NebΔExon55 EDL presented a similar pattern with a reduction of 17% of type 2B isoform while type 2A(X) increased by 22%. On the other hand, soleus and gastrocnemius muscles did not present any change in MyHC isoform composition. These findings indicate that a change toward slow-type MHC isoforms is not likely to explain the force deficit in NebΔexon55 muscle. (Note that previous work on permeabilized fibers of slow-twitch soleus and fast-twitch EDL muscle showed comparable maximal tension [26], further suggesting that a difference in fiber type does not contribute to the reduced contractility of NebΔexon55 muscle.)
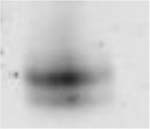
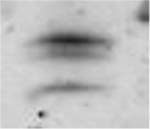
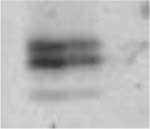
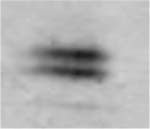
Table 3.
(A) Example of polyacrylamide gel electrophoresis (SDS-PAGE) with a homogenate of murine EDL and soleus mixed together and muscle bundles of diaphragm, soleus, EDL and gastrocnemius; (B) myosin heavy chain isoform composition of permeabilized muscle bundles from Wt and NebΔExon55 mice.
| Diaphragm | Soleus | EDL | Gastrocnemius | |||||
|---|---|---|---|---|---|---|---|---|
| Wt | NebΔExon55 | Wt | NebΔExon55 | Wt | NebΔExon55 | Wt | NebΔExon55 | |
| A | ||||||||
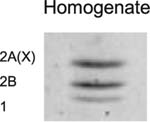 |
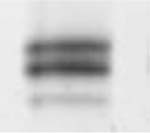 |
 |
 |
 |
 |
 |
 |
 |
| B | ||||||||
| Type 1 (% of total fibers) | 4.9 ± 0.9 | 3.0 ± 0.7 | 30.5 ± 3.9 | 21.5 ± 4.5 | 7.6 ± 2.0 | 4.5 ± 2.9 | 2.3 ± 1.0 | 2.1 ± 1.6 |
| Type 2A(X) (% of total fibers) | 18.1 ± 4.8 | 38.4 ± 1.7* | 15.0 ± 4.0 | 38.5 ± 11.4 | 8.2 ± 3.0 | 30.2 ± 2.8* | 1.8 ± 0.8 | 4.5 ± 2.8 |
| Type 2B (% of total fibers) | 77.0 ± 4.9 | 58.5 ± 1.4* | 54.5 ± 7.3 | 40.0 ± 14.7 | 82.2 ± 3.7 | 65.3 ± 3.4* | 95.9 ± 1.5 | 93.4 ± 4.3 |
p < 0.05 Wt vs. NebΔExon55.
4. Discussion
In the present study, we investigated the effect of deletion of exon 55 in the nebulin gene on the contractility of diaphragm muscle fibers. Diaphragm muscle fibers of NebΔExon55 mice showed severe contractile weakness, a weakness that was more pronounced than that observed in soleus muscle, but not more pronounced than that in EDL and gastrocnemius muscles. The severe diaphragm weakness likely contributes to the development of respiratory failure in NebΔexon55 mice and might explain their early, postnatal death [22].
4.1. Diaphragm weakness in nemaline myopathy
The diaphragm is the main muscle of inspiration, taking up approximately seventy percent of the work of breathing in humans [27]. Contraction of diaphragm muscle fibers creates a negative thoracic pressure resulting in inspiration. Similar to the heart, the diaphragm is highly active with a duty cycle (ratio of active to inactive time) of ~35% [28] compared to 2% for the extensor digitorum longus and 14% for the soleus [29]. The diaphragm is also essential for non-ventilatory behaviors, including expulsive behaviors important for airway clearance. In humans, the diaphragm consists of approximately equal proportions of slow-twitch and fast-twitch muscle fibers [30], fibers which are recruited in an orderly fashion with the slow-twitch fibers being recruited first and the fast-twitch fibers mainly during conditions that require high diaphragm force (e.g. during respiratory failure) [31].
Respiratory failure is the main cause of death in NM, and inspiratory muscle weakness is considered the main contributor to respiratory failure [32]. Although it is generally accepted that the diaphragm is severely affected in NM, perhaps even much more than peripheral muscles [32–35] data on diaphragm contractility in NM patients is scarce and parallel comparison of diaphragm and peripheral muscle strength is, to our knowledge, lacking. Measurements of inspiratory muscle function in NM have been documented in case reports. For instance, Kelly et al. [15] reported significantly reduced maximum inspiratory pressures (MIP, a parameter of in vivo inspiratory muscle function) in a 44-year-old NM-patient resulting in respiratory failure, and a NM-patient with severely reduced MIP was weaned from ventilatory support after inspiratory muscle training improved MIP 2-fold [36], indicating the presence and significance of inspiratory muscle weakness in NM. Furthermore, inspiratory muscle weakness in NM has been assessed using spirometry, which provides only indirect measures of diaphragm function. Thus, the goal of the present study was to quantify the magnitude of diaphragm weakness in NM, its cellular pathophysiology, and to compare this magnitude to that in peripheral skeletal muscles.
Previous work, from our group and from others, on biopsies of peripheral skeletal muscle showed that peripheral muscle weakness in NM patients is, at least partly, caused by dysfunction of sarcomeric proteins. Whether dysfunction of sarcomeric proteins contributes to diaphragm weakness in NM patients has not been investigated, as such studies require biopsy specimens which are difficult to obtain. Thus, in the present study we made use of a mouse model for nebulin-based NM. Mutations in NEB account for ~50% of all NM cases and deletion of exon 55 is one of the most frequent mutations in NEB reported so far [37–39]. The NebΔExon55 mice recapitulate the main features of human NM (including nemaline rods and manifest muscle weakness) with significant sarcomeric dysfunction in tibialis cranialis muscle [22]. In the present study, we investigated the contractility of permeabilized muscle fibers, a preparation which allows us to assess the functioning of contractile proteins in the absence of potential confounding effects of for instance calcium cycling by the sarcoplasmic reticulum. These experiments were performed at a sarcomere length of 2.3 μm. This sarcomere length was chosen because previous work from our group [16] showed that NebΔExon55 muscle has shorter thin filament lengths, and at this length, the overlap of the thin and thick filaments is optimal. Consequently, weakness in NebΔExon55 diaphragm cannot be attributed to shortened thin filament length. Our findings indicate manifest dysfunction of sarcomeric proteins in the diaphragm muscle of NebΔExon55 mice. Maximal tension was reduced to ~25% of Wt levels, a reduction at least partly caused by changes in the kinetics of cross-bridge cycling, resulting in a lower number of bound cross-bridges. Note that the tension reduction is more pronounced at submaximal (in vivo) stimulation as the calcium sensitivity of force was significantly reduced (Fig. 3). Considering that transdiaphragm pressure generation (the gold standard for in vivo diaphragm force) during quiet breathing is ~25% of the maximal transdiaphragm pressure generated during supramaximal bilateral phrenic nerve stimulation (data from small rodents [40,41]), our data suggest that normal breathing in the NebΔExon55 mice requires continuous maximal activation of the diaphragm muscle, a condition that is unlikely to be compatible with life and will likely lead to respiratory failure.
Fig. 3.
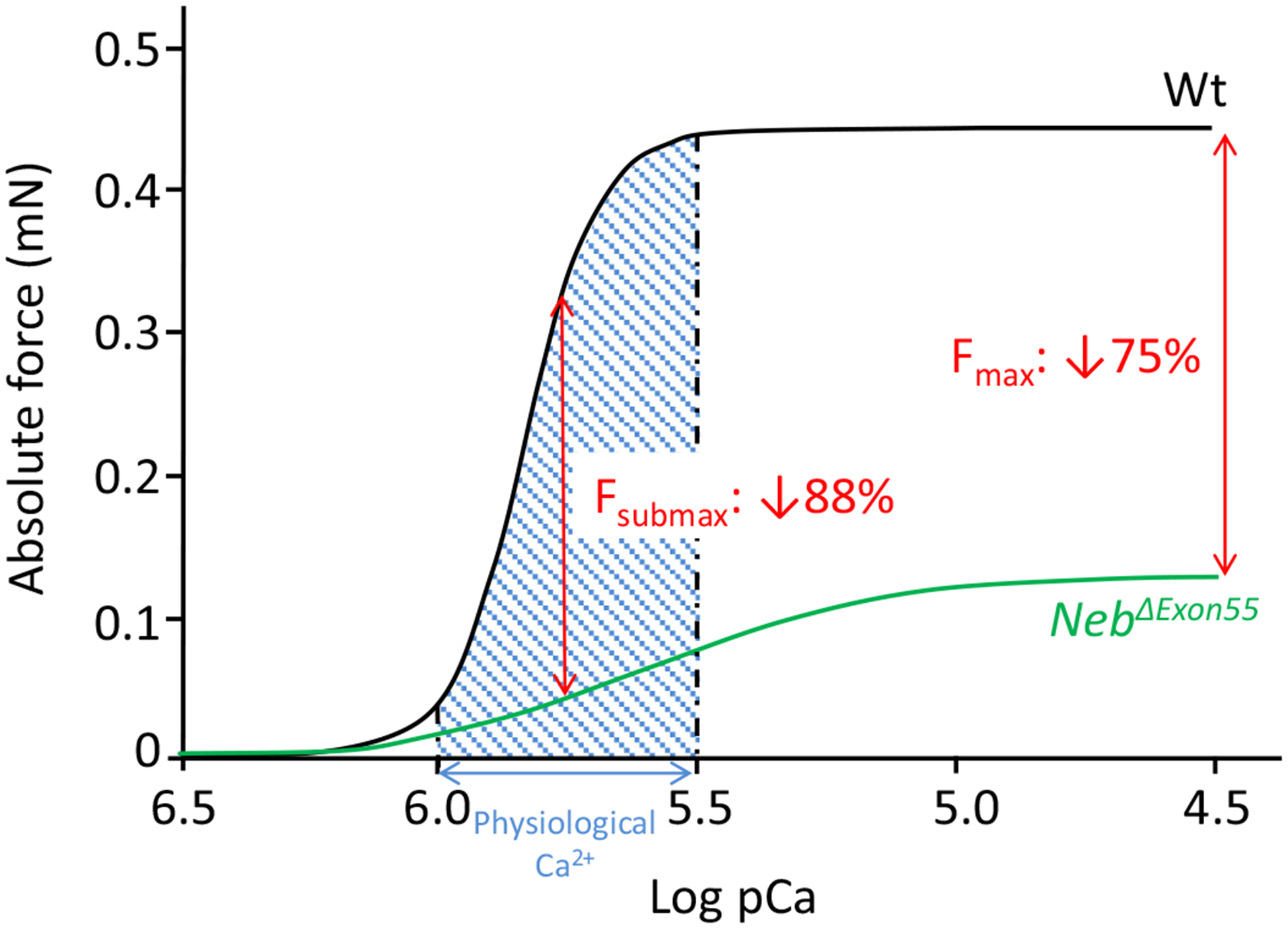
Curves indicate the absolute force–calcium relation of diaphragm bundles of Wt and NebΔExon55 mice. Compared to Wt, at physiological calcium concentration (shade region), the absolute force is reduced by 88%. At pCa 4.5 there is a force decrease of 75% in NebΔExon55 mice compared to Wt.
Previous studies on biopsies of NM patients proposed that the diaphragm is more affected than peripheral skeletal muscles [42], i.e. more nemaline rods were observed in diaphragm than in non-respiratory muscles. The data of the present study do not support this proposition as two out of the three peripheral skeletal muscles tested exhibited a magnitude of weakness that was comparable to that observed in the diaphragm (Table 2). An explanation for this discrepancy might be that diaphragm pathophysiology in NebΔExon55 mice does not resemble that of patients. Alternatively, one could argue that although the number of rods was higher in the diaphragm of patients, the contractility of diaphragm muscle fibers in the patients might have been comparable to that of peripheral muscles fibers (indeed, the number of rods does not seem to correlate with disease severity [42]).
4.2. The diaphragm as therapeutic target in NM
The manifest weakness of the diaphragm and its role in the development of respiratory failure in NM make the inspiratory muscles an important therapeutic target. The use of (moderate) inspiratory muscle strength training has been proven to be beneficial in an adolescent with NM [36]. After two weeks of training (threshold loaded breathing training), the patient made significant gains in inspiratory performance (MIP), accompanied by improvements in resting ventilation. The patient was able to meet her ventilatory requirements, with a reduced perception of respiratory effort, and the day-time ventilatory support was discontinued. Pharmaceutical compounds might be of use as well, more specifically drugs that augment the function of contractile proteins. In vivo administration of levosimendan, a drug that sensitizes troponin to calcium, improved diaphragm contractility in healthy subjects [43] by improving neuromechanical efficiency and by reducing the development of fatigue. Similarly, another class of so-called fast skeletal troponin activators induced a large increase in force generation in diaphragm muscle fibers of critically ill patients [30], illustrating the promise of these compounds.
4.3. Limitations of the study
NebΔExon55 mice die within a week after birth, mimicking the severe congenital form of nemaline myopathy [44]. The severe diaphragm weakness in these mice supports respiratory failure as a cause of death, however, death from insufficient milk intake due to weakness of other muscles (e.g. the facial muscle responsible for suckling) cannot be ruled out. We attempted to isolate hyoid muscles from NebΔExon55 mice for analysis of contractility but were unsuccessful.
In the present study, we measured the contractility of permeablized muscle fibers, a preparation in which only the contractile proteins remain functional. This preparation was used because NM is known to specifically affect the functioning of contractile proteins [22,45], and because measuring the functioning of intact muscle preparations from the diaphragm of six day old mice is challenging and to our knowledge unprecedented. Consequently, the contribution to weakness of upstream processes in excitation–contraction coupling, such as sarcoplasmic reticulum calcium handling, cannot be ruled out, and the 75% reduction in force in permeablized diaphragm fibers might be an underestimation of the reduction in intact diaphragm muscle.
Typically, patients with NM have a predominance of type 1 muscle fibers, a feature that was not apparent in the muscles that were studied in the NebΔExon55 mice. We speculate that this discrepancy is a result of the young age at which the NebΔExon55 mice were studied: the time required for the fiber type shift to occur was too short. Indeed, adult mice with nebulin deficiency do show a predominance of type 1 muscle fibers [26].
Finally, there was a difference in tension developed by the various muscle types of Wt mice (Table 2). The exact cause of this difference is unclear, but might involve differences in the developmental stage of each muscle type and substantial differences in the area occupied by contractile material. Indeed, during isolation of the muscle bundles it was obvious that in Wt mice the gastrocnemius muscle had more connective tissue than the other muscle types. Consequently, a larger fraction of the cross sectional area to which force was normalized was occupied by non-contractile material. Note that based on visual observation during the fiber preparation, we observed no differences in connective tissue within muscle types between Wt and NebΔExon55 mice genotypes.
5. Conclusion
Contractile weakness of the diaphragm is prominent in NebΔExon55 mice, but is not more pronounced than that of most peripheral muscle types. Dysfunction of contractile proteins contributes to diaphragm weakness in NebΔExon55 mice, and provides a therapeutic target to alleviate inspiratory muscle weakness in NM and combat respiratory failure.
Acknowledgments
This work was supported by NWO-VIDI (016.126.319, C.A.C.O.), and National Institutes of Health (5R01AR053897) to H.G.
References
- [1].Shy GM, Engel WK, Somers JE, Wanko T. Nemaline myopathy. A new congenital myopathy. Brain 1963;86:793–810. [DOI] [PubMed] [Google Scholar]
- [2].Nowak KJ, Wattanasirichaigoon D, Goebel HH, et al. Mutations in the skeletal muscle alpha-actin gene in patients with actin myopathy and nemaline myopathy. Nat Genet 1999;23:208–12. [DOI] [PubMed] [Google Scholar]
- [3].Laing NG, Wilton SD, Akkari PA, et al. A mutation in the alpha tropomyosin gene TPM3 associated with autosomal dominant nemaline myopathy NEM1. Nat Genet 1995;10:249. [DOI] [PubMed] [Google Scholar]
- [4].Donner K, Ollikainen M, Ridanpaa M, et al. Mutations in the beta-tropomyosin (TPM2) gene–a rare cause of nemaline myopathy. Neuromuscul Disord 2002;12:151–8. [DOI] [PubMed] [Google Scholar]
- [5].Pelin K, Hilpela P, Donner K, et al. Mutations in the nebulin gene associated with autosomal recessive nemaline myopathy. Proc Natl Acad Sci USA 1999;96:2305–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Yuen M, Sandaradura SA, Dowling JJ, et al. Leiomodin-3 dysfunction results in thin filament disorganization and nemaline myopathy. J Clin Invest 2014;124:4693–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Johnston JJ, Kelley RI, Crawford TO, et al. A novel nemaline myopathy in the Amish caused by a mutation in troponin T1. Am J Hum Genet 2000;67:814–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Thirion C, Stucka R, Mendel B, et al. Characterization of human muscle type cofilin (CFL2) in normal and regenerating muscle. Eur J Biochem 2001;268:3473–82. [DOI] [PubMed] [Google Scholar]
- [9].Ravenscroft G, Miyatake S, Lehtokari V-L, et al. Mutations in KLHL40 are a frequent cause of severe autosomal-recessive nemaline myopathy. Am J Hum Genet 2013;93:6–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Gupta VA, Beggs AH. Kelch proteins: emerging roles in skeletal muscle development and diseases. Skelet Muscle 2014;4:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Gupta VA, Ravenscroft G, Shaheen R, et al. Identification of KLHL41 mutations implicates BTB-Kelch-mediated ubiquitination as an alternate pathway to myofibrillar disruption in nemaline myopathy. Am J Hum Genet 2013;93:1108–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Sambuughin N, Yau KS, Olivé M, et al. Dominant mutations in KBTBD13, a member of the BTB/Kelch family, cause nemaline myopathy with cores. Am J Hum Genet 2010;87:842–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Malfatti E, Böhm J, Lacène E, Romero N, Laporte J. A premature stop codon in MYO18B is associated with severe nemaline myopathy with cardiomyopathy. J Neuromuscul Dis 2015;2:219–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Falgà-Tirado C, Pérez-Pemán P, Ordi-Ros J, Bofill JM, Balcells E. Adult onset of nemaline myopathy presenting as respiratory insufficiency. Respiration 1995;62:353–4. [DOI] [PubMed] [Google Scholar]
- [15].Kelly E, Farrell MA, McElvaney NG. Adult-onset nemaline myopathy presenting as respiratory failure. Respir Care 2008;53:1490–4. [PubMed] [Google Scholar]
- [16].Whitaker J, Love S, Williams AP, Plummeridge M. Idiopathic adult-onset nemaline myopathy presenting with isolated respiratory failure. Muscle Nerve 2009;39:406–8. [DOI] [PubMed] [Google Scholar]
- [17].Maeda MH, Ohta H, Izutsu K, Shimizu J, Uesaka Y. Sporadic late-onset nemaline myopathy as a rare cause of slowly progressive muscle weakness with young adult onset. Muscle Nerve 2015;51:772–4. [DOI] [PubMed] [Google Scholar]
- [18].Taglia A, D’Ambrosio P, Palladino A, Politano L. On a case of respiratory failure due to diaphragmatic paralysis and dilated cardiomyopathy in a patient with nemaline myopathy. Acta Myol 2012;31:201–3. [PMC free article] [PubMed] [Google Scholar]
- [19].Jungbluth H, Sewry CA, Brown SC, et al. Mild phenotype of nemaline myopathy with sleep hypoventilation due to a mutation in the skeletal muscle alpha-actin (ACTA1) gene. Neuromuscul Disord 2001;11:35–40. [DOI] [PubMed] [Google Scholar]
- [20].Shahrizaila N, Kinnear WJM, Wills AJ. Respiratory involvement in inherited primary muscle conditions. J Neurol Neurosurg Psychiatry 2006;77:1108–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].McCool FD, Tzelepis GE. Dysfunction of the diaphragm. N Engl J Med 2012;366:932–42. [DOI] [PubMed] [Google Scholar]
- [22].Ottenheijm CAC, Buck D, Winter JMD, et al. Deleting exon 55 from the nebulin gene induces severe muscle weakness in a mouse model for nemaline myopathy. Brain 2013;136:1718–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Romero NB, Sandaradura SA, Clarke NF. Recent advances in nemaline myopathy. Curr Opin Neurol 2013;26:519–26. [DOI] [PubMed] [Google Scholar]
- [24].de Winter JM, Joureau B, Sequeira V, et al. Effect of levosimendan on the contractility of muscle fibers from nemaline myopathy patients with mutations in the nebulin gene. Skelet Muscle 2015;5:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Oakley BR, Kirsch DR, Morris NR. A simplified ultrasensitive silver stain for detecting proteins in polyacrylamide gels. Anal Biochem 1980;105:361–3. [DOI] [PubMed] [Google Scholar]
- [26].Li F, Buck D, De Winter J, et al. Nebulin deficiency in adult muscle causes sarcomere defects and muscle-type-dependent changes in trophicity: novel insights in nemaline myopathy. Hum Mol Genet 2015;24:5219–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Broaddus VC, Mason RC, Ernst JD, et al. Murray & Nadel’s textbook of respiratory Medicine In: The respiratory system and neuromuscular diseases, ed. disorders S Q R m o e, vol. 2 6th ed. London: Elsevier Health Sciences; 2015. 2064. [Google Scholar]
- [28].Kong FJ, Berger AJ. Firing properties and hypercapnic responses of single phrenic motor axons in the rat. J Appl Physiol 1986;61:1999–2004. [DOI] [PubMed] [Google Scholar]
- [29].Hensbergen E, Kernell D. Daily durations of spontaneous activity in cat’s ankle muscles. Exp Brain Res 1997;115:325–32. [DOI] [PubMed] [Google Scholar]
- [30].Hooijman PE, Beishuizen A, Witt CC, et al. Diaphragm muscle fiber weakness and ubiquitin-proteasome activation in critically ill patients. Am J Respir Crit Care Med 2015;191:1126–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Sieck GC, Fournier M, Enad JG. Fiber type composition of muscle units in the cat diaphragm. Neurosci Lett 1989;97:29–34. [DOI] [PubMed] [Google Scholar]
- [32].Wallgren-Pettersson C, Sewry CA, Nowak KJ, Laing NG. Nemaline myopathies. Semin Pediatr Neurol 2011;18:230–8. [DOI] [PubMed] [Google Scholar]
- [33].Tsujihata M, Shimomura C, Yoshimura T, et al. Fatal neonatal nemaline myopathy: a case report. J Neurol Neurosurg Psychiatry 1983;46:856–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Matsuo T, Tashiro T, Ikeda T, Tsujihata M, Shimomura C. Fatal neonatal nemaline myopathy. Acta Pathol Jpn 1982;32:907–16. [DOI] [PubMed] [Google Scholar]
- [35].Gillies C, Raye J, Vasan U, Hart WE, Goldblatt PJ. Nemaline (rod) myopathy: a possible cause of rapidly fatal infantile hypotonia. Arch Pathol Lab Med 1979;103:1–5. [PubMed] [Google Scholar]
- [36].Smith BK, Bleiweis MS, Zauhar J, Martin AD. Inspiratory muscle training in a child with nemaline myopathy and organ transplantation. Pediatr Crit Care Med 2011;12:e94–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Lehtokari V-L, Greenleaf RS, DeChene ET, et al. The exon 55 deletion in the nebulin gene–one single founder mutation with world-wide occurrence. Neuromuscul Disord 2009;19:179–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Lehtokari VL, Pelin K, Sandbacka M, et al. Identification of 45 novel mutations in the nebulin gene associated with autosomal recessive nemaline myopathy. Hum Mutat 2006;27:946–56. [DOI] [PubMed] [Google Scholar]
- [39].Anderson SL, Ekstein J, Donnelly MC, et al. Nemaline myopathy in the Ashkenazi Jewish population is caused by a deletion in the nebulin gene. Hum Genet 2004;115:185–90. [DOI] [PubMed] [Google Scholar]
- [40].Sieck GC. Physiological effects of diaphragm muscle denervation and disuse. Clin Chest Med 1994;15:641–59. [PubMed] [Google Scholar]
- [41].Mantilla CB, Seven YB, Zhan WZ, Sieck GC. Diaphragm motor unit recruitment in rats. Respir Physiol Neurobiol 2010;173:101–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Ryan MM, Ilkovski B, Strickland CD, et al. Clinical course correlates poorly with muscle pathology in nemaline myopathy. Neurology 2003;60:665–73. [DOI] [PubMed] [Google Scholar]
- [43].Doorduin J, Sinderby CA, Beck J, et al. The calcium sensitizer levosimendan improves human diaphragm function. Am J Respir Crit Care Med 2012;185:90–5. [DOI] [PubMed] [Google Scholar]
- [44].Saunier M, Bonnemann CG, Durbeej M, Allamand V. 212th ENMC International Workshop: animal models of congenital muscular dystrophies, Naarden, The Netherlands, 29–31 May 2015. Neuromuscul Disord 2016;26(3):252–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Ottenheijm CAC, Hooijman P, DeChene ET, Stienen GJ, Beggs AH, Granzier H. Altered myofilament function depresses force generation in patients with nebulin-based nemaline myopathy (NEM2). J Struct Biol 2010;170:334–43. [DOI] [PMC free article] [PubMed] [Google Scholar]