

Posttranslational modifications by phosphorylation can change protein activities, binding partners, or localization. Tyrosine 102 is conserved between delta papillomavirus BPV-1 and alpha papillomavirus HPV-31 E2. We characterized mutations of HPV-31 E2 for interactions with relevant cellular binding partners and replication in the context of the viral genome.
KEYWORDS: E2, HPV, tyrosine phosphorylation, viral replication
ABSTRACT
Several serine and threonine residues of the papillomavirus early E2 protein have been found to be phosphorylated. In contrast, only one E2 tyrosine phosphorylation site in BPV-1 (tyrosine 102) and one in HPV-16/31 (tyrosine 138) site have been characterized. Between BPV-1 and HPV-31 E2, 8 of the 11 tyrosines are conserved in the N-terminal domain, suggesting that phosphorylation of tyrosines has an essential role in E2 biology. In this study, we examine the effect of Y102 phosphorylation on HPV-31 E2 biology. Y102 proteins mutated either to the potential phospho-mimetic glutamic acid (Y102E) or to the nonphosphorylated homologue phenylalanine (Y102F) remain nuclear; however, Y102E is more associated with the nuclear matrix fraction. This is consistent with the inability of Y102E to bind TopBP1. Both BPV-1 and HPV-31 Y102E are similar in that neither binds the C terminus of Brd4, but in all other aspects the mutant behaves differently between the two families of papillomaviruses. BPV-1 Y102E was unable to bind E1 and did not replicate in a transient in vitro assay, while HPV-31 Y102E binds E1 and was able to replicate, albeit at lower levels than wild type. To examine the effect of E2 mutations under more native-like infection conditions, a neomycin-selectable marker was inserted into L1/L2 of the HPV-31 genome, creating HPV-31neo. This genome was maintained in every cell line tested for at least 50 days posttransfection/infection. Y102E in both transfection and infection conditions was unable to maintain high episome copy numbers in epithelial cell lines.
IMPORTANCE Posttranslational modifications by phosphorylation can change protein activities, binding partners, or localization. Tyrosine 102 is conserved between delta papillomavirus BPV-1 and alpha papillomavirus HPV-31 E2. We characterized mutations of HPV-31 E2 for interactions with relevant cellular binding partners and replication in the context of the viral genome.
INTRODUCTION
Human papillomavirus 31 (HPV-31) is a small double-stranded DNA virus that can infect mucosal epithelium and, if persistent, can lead to cervical and oropharyngeal cancer. Regulation of both viral transcription as well as replication relies on the viral early protein E2. The N-terminal 200 amino acids of E2 contain the transcriptional activation domain (TAD), which regulates a multitude of functions during the viral life cycle. More than 70 proteins have been reported to interact with E2 TAD (1, 2). The C-terminal 100 amino acids form a high-affinity, sequence-specific DNA-binding domain (DBD). Separating these two regions is the nonconserved hinge domain.
Posttranslational phosphorylations often alter protein stability, induce a conformational change, cause subcellular redistribution, or modify the repertoire of protein-protein interactions. Several phosphorylation sites have been discovered in E2, the first of which were S298 and S301 in BPV-1 in the DBD (3). Preventing phosphorylation at these sites through mutation to alanine increased E2 protein levels and corresponding transient replication and transcription activities (4), suggesting that phosphorylation at these sites is inhibitory.
The absence of any prior identification of tyrosine (Y) phosphorylation in E2 could be attributed to the method of purification of E2 from insect or mouse cells (3, 5, 6). Our lab recently identified the first phosphorylated tyrosine residue, Y102, through mass spectroscopy of E2 purified from human cervical cells (7). Mutation of this tyrosine to the phospho-mimetic glutamic acid (Y102E) inhibited both the transcription and replication activities of E2. The homologous site in HPV-31 E2 is also tyrosine (Y102); however, given that HPV-31 and BPV-1 are only moderately identical, have different tissue tropisms, and are under different transcriptional control (8), it is possible the role of phosphorylation at Y102 differs between HPV-31 and BPV-1. In this study, HPV-31 E2 tyrosine 102 was mutated to glutamic acid (Y102E) as well as to phenylalanine (Y102F) to mimic the phospho-deficient form, and the mutants were examined for effects on replication, transcription, localization, and genome maintenance.
RESULTS
Identification of tyrosine phosphorylation in HPV-31 E2.
We discovered the first tyrosine phosphorylation site of the papillomavirus E2 protein at residue 102 through mass spectrometry analysis of the BPV-1 E2 protein (7). Y102 is located in the TAD domain (Fig. 1A) and is conserved between BPV-1, HPV-16, and HPV-31 E2 (Fig. 1B). To determine if HPV-31 E2 can be phosphorylated at Y102, we performed mass spectrometry analysis of HPV-31 E2 overexpressed in HEK 293TT cells. The peptides collected covered 95% of the entire E2 protein (Fig. 1C) and a peptide with phosphorylated Y102 was captured multiple times, along with several additional phosphorylated residues (Fig. 1D). The protein sequence of HPV-31 E2 is only 28% identical to BPV-1, but, despite this low homology, most (8 of 11) of the tyrosines in the N-terminal TAD domain are conserved, including Y102. In contrast, only about 30% of the BPV-1 serine and threonine residues are similarly positioned in the HPV-31 TAD. This preference of tyrosine conservation suggests phosphorylation at these sites is essential for E2 function and regulation. Three computer algorithms also predict that Y102 is among the three most likely phosphorylated residues in HPV-31 (NetPhos 3.1 (9), PhosphoSVM (10), and Viral Phos [http://csb.cse.yzu.edu.tw/ViralPhos/index.html]).
FIG 1.

Identification of phosphorylated Y102. (A) Schematic of HPV-31 E2 protein representing the transactivation domain (TAD) and DNA-binding domain (DBD). (B) Conservation of the region surrounding Y102. (C) HPV-31 E2 was purified, digested with trypsin, and submitted for mass spectrometric analysis. Peptide coverage was 95% as highlighted in gray. (D) Phosphorylated amino acids of HPV-31 E2 with high PhosphoRS probabilities (above 75%) are listed on the left. The trypsin fragments carrying the modifications are listed in the middle, with phosphorylation sites in black. HPV-16 and BPV-1 homologue residues are listed on the right, with conserved residues in black and nonconserved in gray. (E) FLAG-HPV-31 E2 (WT or Y102 mutants) was expressed in HEK 293TT cells, immunoprecipitated with custom phospho-specific Y102 antibody, and blotted with FLAG antibody. (F) Y102 mutants localize to the nucleus. N/TERT cells were transfected with FLAG-HPV-31 E2 (WT, Y102E, and Y102F) and HA-HPV-31 E1, and immunostained with FLAG and HA antibodies.
Polyclonal antibodies to phosphorylated HPV-31 E2 Y102 were generated in rabbits as a tool to examine Y102 phosphorylation. We created point mutants in HPV-31 E2 to mimic the phosphorylated state (Y102E) as well as the nonphosphorylated state (Y102F) of E2, and tested their ability to be recognized by the phospho-102 antibody. HEK 293TT cells transfected with the wild-type (WT) as well as mutant FLAG-HPV-31 E2 proteins were immunoprecipitated with the phospho-102 antibody. As expected, Y102E was the predominant form enriched, with minor binding seen for WT and Y102F (Fig. 1E). A synthesized phosphorylated E2 peptide was used as the antigen, but the phosphate group will get cleaved upon injection into the rabbit. As a result, the resultant polyclonal antibody stock will detect nonphosphorylated as well as phosphorylated protein as, indeed, we see in Fig. 1E. Robust recognition of Y102E by the phospho-102 antibody also suggests that this glutamic acid mutation is a close homologue in structure to the true phosphorylated form of Y102. Unfortunately, this antibody was not effective for E2 localization or coimmunoprecipitation experiments and, while useful for immunoblotting, detection of native E2 in infected cells was not reliable.
Nuclear localization of HPV-31 E2 Y102 mutants.
E2 resides in the nucleus, which coincides with its role in genome replication and transcription. Two basic-rich nuclear localization signals (NLS) were identified in BPV-1: N-terminal 107 KRCFKKGAR 115 and C-terminal 339 KCYRFRVKKNHRHR 352 (11). As this N-terminal NLS is close to the Y102 phosphorylation site, and since nuclear shuttling often is modulated by adjacent phosphorylation (12, 13), HPV-31 Y102 mutants might have altered localization. Examination of the HPV-31 E2 sequence, however, reveals conservation of only the C-terminal NLS: 306 KCLRYRLSKYK 316. The N-terminal NLS region contains very few basic amino acids, making it unlikely this region confers localization, and, therefore, phosphorylation of Y102 should not interfere with nuclear positioning. To determine if Y102 affects E2 localization, FLAG-tagged WT, Y102E, and Y102F HPV-31 E2 mutants were cotransfected with HA-HPV-31 E1 into N/TERT cells (human foreskin keratinocytes immortalized with H-tert) (14), fixed with paraformaldehyde, and immunostained with M2 (FLAG) and HA antibodies. Localization of WT E2 as well as both mutants was nuclear (Fig. 1F).
The E2-interacting protein topoisomerase II binding protein 1 (TopBP1) is a large scaffolding protein with nine breast cancer gene C-terminal (BRCT) domains (15) that mediate phospho-dependent protein interactions required for the initiation of human DNA replication, initial viral replication, and segregation of the viral genome during mitosis (16–18). Changing the phosphorylation state of E2 could therefore modulate its ability to bind TopBP1. To determine if binding is altered, FLAG-HPV-31 E2 WT and mutants were transfected into HEK 293TT cells, which were then lysed and subjected to immunoprecipitation (IP) with TopBP1 antibody. Immunoblot analysis of the precipitates shows that WT and Y102F bind endogenous TopBP1, while Y102E binding was greatly reduced (Fig. 2A). Performing the reverse IP with FLAG antibody similarly showed that WT and Y102F, but not Y102E, coimmunoprecipitate TopBP1 (Fig. 2B). Previous studies on HPV-16 E2 demonstrated that in the absence of TopBP1, E2 associates with a nuclear matrix fraction (18). With Y102E unable to bind TopBP1, a similar relocalization would be expected to occur. E2 from transfected cells was sequentially extracted according to an established protocol (Fig. 2C), and Western blot analysis showed that, as predicted, the majority of Y102E associated with the nuclear matrix fraction, much more so than WT E2 (Fig. 2D and E). To control for fractionation, lysates were analyzed for tubulin, histone, and p53: cytoplasmic tubulin was present in the cytoplasmic fraction (lane 1); nuclear histones were released upon DNase and moderate salt treatment (lanes 3 and 4); and p53 was present as previously described in both soluble (lane 1) and nuclear matrix fractions (lane 5) (19) (Fig. 2D).
FIG 2.

HPV-31 E2 Y102E does not bind TopBP1. (A and B) Lysates of cells transfected with FLAG-HPV-31 E2 (WT, Y102E, and Y102F) were immunoprecipitated either with an antibody to TopBP1 (A) or FLAG (B). (C) Diagram of lysate fractionation performed in parts D and E. (D) HEK 293TT cells transfected with FLAG-HPV-31 E2 WT or Y102E were lysed and fractionated according to the scheme shown in C, run on a gel, and blotted with FLAG, tubulin, histone, and p53 antibodies. (E) Ratio of nuclear matrix bound (lane 4 in D) over soluble (lane 1) HPV-31 E2 from three experiments, normalized to WT. Y102E was significantly different from WT with P < 0.005, and means are expressed ± SEM.
E2 Y102E transcription and DNA replication activities.
Transcription of papillomavirus genes is mainly regulated by E2 and its ability to recruit cellular transcription factors to the viral genome. One such factor is the activation domain modulating factor (Amf1) protein, also known as G-protein pathway suppressor (GPS2). Previously, our lab has shown that the beta-sheet region of BPV-1 E2 binds to GPS2, and mutants that cannot bind GPS2 are defective for transcriptional activation (20). To determine if mutation of Y102 effects binding, soluble lysate from HEK 293TT cells transfected with FLAG-HPV-31 E2 constructs and HA-GPS2 was immunoprecipitated with M2 FLAG antibody. GPS2 bound to wild type and Y102F efficiently, while Y102E had significantly decreased binding (Fig. 3A).
FIG 3.
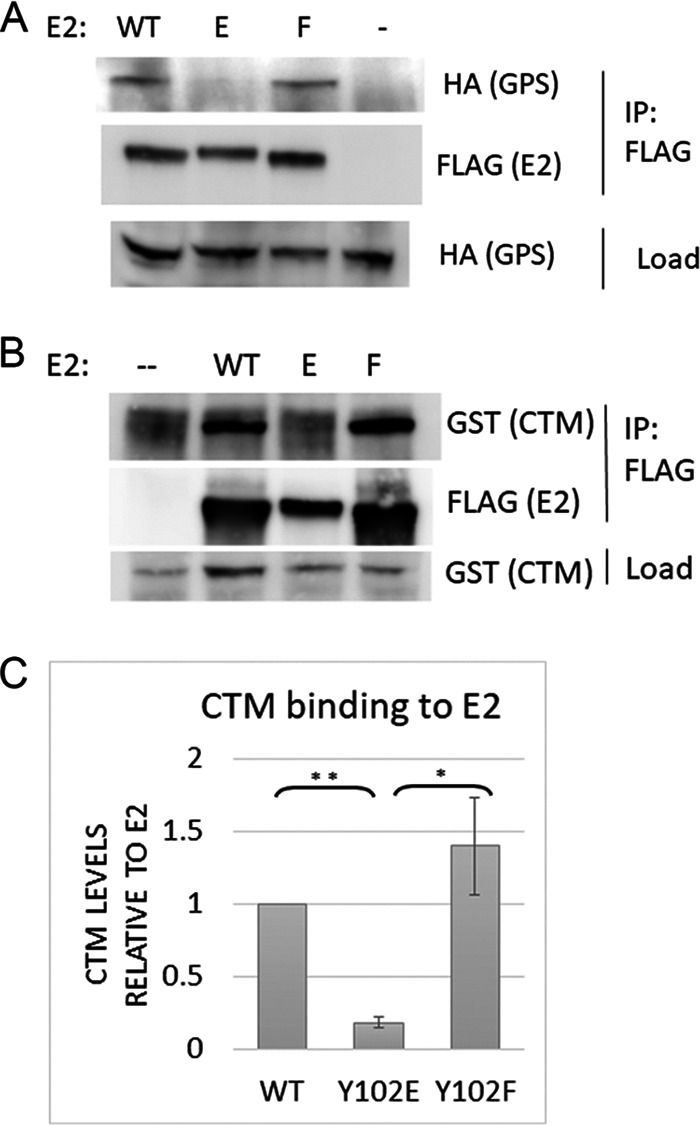
Y102E prevents binding to GPS2 and Brd4-CTM. (A) HEK 293TT cells transfected with FLAG-HPV-31 E2 (WT, Y102E, and Y102F) and HA-GPS2 were lysed and immunoprecipitated with HA antibody. (B) Same as A, except GST-CTM was cotransfected instead of GPS2. (C) Ratio of immunoprecipitated GST-CTM normalized to E2 from three experiments. Values are expressed as means ± SEM. *, P < 0.05; **, P < 0.0001.
Brd4 binding to E2 also regulates the transcriptional activities of E2. Since the BPV-1 Y102E mutant was unable to bind the C-terminal motif (CTM) of Brd4, we sought to check if this occurs with HPV-31 E2. GST-tagged CTM was coexpressed with FLAG-HPV-31 E2 WT or Y102 mutants and immunoprecipitated with FLAG antibody. WT and Y102F bound efficiently (Fig. 3B), while Y102E binding was decreased ∼80% compared to WT, as calculated from three replicates (Fig. 3C).
HPV DNA replication requires E2 binding to the viral E1 DNA helicase. E2 transports E1 to binding palindromes at the origin of replication (ori) (nucleotides [nt] 7721 to 100) (21). To determine if Y102 mutants alter E1 binding, lysates from HA-HPV-31 E1 and FLAG-HPV-31 E2 WT/mutant transfected HEK 293TT cells were immunoprecipitated with FLAG antibody. WT, Y102E, and Y102F all bound E1 to similar levels (Fig. 4A). In contrast, BPV-1 E2 Y102E was unable to bind E1, preventing replication of the BPV-1 genome (7). With HPV-31 E2 Y102E still competent for binding to E1, HPV-31 Y102E might retain replication activities. We utilized a luciferase reporter system in which firefly luciferase is encoded on a plasmid in cis with the HPV-31 origin (22). Cotransfection of this reporter together with E1 and E2 plasmids resulted in increased luciferase expression. Since E2 can also transcriptionally activate gene expression, a control plasmid encoding Renilla luciferase was included. Replication activity can then be calculated as firefly expression divided by Renilla expression. Y102F did not affect transient replication (Fig. 4B), while Y102E retained replication, though at 70% of WT.
FIG 4.
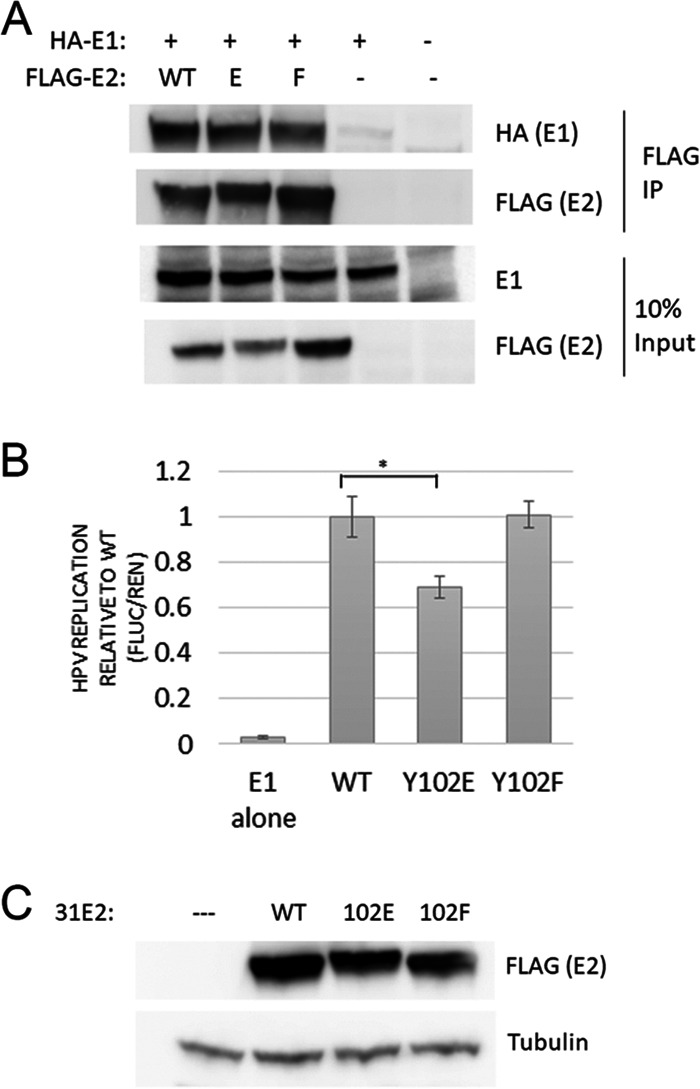
Y102E supports transient replication. (A) Y102E/F mutants bind E1. HEK 293TT cells transfected with HA-HPV-31 E1 and FLAG-HPV-31 E2 (WT, Y102E, and Y102F) were lysed and immunoprecipitated with FLAG antibody. (B) In vitro luciferase-based transient replication assay. WT/mutant E2 along with E1, pRenilla, and luciferase plasmid with E2-binding sites were transfected into C33a cells. Following 2 days of transfection, luciferase signals analyzed with DuoGlo kit were normalized against Renilla. Replication levels are an average from 6 replicates. Y102E replicates at significantly lower levels than WT (*, P < 0.005), and means are expressed ± SEM. (C) Expression of E2 and actin loading control for part B.
Upon infection of cells with HPV-31, the genome either is maintained as an extrachromosomal DNA entity called an episome that segregates during mitosis, or integrates into the host chromosomes with a breakpoint often in the E2 region, disrupting E2 regulatory functions. While Y102E and Y102F transiently replicate in C33a cells, the abnormally high levels of transfected E1/E2 in this system may not accurately reflect their ability to initiate DNA replication, maintain the entire genome, and segregate. To examine maintenance of genomes in cells, we created HPV-31 quasiviruses and infected HaCaT and N/TERT cells. Isolation of DNA from these cells at 2, 4, and 7 days postinfection revealed that the cells consistently failed to maintain genomes past day 4 (Fig. 5A). Given the success with which addition of a neo cassette within the HPV-18 genome required cells to retain the genome (23), we sought to create a similar selectable HPV-31 genome in which the L1/L2 region was replaced with the SV40-promoter-intron-Neo-β-actin-polyA cassette, creating HPV-31neo (Fig. 5B). N/TERT cells were transfected with HPV-31neo WT, Y102E, and Y102F constructs, maintained for 50 days in Geneticin, and harvested by Hirt extraction, which specifically isolates small episomal DNA. As shown in Fig. 5B, WT and Y102F replicated the genome and were maintained as episomes, but not Y102E. Genomic DNA isolated from these same cell lines show that all three genomes are present. E1̂E4 RNA was produced in all three cell lines (Fig. 5D). Together these data suggest that the E2 Y102E genome was unable to be maintained episomally and integrated early after infection.
FIG 5.
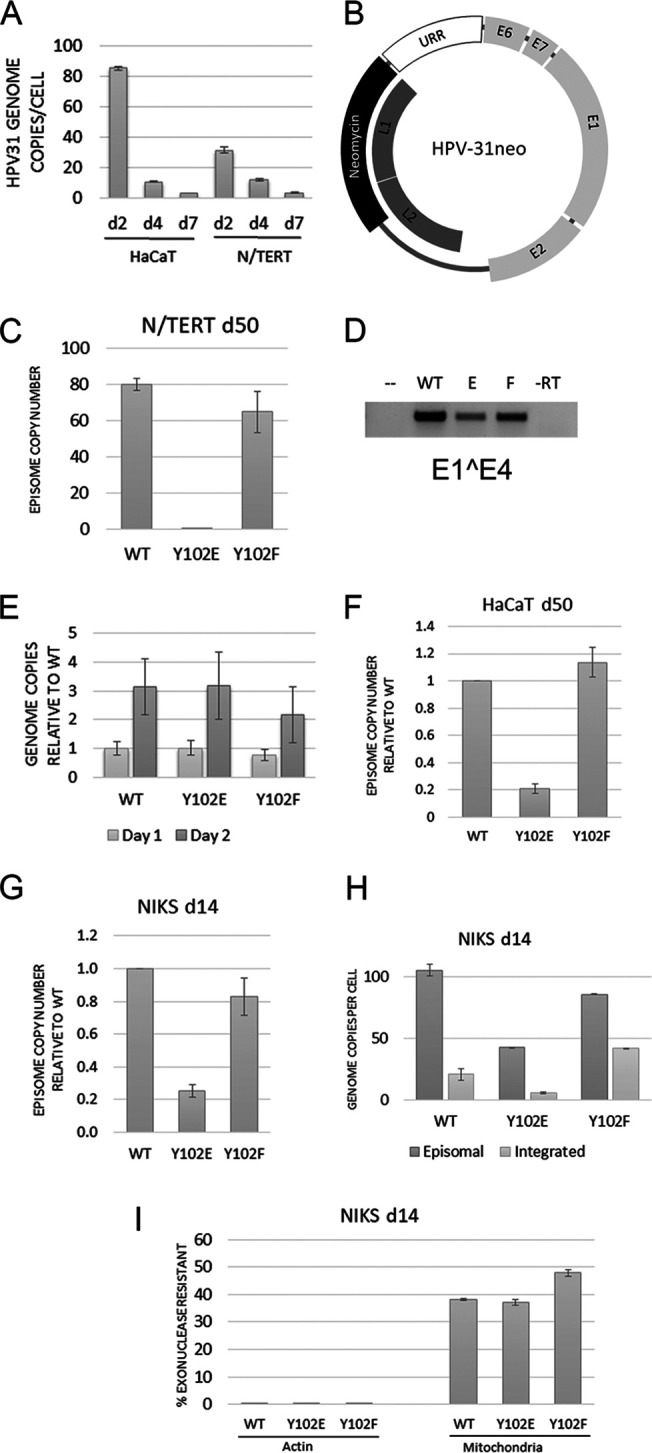
HPV-31 E2 Y102E prevents episomal maintenance. (A) HaCaT and N/TERT cells infected with HPV-31 quasivirus were harvested at days 2, 4, and 7 postinfection. Total DNA was isolated and amplified with primers to the long control region (LCR). (B) Schematic of HPV-31neo genome. The inserted neomycin cassette replaced parts of the L1 and L2 genes in the HPV-31 genome. (C) N/TERT cells transfected with WT, Y102E, or Y102F HPV-31neo genomes were stably maintained through Geneticin selection. At 50 days posttransfection, episomal DNA was isolated through Hirt extraction and the copy number was established through qPCR with LCR primers. (D) RNA was extracted from the cells in B and amplified with primers to E1̂E4. (E) Total DNA was isolated from HaCaT cells at 1 and 2 days postinfection with quasivirus, amplified with primers to LCR, and normalized to the WT genome. (F) Hirt extraction of HaCaT cells infected with quasivirus and stably maintaining genome for 50 days. (G) Same as E except NIKS cells were infected for 14 days. (H) Episomal DNA from 14-day NIKS infection isolated through the exonuclease V method. (I) Controls for exonuclease digestion of part H. Linear genomic actin DNA should be completely digested by exonuclease, while circular mitochondrial DNA is more resistant to digestion. Each graph is representative of multiple infections, and means are expressed ± SEM.
In additional experiments, quasivirions made with HPV-31neo genomes packaged into L1/L2 capsid proteins were infected into NIKS and HaCaT cells to similarly examine episomal maintenance, but in a context with more physiologically accurate copy numbers. Initial infections were performed with pseudoviruses packaging a plasmid encoding mCherry to quantify infection rate. Infection of both NIKS and HaCaTs initially only resulted in 25% red-fluorescing cells. More virus could be used to increase infectivity, but we preferred to use a more normal physiological range of infection with less than 50 viral particles/cell. Since several reports suggest that binding of virus first to the extra cellular matrix (ECM) before the cell surface yields efficient infection (24–26), we tried HaCaT ECM for increasing infectivity. First, HaCaT cells were grown to confluence, the cells but not the deposited ECM were removed with ammonium hydroxide/Triton-X, then virus was allowed to bind overnight. The next day, unbound virus was removed, fresh medium was added along with either HaCaT or NIKS cells. Following 2 days of infection, >95% of cells expressed mCherry and fluoresced red, suggesting that HaCaT ECM drastically enhanced infectivity of these cell lines.
With efficient infection conditions established, we infected HaCaTs with quasivirus encoding WT, Y102E, and Y102F HPV-31 genomes. At days one and two, total DNA was isolated through phenol chloroform extraction and quantified through quantitative PCR (qPCR). All three infections showed similar genome content, though Y102F was present at slightly lower levels (Fig. 5E). At 50 days postinfection, when the cells were well established, DNA was extracted from cell pellets using the Hirt method, and taking into consideration that Y102F had slightly lower initial genomes, we found that again Y102E was unable to maintain high episomal copy number compared to WT and Y102F (Fig. 5F). Infection of NIKS cells similarly resulted in low Y102E episomal copy number (Fig. 5G). Primary HFKs infected with Y102E virus did not result in any proliferating colonies.
Hirt separation of DNA into episomal and cellular genomic fractions has been a widely accepted technique, but some of one fraction frequently contaminates the other. A new technique for distinguishing episomal from integrated HPV involves isolating total DNA with a kit, digesting any linear cellular DNA with an exonuclease, and performing qPCR for the HPV genome, a nuclear gene, and a mitochondrial gene (27). Exonuclease III and V were first compared for DNA digestion. Since exonuclease V digestion was more consistent, more efficient, and does not digest nicked DNA (unlike exonuclease III), all experiments were conducted with exonuclease V. The infected NIKS cells in Fig. 5G were analyzed by this exonuclease protocol for comparison to the Hirt method. HPV genomic qPCR of exonuclease-reacted DNA showed episomal DNA levels consistent with Hirt extraction and that Y102E cells did not contain high episomal copy numbers (Fig. 5H). By subtracting uncut from cut DNA, the integrated DNA content can be estimated. As an internal control for exonuclease, actin DNA levels were monitored to confirm that linear genomic DNA is efficiently digested with exonuclease, and mitochondrial DNA levels should remain relatively unchanged, since the circular mitochondrial genome is not a substrate of exonuclease (Fig. 5I).
DISCUSSION
Several serine/threonine phosphorylation sites have been identified in the papillomavirus protein E2 (3, 5, 6, 28, 29), but only two tyrosine phosphorylation sites have been examined: Y102 and Y138 (7, 30). Phosphorylation at Y102 was originally identified in BPV-1 E2 through mass spectrometric analysis (7) and subsequently confirmed in the current work for HPV-31 E2. Mutation of Y102 to the phospho-mimetic residue glutamic acid (Y102E) blocked viral genome replication. Since HPV-31 also has a tyrosine at residue 102, we sought to determine if this inhibitory phenotype is conserved between BPV-1 of the delta papillomavirus family and HPV-31 of the alpha papillomavirus family.
Glutamic acid (E) or phenylalanine (F) mutations were introduced at position Y102 to mimic either the phosphorylated or unphosphorylated state, respectively, of the tyrosine. When introduced into either BPV-1 or HPV-31, these mutations did not alter the normal nuclear localization of E2 and both the Y102E mutants no longer bound the C terminus of Brd4. These are the extent of the similarities. As summarized in Table 1, BPV-1 Y102E mutant was defective while HPV-31 E2 retained binding to E1, directly correlating with the results observed in transient DNA replication assays. Given that the TAD domain of BPV-1 and HPV-31 only share 28% identity, it is possible that the variations in the interface between E2 and its binding partners results in the differences in BPV-1 versus HPV-31 Y102E binding to TopBP1, GPS2, and E1. Other labs have also noted differences between BPV-1 and HPV-31 binding; for example, BPV-1 but not alpha papillomavirus E2 proteins segregate tightly with chromosomes during mitosis (31), and HPV-16 and BPV-1 may have different segregation receptors (18).
TABLE 1.
Summary of BPV-1 and HPV-31 E2 Y102E mutant phenotypes
| Binds E1 | Binds Amf | Binds CTM | Binds TopBP1 | Nuclear | Transient replication | Stable episome | |
|---|---|---|---|---|---|---|---|
| BPV-1 | − | + | − | + | + | − | − |
| HPV-31 | + | − | − | − | + | + | − |
Initial quasivirus infections utilized HPV-31 genome without any selection. Regardless of the cell line used, the high genome copy numbers found at 1 or 2 days posttransfection became negligible by days 4 and 7. One method to selectively maintain HPV in cell lines is to cotransfect the HPV-31 genome with another plasmid containing a selection gene, but in our experience the resulting cell lines preferentially retain the selectable plasmid and lose the HPV genome. Only upon substituting the neo cassette for L1/L2 sequences was HPV genome copy number reliably maintained in cells. HPV-31 L2 and L1 genes contain multiple CCCTC DNA sequences which confer CTCF binding (32). Mutation of these sites in L2 restricted episomal maintenance of the HPV-31 genome, as well as genome amplification (32); however, the effect of mutation of the L1 sites has not been examined. HPV-31neo retained the three L2 CCCTC sequences and deleted the two L1 sites. Our observed amplification and episomal maintenance of HPV-31neo genome suggests that these CCCTC sites within L1 are not required for HPV replication.
HPV-31 E2 Y102E no longer bound TopBP1, and, as a result, associated very tightly with the nuclear matrix. Other residues in the same region, N89/E90, have also been characterized as necessary for TopBP1 association (33). While TopBP1 is not essential in transcriptional activation assays, it has been implicated as crucial for either initiation of replication in early infection or segregation during mitosis (16–18). The reduced interaction of Y102E with TopBP1 may prevent episomal maintenance and mitotic segregation in cells infected with HPV-31neo Y102E. This could explain our observation that Y102E always formed at least 10-fold fewer colonies than WT when equal amounts of quasivirus were used for infections. Infections were successfully performed in multiple cell lines, and even though HaCaT cells are notorious for being unable to support long-term HPV episomes (34–36), HaCaT cells can do so with a cis-linked neo selection gene.
Early studies on E2 phosphorylation concluded that at least 20 phosphorylation sites are present in the N terminus (5), none of which were on a tyrosine (3). These experiments were based upon E2 expressed in insect cells that are not capable of fully modifying human proteins; there are only 3 tyrosine kinases in the Spodoptera frugiperda genome, instead of the full complement of 90 human kinases. Also limiting detection in these studies was the absence of tyrosine phosphatase inhibitors in the lysates. Tyrosine phosphorylations are notoriously short-lived, often with a half-life of only a few seconds due to highly active phosphatases (37). Unless the phospho-tyrosine site is bound to SH2/PTP domains, the site goes through hundreds of rounds of phosphorylation/dephosphorylation, which makes detection difficult in the absence of tyrosine phosphatase inhibitors. There may be a bias also to detection of serine/threonine phosphorylations, as 86% of all phosphorylation occurs on serines, 12% on threonine, and only 2% on tyrosines (38). It is therefore possible that additional tyrosines besides Y102 and Y138 can be phosphorylated, and these might regulate replication and genome maintenance.
Through mass spectrometry of HPV-31 E2, we identified tyrosine phosphorylation not only on Y102, but also on Y87 (Fig. 1D). Further studies are required to determine what effect phosphorylation has at this site. Additionally, we found phosphorylation at serine 269, the homologue of which was previously characterized as a minor phosphorylation site in HPV-1 (S281) (39). We have also detected phosphotyrosine at positions Y131 and Y138 in HPV-31 E2, though with lower than 75% probability scores. Our mass spectrometric analysis of BPV-1 E2 has also expanded the list of E2 phosphorylation sites that are fibroblast growth factor receptor 2 (FGFR2) dependent: Y32, Y44, Y131, Y158, Y159, Y169, Y170, and Y262 (40).
Another tyrosine mutant, Y131A, has a very similar phenotype to HPV-31 Y102E. HPV-16 Y131A prevents phosphorylation, increases nuclear matrix association, has no effect on transient replication, and cannot be maintained episomally in HFKs (41). A more direct comparison would require Y131F and Y131E mutations to be generated, but it appears phosphorylation of Y102 mimics nonphosphorylated Y131A. Phosphorylation at one site may restrict phosphorylation at the other, each the target of specific kinases that are activated through different pathways. The hydroxyl of Y102 and tyrosine 159 are only 11 Å apart and face the same direction in the crystal structure of 16E2 (PDB: 2NNU), so it is possible phosphorylation at one site influences subsequent phosphorylation at the other residue.
The kinase which phosphorylates at position 102 has yet to be identified. We previously published that that the FGFRs associate with E2 (40, 42), though none of these phosphorylate at Y102. Of the 90 known tyrosine kinases, a consensus sequence is only recognized for LCK, Abl, Src, ALK, and EGFR (38). HPV-31 E2 Y102 fits the epidermal growth factor receptor (EGFR) consensus sequence, but our previous observations that E2 does not bind EGFR and that EGFR does not inhibit replication (40) imply that EGFR is not the kinase for Y102. The kinase that phosphorylates BPV-1 may differ from HPV-31, as the sequence flanking Y102 differs significantly (Fig. 1B).
MATERIALS AND METHODS
Plasmids.
For the replication assay, pFLORI-31, containing the firefly luciferase as well as HPV-31 origin of replication, and pRenilla, containing the Renilla luciferase behind a CMV promoter, were used (43). For immunoprecipitations, the following plasmids were used: 3×FLAG-HPV-31 E1 (codon optimized) in pCMV 3Tag 1a vector (43), pcDNA3-FLAG-HPV-31 E2 (codon optimized) (42), pHA-GPS2 (20), pCN-GST-hBrd4 1224–1362 (CTM) (44), FLAG-TopBP1 (45), p16LL (46), and pBR322-HPV-31, HA-HPV-31 E1.
Cloning.
HPV-31neo. First, 31neoA, in which an AgeI restriction site was added to the L1 region (4573 bp) of pBR322-HPV-31 through site directed mutagenesis using primers designed by PrimerX (F: AGCATCTACCGGTACACCAGCA; R: TGCTGGTGTACCGGTAGATGCT). The neo cassette, encompassing the SV40promoter-intron-neomycin-βactin-poly-A-tail, was PCR amplified from the template pBR322-HPV-18neo (23) using primers which add an Sbf1 site (in lower case) to the 5′ end and an AgeI site to the 3′ end (F: GATCcctgcaggGGCCTGAAATAAC; R: GTACaccggtTAAAATACAGCATAGCAA), and inserted into Age/Sbf1-digested 31neoA. The neo cassette contains a HindIII site which was to be removed to permit release of the HPV-31neo genome from the pBR322 backbone through HindIII digestion. Similarly, the neo cassette contains a CsiI site that was removed to allow cut-and-paste insertion of E2 mutants into the HPV-31neo plasmid. Primers for HindIII removal (F: GCTTTTGCAAATCGCTTCTGCCTTC, R: GAAGGCAGAAGCGATTTGCAAAAGC) and CsiI removal (F: TCCACACCTGCTTGCTGACTA, R: TAGTCAGCAAGCAGGTGTGGA) were designed in the PrimerX website. The resulting pBR322-HPV-31neoHC was sequenced to confirm no mutations were incorporated during cloning. This plasmid is referred to as HPV-31neo and is available in the Addgene repository (Plasmid number 153283).
Mutants 31E2 Y102E/F. Site directed mutagenesis was performed following the QuikChange II protocol (Agilent Technologies) to generate HPV-31 E2 Y102E and Y102F mutants in codon-optimized E2 of pcDNA3-FLAG 31 E2, noncodon-optimized E2 of pSB-HA-HPV-31 E2 (8), and endogenous E2 of HPV-31neo. Primers were designed using PrimerX website (Y102E F: CAAACAAGTCTTGAACTGGAGTTAACTGCACCTACAGGG, R: CCCTGTAGGTGCAGTTAACTCCAGTTCAAGACTTGTTTG; Y102F F: CAAACAAGTCTTGAACTGTTCTTAACTGCACCTACAGGG, R: CCCTGTAGGTGCAGTTAAGAACAGTTCAAGACTTGTTTG; codon-optimized Y102E [+XcmI silent mutation for selection] F: CAGCCTCGAGCTGGAGCTGACCGCTCCCA, R: TGGGAGCGGTCAGCTCCAGCTCGAGGCTG; codon-optimized Y102F F: GACCAGCCTCGAGCTGTTCCTGACCGC, R: GCGGTCAGGAACAGCTCGAGGCTGGTC).
Cells and transfections.
HEK 293TT, TTF, and C33a cells were cultured in Dulbecco’s modified Eagle medium (DMEM) with 10% fetal bovine serum (FBS; Atlas Biologicals). HaCaT cells were cultured in calcium-free DMEM supplemented with 5% FBS, which was calcium depleted for 1 h in Chelex-100 resin, then supplemented with CaCl2 added to a final concentration of 0.02 mM. N/TERT cells were grown in KSFM, and NIKS cells were grown in F-media. Polyethylenimine (PEI) was used for transfections at a ratio of 2 μg PEI to 1 μg DNA, which is equivalent to an N/P ratio of 15:1. PEI and DNA were diluted in Opti-MEM serum-free medium before placing on cells. At 20 h posttransfection, cells were harvested for lysis or fixed for immunofluorescence. For viral production in HEK 293 TTF cells, lipofectamine 2000 was used at a ratio of 2:1, as PEI tends to precipitate the large DNA. HEK 293TT cells were transfected with FLAG-HPV-31 E2 WT or Y102E and harvested 24 h later. Cell pellets were fractionated as previously described (47).
Transient DNA replication assays were performed with C33a cells in triplicate using Dual-Glo luciferase assay system (Promega) (22), with one modification: 20 μl Dual-Glo reagent was used instead of 50 μl. At 3 days posttransfection, firefly and Renilla luminescence was measured with a PHERAstar plate reader and software.
Sample preparation for mass spectrometry.
HEK293 TT cells transfected with FLAG-HPV-31 E2 were grown on 6 × 15-cm dishes, washed in HBSS buffer (10 mM HEPES pH 7.5, 140 mM NaCl, 0.5 mM CaCl2, 0.8 mM MgCl2), and incubated with the irreversible tyrosine phosphatase inhibitor pervanadate (30 μM) for 1 h at 37°C. Cells were lysed in 20 mM HEPES pH 7.5, 150 mM NaCl, 30 μM pervanadate, 7.5 μM trichostatin A (TSA), and 0.525 mg/ml NaF and immunoprecipitated overnight with B201 antibody. Bands were excised from Coomassie-stained polyacrylamide gels for tandem mass spectrometry (MS-MS). The gel bands were subjected to reduction (10 mM DTT) and alkylation (55 mM iodoacetamide) and digested with trypsin (Promega) overnight at 37°C and injected into a C18 column. Peptide spectra were recorded in Orbitrap Velos Pro (Thermo Fisher Scientific) and Dionex UltiMate 3000 RSLC nanoflow system (Thermo Fisher Scientific) and database searches were carried out using Sequest algorithm. From the MS-MS data, individual probability values for each phosphorylation site were calculated using Proteome Discoverer V1.3 equipped with phospho RS 2.0.
Immunoprecipitations and immunoblotting.
At 24 h posttransfection, cells were washed in PBS, harvested, and then lysed in lysis buffer (0.5% NP-40, 150 mM NaCl, 20 mM Tris pH 7.5). Insoluble protein was separated by centrifugation, and soluble lysate was quantitated using BCA reagent (Pierce Thermo Scientific). Equal amounts of lysate per immunoprecipitation (IP) were diluted to 500 μl in lysis buffer and incubated for 3 h at 4°C with 20 μl of 50% slurry M2 affinity gel (Sigma), 20 μl of 50% slurry glutathione beads, or 1 μl of TopBP1 antibody + 25 μl of 50% slurry protein A/G Sepharose beads. E1 and Brd4-CTM were also treated with 0.1 μg/μl ethidium bromide as previously described during immunoprecipitation (44). Beads were washed 5× in lysis buffer, boiled in 2× protein sample buffer, run on SDS-PAGE gels, and transferred onto 0.45-μm PVDF membranes (Millipore) in semidry transfer boxes (Bio-Rad). Membranes were blocked in 5% nonfat milk/TBST overnight and incubated with primary antibodies for 1 h at the following dilutions: FLAG: M2 (Sigma; 1:5,000), HA: 12CA5 supernatant 1:1,000, TopBP1 (Bethyl; 1:2,000), or GST (Arbor Vitae; 1:5,000). Light-chain-specific secondary antibodies diluted 1:5,000 were added following three TBST washes. Signals were detected with ECL (Amersham) and ImageQuant LAS 4000 system (GE Healthcare).
For phospho-Y102 antibody production, New Zealand white rabbits were immunized with an HPV-31 E2 phospho-tyrosine peptide (amino acids [aa] 97 to109; TSLELYLTAPTGC) formulated in complete Freund’s adjuvant followed by four injections in incomplete Freund’s adjuvant. Serum was analyzed by enzyme-linked immunosorbent assay (ELISA) to confirm antibody production. The rabbit 10008 antiserum predominantly recognizes phosphorylated Y102, but also detects several cellular phosphotyrosine proteins by immunoblot. This antiserum can be utilized for E2 chromatin immunoprecipitation (ChIP) assays (48).
Cell fractionation.
Based upon published methods (47), HEK293 TT cells transfected with FLAG-HPV-31 E2 WT or Y102E were lysed in lysis buffer (15 mM Tris pH 8.0, 150 mM NaCl, 0.5% Triton-x) for 20 min, and then spun at 12,000 rpm for 10 min. The soluble fraction was kept on ice while the pellet was resuspended in lysis buffer and divided into two equal parts, A and B. The A portion was solubilized in an equal volume of 0.8 M NaCl for a final 0.4 M concentration and spun at 12,000 rpm. The soluble fraction was kept on ice. The B portion was digested with Benzonase at room temperature for 30 min, then spun at 7,000 rpm. The soluble fraction was kept on ice, while the pellet was resuspended in lysis buffer with 2 M NaCl and spun at 12,000 rpm. The soluble fraction was kept and the pellet was resuspended in 8 M Urea, 100 mM Tris pH 8.0, 100 mM NaCl.
Immunofluorescence.
N/TERT cells were plated onto UV-sterilized glass coverslips at 3 × 105 cells per well of a 12-well plate. Cells were transfected with FLAG-HPV-31 E2 and HA-HPV-31 E1 constructs. After 24 h, these were fixed with 4% paraformaldehyde/phosphate-buffered saline (PBS) and permeabilized in blocking solution (5% goat serum, 0.25% Triton X-100, PBS). Primary M2 FLAG antibody diluted 1:5,000 and rat 3F10 HA antibody diluted 1:500 in blocking solution and incubated for 1 h at room temperature (RT). Secondary antibody was 594 Alexa-Fluor mouse and 488 Alexa-Fluor rat antibody diluted 1:1,000. Slides were mounted using Prolong Gold with DAPI (4′,6-diamidino-2-phenylindole), and visualized using a Nikon Microphot-SA microscope.
Quasivirus preparation and infection.
Viral stocks were made according to a modified version of the original method (https://ccrod.cancer.gov/confluence/display/LCOTF/Home). A more detailed description of the method will be reported separately. To generate virus genomes, pBR322-HPV-31neoHC was digested with HindIII (Anza) for 2 h, gel purified to separate from the vector, and religated with T4 DNA ligase for 3 h. HEK293 TTF cells (49) were transfected with 5 μg of p16sheLL (46) and 5 μg of recircularized HPV-31neo. Two days after transfection, cells were resuspended in viral maturation buffer (0.5% Triton-X, 10 mM MgCl2, 5 mM CaCl2, PBS) at 1.5 cell pellet volume equivalents and incubated at 37°C for 2 days. Lysate was spun at 7,000 rpm and the soluble viral preparation was stored at 4°C and used within 1 month.
HaCaT cells were grown to confluence and treated with extracellular matrix (ECM) buffer (170 mM NH4OH, 0.5% Triton, PBS), which removes the cell body but leaves behind the ECM (25). ECM was washed 3× in PBS to remove buffer, then HaCaT medium was added along with quasivirus/pseudovirus (10 multiplicity of infection [MOI] for HaCaT, 40 MOI for NIKS). After a 4-h incubation of virus on ECM, HaCaT or NIKS cells were added. Two days after infection, Geneticin was added at 500 μg/ml for HaCaT or 200 μg/ml for NIKS to select for infected cells. For transfection, 1 μg recircularized HPV-31 genomes as prepared for viral production was transfected into N/TERT cells. Two days posttransfection, cells were cultured in 200 μg/ml Geneticin.
For quantitation of viral titer, 5 μl of virus was diluted 10× in 15 mM Tris pH 7.5, 1 mM MgCl2, 1 μl Benzonase, and digested at 37°C for 2 h. Samples were heated for 5 min to inactivate the DNase and break open the capsids. Proteinase K was added to degrade the proteins at 55°C for 30 min, followed by inactivation with 5 mM phenylmethylsulfonyl fluoride (PMSF). Quantitative PCR was then performed on the viral preparation, along with 500 pg to 0.05 pg genome standards for calibration.
DNA and RNA isolation.
Trypsinized cells were counted and washed in PBS, and episomal DNA was isolated using the standard Hirt protocol (50). qPCR primers were used to amplify the HPV genome (F: ACACCATGCATTATACTAAC; and R: TCACTGCTAAATACAGATTC) using Ssofast Evagreen Supermix (Bio-Rad). RNA was isolated using standard TRIzol protocol, converted into cDNA using standard SuperScript III reverse transcriptase (Thermo Fisher), and qPCR primers to E1̂E4 for amplification. For DNA extraction and digestion with exonuclease V, previously described methods were used (27). In brief, DNA was isolated using the Qiagen DNeasy blood and tissue kit according to the standard protocol, and 10 μl of DNA was digested with 1 μl exonuclease V in NEB Cutsmart buffer supplemented with ATP for 3 h at 37°C. Exonuclease was heat-inactivated at 70°C for 30 min, and 1 μl of cut or uncut DNA was amplified using qPCR primers for the HPV genome as described earlier: actin (F: GAGGCACTCTTCCAGCCTTC; R: CGGATGTCCACGTCACACTT) (48), and mitochondria (F: CAGGAGTAGGAGAGAGGGAGGTAAG; R: TACCCATCATAATCGGAGGCTTTGG) (27).
Statistical analysis.
For all experiments, means are expressed ± standard error of the mean (SEM). P values of ≤0.05 were considered significant. Student’s t test was performed for E2 fractionation comparing wild type to mutant. Each experiment was performed multiple times.
ACKNOWLEDGMENTS
We appreciate the generosity of the following people for cells and plasmids: Alison McBride (NIAID/NIH) for the codon-optimized HPV-31 E2 and selectable HPV-18 genome, Jacques Archambault (McGill University) for HPV-31 ori luciferase plasmids, John Schiller (NCI) for HEK 293TT cells, Richard Roden for HEK293 TTF cells, Iain Morgan (Virginia Commonwealth University) for N/TERT cells, and Cheng-Ming Chiang (University of Texas Southwestern Medical Center) for the Brd4 constructs. This research was supported by the National Cancer Institute (R01 CA058376 to E.A.) and the National Institute of Allergy and Infectious Diseases (T32 AI007637 and T32 AR062495 to T.G., T32 AI060519 to M.D., F30 AI114284 to S.P.C.). M.D. was also supported by an award from the Ralph W. and Grace M. Showalter Research Trust. The content is solely the responsibility of the authors and does not represent the official views of the NIH.
REFERENCES
- 1.McBride AA. 2013. The papillomavirus E2 proteins. Virology 445:57–79. doi: 10.1016/j.virol.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bellanger S, Tan CL, Xue YZ, Teissier S, Thierry F. 2011. Tumor suppressor or oncogene? A critical role of the human papillomavirus (HPV) E2 protein in cervical cancer progression. Am J Cancer Res 1:373–389. [PMC free article] [PubMed] [Google Scholar]
- 3.McBride AA, Bolen JB, Howley PM. 1989. Phosphorylation sites of the E2 transcriptional regulatory proteins of bovine papillomavirus type 1. J Virol 63:5076–5085. doi: 10.1128/JVI.63.12.5076-5085.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McBride AA, Howley PM. 1991. Bovine papillomavirus with a mutation in the E2 serine 301 phosphorylation site replicates at a high copy number. J Virol 65:6528–6534. doi: 10.1128/JVI.65.12.6528-6534.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sanders CM, Stern PL, Maitland NJ. 1995. Characterization of human papillomavirus type 16 E2 protein and subdomains expressed in insect cells. Virology 211:418–433. doi: 10.1006/viro.1995.1424. [DOI] [PubMed] [Google Scholar]
- 6.Lehman CW, King DS, Botchan MR. 1997. A papillomavirus E2 phosphorylation mutant exhibits normal transient replication and transcription but is defective in transformation and plasmid retention. J Virol 71:3652–3665. doi: 10.1128/JVI.71.5.3652-3665.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Culleton SP, Kanginakudru S, DeSmet M, Gilson T, Xie F, Wu S-Y, Chiang C-M, Qi G, Wang M, Androphy EJ. 2017. Phosphorylation of the bovine papillomavirus E2 protein on tyrosine regulates its transcription and replication functions. J Virol 91:e01854-16. doi: 10.1128/JVI.01854-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stubenrauch F, Colbert AM, Laimins LA. 1998. Transactivation by the E2 protein of oncogenic human papillomavirus type 31 is not essential for early and late viral functions. J Virol 72:8115–8123. doi: 10.1128/JVI.72.10.8115-8123.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blom N, Gammeltoft S, Brunak S. 1999. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J Mol Biol 294:1351–1362. doi: 10.1006/jmbi.1999.3310. [DOI] [PubMed] [Google Scholar]
- 10.Dou Y, Yao B, Zhang C. 2014. PhosphoSVM: prediction of phosphorylation sites by integrating various protein sequence attributes with a support vector machine. Amino Acids 46:1459–1469. doi: 10.1007/s00726-014-1711-5. [DOI] [PubMed] [Google Scholar]
- 11.Skiadopoulos MH, McBride AA. 1996. The bovine papillomavirus type 1 E2 transactivator and repressor proteins use different nuclear localization signals. J Virol 70:1117–1124. doi: 10.1128/JVI.70.2.1117-1124.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rihs HP, Jans DA, Fan H, Peters R. 1991. The rate of nuclear cytoplasmic protein transport is determined by the casein kinase II site flanking the nuclear localization sequence of the SV40 T-antigen. EMBO J 10:633–639. doi: 10.1002/j.1460-2075.1991.tb07991.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leemann-Zakaryan RP, Pahlich S, Grossenbacher D, Gehring H. 2011. Tyrosine phosphorylation in the C-terminal nuclear localization and retention signal (C-NLS) of the EWS protein. Sarcoma 2011:218483. doi: 10.1155/2011/218483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dickson MA, Hahn WC, Ino Y, Ronfard V, Wu JY, Weinberg RA, Louis DN, Li FP, Rheinwald JG. 2000. Human keratinocytes that express hTERT and also bypass a p16(INK4a)-enforced mechanism that limits life span become immortal yet retain normal growth and differentiation characteristics. Mol Cell Biol 20:1436–1447. doi: 10.1128/mcb.20.4.1436-1447.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garcia V, Furuya K, Carr AM. 2005. Identification and functional analysis of TopBP1 and its homologs. DNA Repair (Amst) 4:1227–1239. doi: 10.1016/j.dnarep.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 16.Boner W, Taylor ER, Tsirimonaki E, Yamane K, Campo MS, Morgan IM. 2002. A Functional interaction between the human papillomavirus 16 transcription/replication factor E2 and the DNA damage response protein TopBP1. J Biol Chem 277:22297–22303. doi: 10.1074/jbc.M202163200. [DOI] [PubMed] [Google Scholar]
- 17.Kanginakudru S, DeSmet M, Thomas Y, Morgan IM, Androphy EJ. 2015. Levels of the E2 interacting protein TopBP1 modulate papillomavirus maintenance stage replication. Virology 478:129–135. doi: 10.1016/j.virol.2015.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Donaldson MM, Boner W, Morgan IM. 2007. TopBP1 regulates human papillomavirus type 16 E2 interaction with chromatin. J Virol 81:4338–4342. doi: 10.1128/JVI.02353-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiang M, Axe T, Holgate R, Rubbi CP, Okorokov AL, Mee T, Milner J. 2001. p53 binds the nuclear matrix in normal cells: binding involves the proline-rich domain of p53 and increases following genotoxic stress. Oncogene 20:5449–5458. doi: 10.1038/sj.onc.1204705. [DOI] [PubMed] [Google Scholar]
- 20.Breiding DE, Sverdrup F, Grossel MJ, Moscufo N, Boonchai W, Androphy EJ. 1997. Functional interaction of a novel cellular protein with the papillomavirus E2 transactivation domain. Mol Cell Biol 17:7208–7219. doi: 10.1128/mcb.17.12.7208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mohr IJ, Clark R, Sun S, Androphy EJ, MacPherson P, Botchan MR. 1990. Targeting the E1 replication protein to the papillomavirus origin of replication by complex formation with the E2 transactivator. Science 250:1694–1699. doi: 10.1126/science.2176744. [DOI] [PubMed] [Google Scholar]
- 22.Gagnon D, Fradet-Turcotte A, Archambault J. 2015. A quantitative and high-throughput assay of human papillomavirus DNA replication. Methods Mol Biol 1249:305–316. doi: 10.1007/978-1-4939-2013-6_23. [DOI] [PubMed] [Google Scholar]
- 23.Van Doorslaer K, Porter S, McKinney C, Stepp WH, McBride AA. 2016. Novel recombinant papillomavirus genomes expressing selectable genes. Sci Rep 6:37782. doi: 10.1038/srep37782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Day PM, Lowy DR, Schiller JT. 2008. Heparan sulfate-independent cell binding and infection with furin-precleaved papillomavirus capsids. J Virol 82:12565–12568. doi: 10.1128/JVI.01631-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Day PM, Pang Y-Y, Kines RC, Thompson CD, Lowy DR, Schiller JT. 2012. A human papillomavirus (HPV) in vitro neutralization assay that recapitulates the in vitro process of infection provides a sensitive measure of HPV L2 infection-inhibiting antibodies. Clin Vaccine Immunol 19:1075–1082. doi: 10.1128/CVI.00139-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Culp TD, Budgeon LR, Christensen ND. 2006. Human papillomaviruses bind a basal extracellular matrix component secreted by keratinocytes which is distinct from a membrane-associated receptor. Virology 347:147–159. doi: 10.1016/j.virol.2005.11.025. [DOI] [PubMed] [Google Scholar]
- 27.Bienkowska-Haba M, Luszczek W, Myers JE, Keiffer TR, DiGiuseppe S, Polk P, Bodily JM, Scott RS, Sapp M. 2018. A new cell culture model to genetically dissect the complete human papillomavirus life cycle. PLoS Pathog 14:e1006846. doi: 10.1371/journal.ppat.1006846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sekhar V, McBride AA. 2012. Phosphorylation regulates binding of the human papillomavirus type 8 E2 protein to host chromosomes. J Virol 86:10047–10058. doi: 10.1128/JVI.01140-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chang S-W, Liu W-C, Liao K-Y, Tsao Y-P, Hsu P-H, Chen S-L. 2014. Phosphorylation of HPV-16 E2 at serine 243 enables binding to Brd4 and mitotic chromosomes. PLoS One 9:e110882. doi: 10.1371/journal.pone.0110882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.DeSmet M, Jose L, Isaq N, Androphy EJ. 2019. Phosphorylation of a conserved tyrosine in the papillomavirus E2 protein regulates Brd4 binding and viral replication. J Virol 93:e01801-18. doi: 10.1128/JVI.01801-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oliveira JG, Colf LA, McBride AA. 2006. Variations in the association of papillomavirus E2 proteins with mitotic chromosomes. Proc Natl Acad Sci U S A 103:1047–1052. doi: 10.1073/pnas.0507624103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mehta K, Gunasekharan V, Satsuka A, Laimins LA. 2015. Human papillomaviruses activate and recruit SMC1 cohesin proteins for the differentiation-dependent life cycle through association with CTCF insulators. PLoS Pathog 11:e1004763. doi: 10.1371/journal.ppat.1004763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Donaldson MM, Mackintosh LJ, Bodily JM, Dornan ES, Laimins LA, Morgan IM. 2012. An interaction between human papillomavirus 16 E2 and TopBP1 is required for optimum viral DNA replication and episomal genome establishment. J Virol 86:12806–12815. doi: 10.1128/JVI.01002-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brune W, Dürst M. 1995. Epithelial differentiation fails to support replication of cloned human papillomavirus type 16 DNA in transfected keratinocytes. J Invest Dermatol 104:277–281. doi: 10.1111/1523-1747.ep12612814. [DOI] [PubMed] [Google Scholar]
- 35.Culp TD, Christensen ND. 2004. Kinetics of in vitro adsorption and entry of papillomavirus virions. Virology 319:152–161. doi: 10.1016/j.virol.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 36.Wang Y, Li X, Song S, Wu J. 2014. Development of basal-like HaCaT keratinocytes containing the genome of human papillomavirus (HPV) type 11 for screening of anti-HPV effects. J Biomol Screen 19:1154–1163. doi: 10.1177/1087057114536987. [DOI] [PubMed] [Google Scholar]
- 37.Hunter T. 2014. The genesis of tyrosine phosphorylation. Cold Spring Harb Perspect Biol 6:a020644. doi: 10.1101/cshperspect.a020644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Olsen JV, Blagoev B, Gnad F, Macek B, Kumar C, Mortensen P, Mann M. 2006. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127:635–648. doi: 10.1016/j.cell.2006.09.026. [DOI] [PubMed] [Google Scholar]
- 39.Prescott EL, Brimacombe CL, Hartley M, Bell I, Graham S, Roberts S. 2014. Human papillomavirus type 1 E1̂E4 protein is a potent inhibitor of the serine-arginine (SR) protein kinase SRPK1 and inhibits phosphorylation of host SR proteins and of the viral transcription and replication regulator E2. J Virol 88:12599–12611. doi: 10.1128/JVI.02029-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.DeSmet M, Kanginakudru S, Jose L, Xie F, Gilson T, Androphy EJ. 2018. Papillomavirus E2 protein is regulated by specific fibroblast growth factor receptors. Virology 521:62–68. doi: 10.1016/j.virol.2018.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Harris L, McFarlane-Majeed L, Campos-León K, Roberts S, Parish JL. 2017. The cellular DNA helicase ChlR1 regulates chromatin and nuclear matrix attachment of the human papillomavirus 16 E2 protein and high-copy-number viral genome establishment. J Virol 91:e01853-16. doi: 10.1128/JVI.01853-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xie F, DeSmet M, Kanginakudru S, Jose L, Culleton SP, Gilson T, Li C, Androphy EJ. 2017. Kinase activity of fibroblast growth factor receptor 3 regulates activity of the papillomavirus E2 protein. J Virol 91:e01066-17. doi: 10.1128/JVI.01066-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fradet-Turcotte A, Morin G, Lehoux M, Bullock PA, Archambault J. 2010. Development of quantitative and high-throughput assays of polyomavirus and papillomavirus DNA replication. Virology 399:65–76. doi: 10.1016/j.virol.2009.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu S-Y, Lee A-Y, Lai H-T, Zhang H, Chiang C-M. 2013. Phospho switch triggers Brd4 chromatin binding and activator recruitment for gene-specific targeting. Mol Cell 49:843–857. doi: 10.1016/j.molcel.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu K, Luo Y, Lin F-T, Lin W-C. 2004. TopBP1 recruits Brg1/Brm to repress E2F1-induced apoptosis, a novel pRb-independent and E2F1-specific control for cell survival. Genes Dev 18:673–686. doi: 10.1101/gad.1180204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Buck CB, Thompson CD, Roberts JN, Müller M, Lowy DR, Schiller JT. 2006. Carrageenan is a potent inhibitor of papillomavirus infection. PLoS Pathog 2:e69. doi: 10.1371/journal.ppat.0020069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sankovski E, Karro K, Sepp M, Kurg R, Ustav M, Abroi A. 2015. Characterization of the nuclear matrix targeting sequence (NMTS) of the BPV1 E8/E2 protein—the shortest known NMTS. Nucleus 6:289–300. doi: 10.1080/19491034.2015.1074359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.DeSmet M, Kanginakudru S, Rietz A, Wu W-H, Roden R, Androphy EJ. 2016. The replicative consequences of papillomavirus E2 protein binding to the origin replication factor ORC2. PLoS Pathog 12:e1005934. doi: 10.1371/journal.ppat.1005934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang JW, Jagu S, Kwak K, Wang C, Peng S, Kirnbauer R, Roden R. 2014. Preparation and properties of a papillomavirus infectious intermediate and its utility for neutralization studies. Virology 449:304–316. doi: 10.1016/j.virol.2013.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hirt B. 1967. Selective extraction of polyoma DNA from infected mouse cell cultures. J Mol Biol 26:365–369. doi: 10.1016/0022-2836(67)90307-5. [DOI] [PubMed] [Google Scholar]