
Figure 1. Defining novel RAS vulnerabilities for the development of anti-RAS strategies.
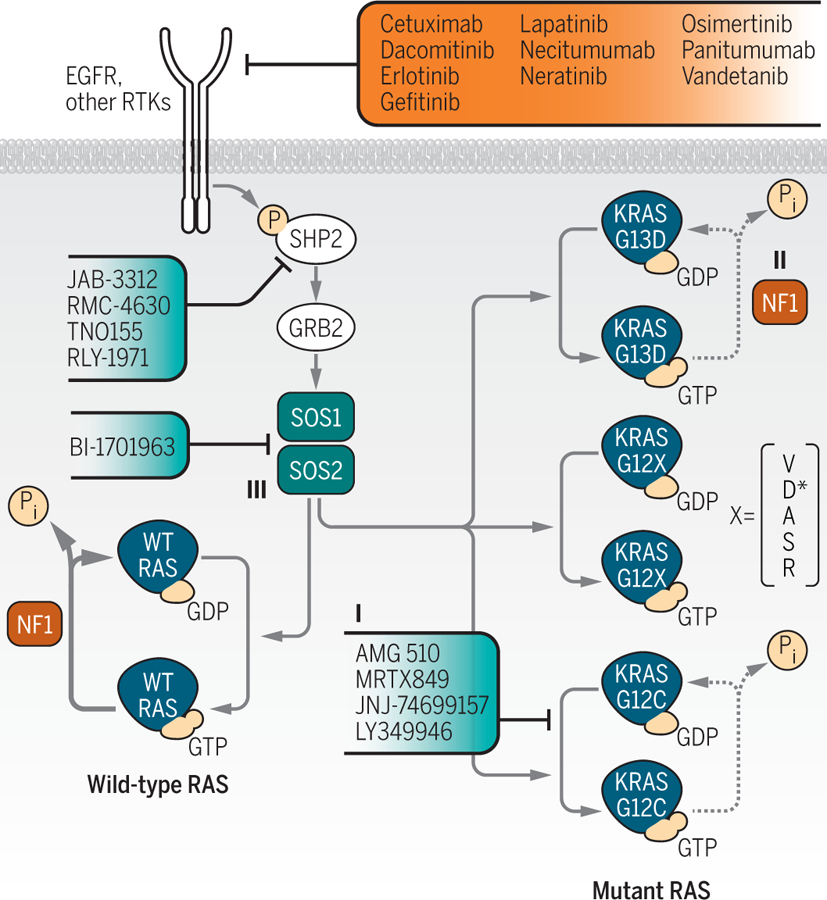
A series of recent studies, addressing different aspects of RAS function, have provided potential new clues to targeting RAS for cancer treatment. Where appropriate, clinically approved (red) and investigational (blue) therapeutics indicated for targeting RAS activation and signaling in cancer are noted. Lou et al. applied a CRISPR genetic loss-of-function screen to identify signaling modulators of a small molecule inhibitor selective for the KRASG12C mutant (I) found predominantly in lung cancer7. In contrast to other G12 mutants, KRASG12C retains intrinsic GTP-hydrolysis (*with the exception of G12D which retains minor GTP-hydrolysis), thereby enabling its’ targeting by GDP-KRASG12C-specific small molecules. The findings revealed various pathways to target to enhance the potency and durable efficacy of the inhibitor. McFall et al. proposed a mechanism to explain the EGFR-dependence of KRASG13D-mutant colorectal cancer22. Decreased affinity for the RAS-GAP NF1 (II) by the G13D mutant protein enables GAP- and EGFR-dependent regulation of wild-type RAS, thereby retaining sensitivity to EGFR inhibitors in KRASG13D-mutant cells. And Sheffels et al. identified a role for the RAS-GEF SOS2 (III) in promoting WT HRAS activation of AKT to support mutant KRAS-induced transformation of mouse fibroblasts in 3D growth culture conditions.