

Résumé
Les dommages causés à l’ADN par l’attachement dissociatif d’électrons ont été bien étudiés dans les phases gazeuse et solide. Cependant, la compréhension à un niveau fondamental de ce processus dans l’environnement aqueux de l’ADN constitue encore un défi. Nous avons utilisé la radiolyse impulsionnelle picoseconde pour générer des électrons dans l’eau ou le diéthylène-glycol (DEG) liquides afin d’observer la dynamique de capture de ces électrons par les bases ADN/ARN. Dans un liquide, il faut au moins distinguer trois états différents d’électron : un électron quasi-libre (eql−) qui est un électron avec une certaine énergie cinétique dans le liquide, un électron présolvaté qui est un électron ayant perdu son énergie cinétique et est en cours de solvatation et un électron solvaté. Contrairement à certaines hypothèses de la littérature, nous montrons d’abord que l’électron quasi-libre (eql−) dans l’eau ne peut pas être capté par la guanine et l’adénine à très longue distance dans des solutions aqueuses avec des concentrations ≤ 50 mM. Cependant, l’observation d’une diminution substantielle du rendement initial de l’électron hydraté en fonction de la concentration en nucléosides/nucléotides, accompagné de la formation d’un radical anionique nucléotidique, présente une preuve directe d’une étape ultrarapide de dommages à l’ADN par le rayonnement ionisant via l’électron. Le canal de dissociation des ions négatifs transitoires dans les solutions nucléotidiques ne se produit pas, même pour des solutions contenant 0,25 M de nucléotides. En revanche, dans le diéthylène-glycol, nous démontrons que contrairement à l’électron solvaté (eDEG−) et l’électron présolvaté, l’électron quasi-libre se fixe efficacement à la ribothymidine, en formant juste après le dépôt d’énergie un radical anionique à l’état excité qui subit une dissociation de la liaison glycosidique N1-C1′. Grâce à la dissociation, ce processus, induit par un électron quasi-libre dans l’eau, constitue de fait une oxydation de la molécule qui aboutit à la même coupure de brin provoquée par une oxydation induite par le radical •OH.
Keywords: Radiolyse impulsionnelle picoseconde, bases d’ADN, attachement dissociatif, dommage oxydatif, radical anionique
Keywords: Picosecond pulse radiolysis, DNA bases, dissociative electron attachment, oxidative damage, anionic radical
Abstract
DNA damage caused by the dissociative electron attachment (DEA) has been well-studied in the gas and solid phases. However, understanding of this process at the fundamental level in solution is still a challenge. The electrons, after losing their kinetic energy via ionization and excitation events, are thermalized and undergo a multistep hydration process with a time constant of ca. ≤1 ps, to becoming fully trapped as a hydrated or solvated electron (esol− or eaq−). Prior to the formation of esol−, the electron exists in its presolvated (or prehydrated) state (epre−) with no kinetic energy. We used picosecond pulse radiolysis to generate electrons in water or in liquid diethylene glycol (DEG) to observe the dynamics of capture of these electrons by DNA/RNA bases, nucleosides, and nucleotides. Contrary to the hypotheses in the literature that the presolvated electrons (epre−) are captured well by the DNA-nucleosides/tides and the transient negative ions (TNIs) cause strand breaks, we first show that the quasi-free electrons with kinetic energy (eqf−) or epre−cannot be captured by guanine and adenine at very long distances in aqueous solutions with concentrations lower than 50 mM. However, the observation of a substantial decrease in the initial yield of esol− as a function of nucleoside/nucleotide concentrations accompanied by the formation of the nucleotide anion radicals provides direct evidence of an ultrafast step involving radiation-produced electron-mediated DNA damage via DEA. Transient signal analysis suggests that the dissociation channel of TNIs in nucleotide solutions is not even probable up to 0.25 M. On the other hand, in diethylene glycol, we demonstrate that unlike esol− and epre−, eqf− effectively attaches itself to the RNA-nucleoside, ribothymidine, forming the TNI in the excited state (TNI*) that undergoes the N1-C1′ glycosidic bond dissociation. Thanks to DEA, this process induced by eqf−, in fact, leads to an oxidation of the parent molecule similar to the hydroxyl radical (•OH) leading to the same glycosidic bond (N1-C1′) cleavage.
Les différentes voies de dommage à l’ADN
L’acide désoxyribonucléique, ADN, est une macromolécule présente dans toutes les cellules. L’ADN contient toute l’information génétique, permettant le développement, le fonctionnement et la reproduction des êtres vivants. L’ADN est constitué des 4 bases adénine, thymine, guanine et cytosine (A, T, G, C) qui sont reliées par des groupements phosphate O2P(OR)2 et sucre (R : deoxyribose).
L’ADN peut être irradié par des rayonnements ionisants, soit accidentellement, soit lors de la radiothérapie afin d’éliminer les cellules cancéreuses. En raison de son rôle primordial dans le fonctionnement des êtres vivants, il est important de connaître de manière précise la nature des dommages causés quand l’ADN est soumis à l’irradiation. Il est bien établi que l’interaction des radiations ionisantes de grande énergie (rayons X et gamma, particules chargées accélérées) avec le milieu environnant l’ADN génère divers dommages à l’ADN, y compris les dommages sur les bases, la libération des bases, la rupture des brins et l’établissement de liaisons transversales ADN-ADN ou ADN-protéine, tous ces processus pouvant être irréversibles et souvent irréparables[1].
Le mécanisme des dommages causés à l’ADN par les rayonnements ionisants est très complexe et implique de nombreuses espèces intermédiaires. Dans un milieu biologique, l’eau est une partie de l’environnement naturel de l’ADN. En fait, la fraction molaire de l’ADN dans l’eau est très importante, ce qui impose des réactions très rapides entre les radicaux issus de l’eau et l’ADN[2,3]. De nombreuses études résolues en temps à l’échelle de la microseconde ont été effectuées pour comprendre ces réactions impliquées lors de l’oxydation de l’ADN [4].
Au cours des deux dernières décennies, des études sur les dommages à l’ADN ont été menées pour élucider le rôle des électrons. Léon Sanche et ses collaborateurs ont effectué les premières observations expérimentales du clivage des brins d’ADN induit par les électrons de basse énergie (0–20 eV) [5,6]. Les études ont été effectuées sur ces molécules adsorbées sur une surface à très basse température en les bombardant par des électrons dans le vide avec une énergie cinétique bien maîtrisée. Ces travaux ont conduit à de nombreuses études mécanistiques sur les dommages induits par des électrons de basse énergie cinétique dans les systèmes modèles d’ADN (blocs de construction d’ADN à l’état gazeux, oligomères simples et doubles brins de séquences définies), de l’ADN plasmidique et de l’ADN de diverses cellules. Ces études ont établi que les électrons de basse énergie dans le vide conduisent tout d’abord à la formation d’anions transitoires. La fragmentation efficace subséquente de ces anions se produit dans un état énergétiquement excité. Cette dissociation induit une dégradation irréversible, et donc très sévère, de l’ADN. Toutefois, les méthodes de mesures (spectroscopie d’électron, spectrométrie de masse, …) restreignirent ces études à des molécules dans le vide ou bien déposées sur des substrats à basse température [7,8], alors que tous les processus chimiques et biologiques nécessaires à la vie se déroulent à température ambiante dans un milieu aqueux. Il est donc très important d’étudier les dommages aux bases d’ADN dans l’eau puisqu’elle joue un rôle-clé dans la voie indirecte des lésions de l’ADN induites par le rayonnement. Il est donc indispensable de mener des recherches sur les dommages à l’ADN induits par les électrons à température ambiante et dans l’eau.
Les réactions plus lentes avec eaq− ont été étudiées à faible concentration des bases de l’ADN. Mais l’objectif de notre étude a été de suivre à température ambiante les processus très rapides qui se produisent comme en milieu biologique où la fraction molaire de l’ADN dans l’eau est très importante. Pour cela nous avons utilisé l’outil de choix qui est la radiolyse impulsionnelle avec des impulsions d’électrons les plus courtes disponibles [encadré].
Dans la radiolyse des solutions aqueuses à température ambiante, la majorité du rayonnement ionisant est initialement absorbée par l’eau environnante du soluté et un électron secondaire de grande énergie cinétique (Ec) est éjecté, en laissant un radical cationique de l’eau, H2O•+ (ou le trou). Dans un laps de temps sub-picoseconde, le trou H2O•+ donne naissance à un radical hydroxyle (•OH) par transfert de protons à la molécule d’eau proche [10, 11, 12, 13]. L’électron secondaire énergétique provoque d’autres ionisations conduisant à une cascade d’électrons avec des énergies cinétiques de plus en plus faibles. Après avoir perdu son énergie cinétique, par des phénomènes d’ionisation et d’excitation des molécules du solvant, l’électron est thermalisé (avec une énergie cinétique égale à kBT), et il subit ensuite un processus d’hydratation avec une constante de temps d’environ 300 fs. Il devient ainsi entièrement piégé sous forme d’électron hydraté (eaq−) qui est une espèce thermodynamiquement stable, mais très réactive [14].
Pour plus de clarté, dans le présent travail, nous utilisons le terme d’électrons pré-hydratés epre− pour les précurseurs de eaq– qui n’ont plus d’énergie cinétique et qui sont en cours de solvatation et nous nommons électrons quasi-libres eql–, des électrons qui ne sont pas thermalisés et qui possèdent encore une énergie cinétique dans l’eau (figure 1).
Figure 1 -.
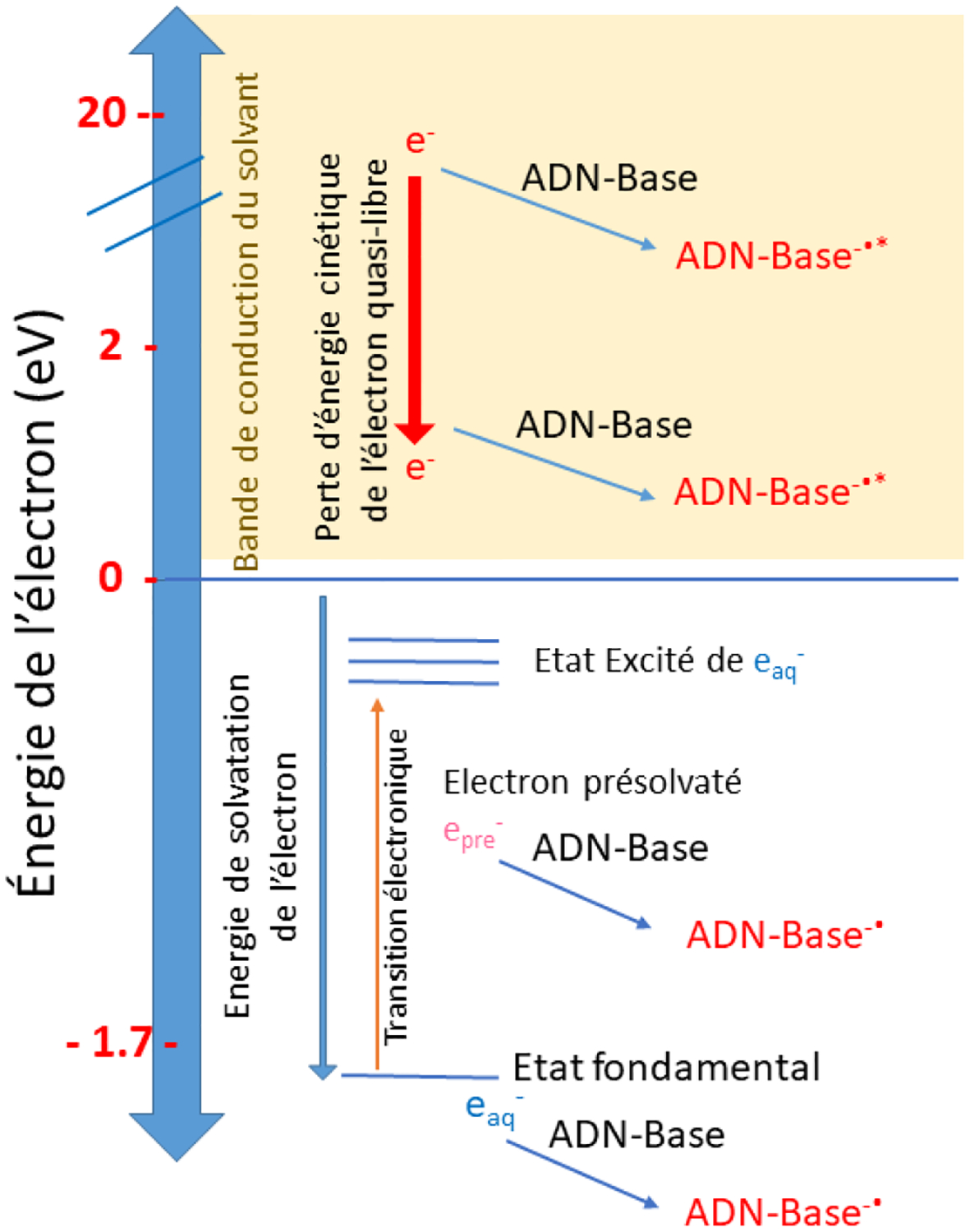
Schéma représentant le cycle énergétique de la solvatation des électrons et la réaction avec la base d’ADN. Un électron éjecté dans l’eau perd son énergie cinétique en quelques femtosecondes, pour devenir thermalisé. Ensuite cet électron subit la solvatation qui est un processus de réarrangement des molécules du solvant autour de l’électron en excès. Le zéro d’énergie est celui d’un électron thermalisé dans l’eau. L’électron en excès dans le solvant peut réagir avec un soluté, notamment avec une base d’ADN. La thermodynamique et le produit de cette réaction dépendent de l’état énergétique et de l’état de solvatation de l’électron.
Dans l’environnement aqueux, eql−, epre− et eaq− sont des entités qui peuvent effectivement réagir avec l’ADN. L’étude de la réduction ultra-rapide de modèles simples de biomolécules (p. ex. nucléobases et nucléosides/nucléotides) par un électron en compétition avec son processus de thermalisation et d’hydratation, est encore un sujet de défi expérimental. En effet, la durée de vie de l’électron quasi-libre dans l’eau est extrêmement courte, estimée inférieure à une femtoseconde. D’après la littérature existante, il est admis qu’en milieu aqueux l’étape initiale pour la réaction avec l’ADN soit la capture d’un électron par la purine (adénine et guanine) ou par la pyrimidine (thymidine et cytosine) [15,16]. Cette réaction conduirait à la formation d’un anion radicalaire transitoire à l’état excité (figure 1).
Par la suite, cet anion génère des dommages complexes et souvent irréversibles de l’ADN qui détermineraient l’action biologique ultérieure [17]. C’est pourquoi, dans un premier temps, il est très important d’examiner expérimentalement les valeurs des constantes de vitesse et l’ampleur des réactions des blocs de construction de l’ADN avec eql−. Dans un second temps, la nature des espèces transitoires à courte durée de vie (anions radiolytiques), qui sont produites immédiatement après la capture des électrons, doit être clarifiée pour mieux comprendre le clivage de la liaison ADN induit par les électrons. Nous étudions donc ici l’effet de l’état de l’électron dans un solvant (dans ses trois états eql−, epre− et eaq−) sur sa réactivité avec des bases d’ADN.
Capture de l’électron dans ses différents états en solution
Capture de eql− et eaq− dans l’eau
Par les bases d’ADN
Les réactions de l’électron avant solvatation sont extrêmement rapides car il possède une très grande mobilité et de plus son hydratation a lieu en moins de 300 fs [15]. En revanche, la mobilité de eaq− est beaucoup plus faible et ses réactions ont lieu bien au-delà de quelques picosecondes. Tout d’abord, nous avons considéré le cas des bases de l’ADN. Les bases de l’ADN sont très peu solubles dans l’eau et à ces faibles concentrations, les réactions sont trop lentes. Par conséquent, il n’est pas possible d’observer la capture de eql−, trop fugace, par ces bases. Seul l’électron hydraté réagit avec les bases d’ADN à l’échelle de la nanoseconde pour former leur anion.
Par les nucléosides et les nucléotides
Nous avons donc examiné les réactions avec des nucléotides et des nucléosides qui sont bien plus solubles que les bases dans l’eau. La diminution du rendement initial de la formation de eaq− observé à 5 ps, par rapport à celui dans l’eau pure, indique qu’une réaction de l’électron quasi-libre, précurseur de eaq−, a eu lieu. Cette réaction chimique est terminée en moins d’une picoseconde et peut être considérée comme l’événement de dommage indirect le plus précoce. La solubilité intrinsèque plus élevée dans l’eau des nucléotides par rapport aux nucléosides nous a permis d’étudier la capture de eql− ainsi que de eaq− à des concentrations assez élevées. Comme le montre la figure 2, les analyses globales des données d’absorption transitoire, pour des solutions concentrées de nucléotides de 250 mM, montrent qu’outre la large bande d’absorption typique de eaq−, il existe une espèce absorbante supplémentaire qui est observée juste après l’impulsion (Encart). L’observation du produit de la réaction est très délicate car sa très faible absorbance est masquée par la forte absorbance de l’électron hydraté. L’espèce transitoire formée pendant l’impulsion de 5 ps (Figure 2, à droite) est identifiée comme le radical anionique nucléotidique. Cette attribution repose sur le fait que le spectre observé est identique à celui des spectres de radicaux anioniques précédemment rapportés après réaction de eaq− avec des nucléotides (36, 37). Parmi les nucléotides, la guanosine monophosphate (GMP) présente la réactivité la plus faible vis-à-vis de eql− (seulement 7 % des électrons sont capturés), tandis que la thymidine monophosphate (TMP, dans ce cas désoxythymidine monophosphate est utilisé) a la plus grande efficacité de capture (40 % des électrons). Nous avons pu estimer par la méthode de compétition (Encart) les vitesses de réaction de eql− avec les nucléotides à ~ 5 × 1012 pour TMP, 4 × 1012 pour CMP, 3 × 1012 pour AMP, et 0,6 × 1012 M−1 s−1 pour GMP [18].
Figure 2 –
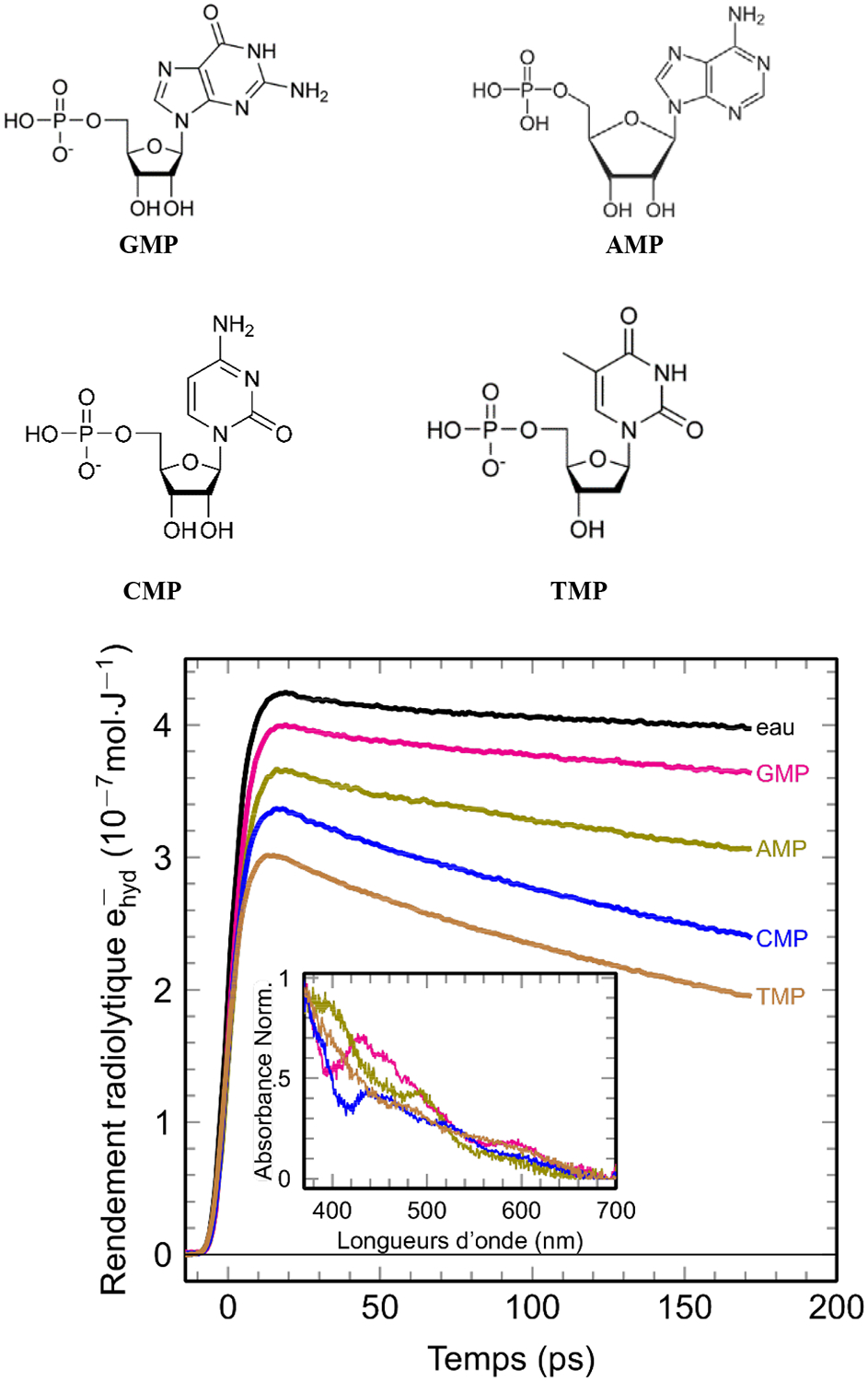
Rendement radiolytique de eaq− en fonction du temps en présence de nucléotides 250 mM (GMP, AMP, CMP, et TMP, voir les formules chimiques en haut) dans l’eau. en encart : Les spectres d’absorption des radicaux (GMP•-, AMP•-, CMP•-, et TMP•-) observés à 5 ps sont obtenus par soustraction appropriée du spectre d’absorption de eaq−, du spectre total enregistré expérimentalement, en utilisant une analyse globale des données [18]. Adaptation à partir de la Ref. [18] Copyright 2017 American Association for the Advancement of Science.
L’identification des sites où les électrons sont initialement piégés dans l’ADN pour produire les radicaux anioniques est une question importante pour élucider les voies de dégradation de l’ADN par la voie de la réaction avec des électrons. Les études par résonance paramagnétique électronique de Bernhard [19] et Sevilla et al. [20] ont conclu que les pyrimidines ont une plus grande affinité électronique que les purines. Steenken et ses collaborateurs [21] ont constaté que la thymine a le potentiel de réduction le plus élevé tandis que la guanine a le potentiel de réduction le plus faible. Nos résultats présentés sur la figure 2 montrent que la capture de eql− confirme les modèles expérimentaux et théoriques selon lesquels les bases de pyrimidine entièrement solvatées ont une affinité électronique supérieure à celle des purines. Les conclusions de ces travaux obtenus par radiolyse pulsée sur des bases isolées indiquent que, parmi les radicaux anioniques de base, le radical anionique guanine (G•-) subit une protonation très rapide. Cependant, cette réaction ne survient pas dans l’ADN à cause du transfert rapide des électrons de G•- à C et T, formant des radicaux anioniques de cytosine et de thymine (C•-, T•-) [22].
Les bases pyrimidines se révèlent être des capteurs d’électrons plus efficaces, avec une réactivité en ordre décroissant : T > C > A > G, en accord avec les résultats précédemment cités. En comparant le rendement initial de formation de eaq−, et par conséquent la quantité d’anions formés de ces bases, nos résultats suggèrent que la voie de dissociation de GMP*.- ne se produit pas dans les conditions de nos expériences. Ainsi, nos études montrent que ni eql− ni eaq− ne peuvent induire une dissociation directe des bases de l’ADN par la voie de l’attachement dissociatif en accord avec ce que l’on savait sur la réaction de eql−. De plus, nos résultats démontrent que la réaction d’un nucléotide avec eql− ou eaq− en solution neutre donne essentiellement les mêmes radicaux anioniques.
Pourquoi dans l’eau l’anion excité se relaxe-t-il sans se dissocier? Est-ce que la dissipation d’énergie de l’état excité est plus rapide que la dissociation (figure 3) ? Sur un système simple comme le soluté inorganique Ag(CN)2−, nous avons observé au contraire que la réaction de l’électron quasi libre forme un état anionique excité qui se dissocie rapidement avant relaxation [22].
Figure 3 -.
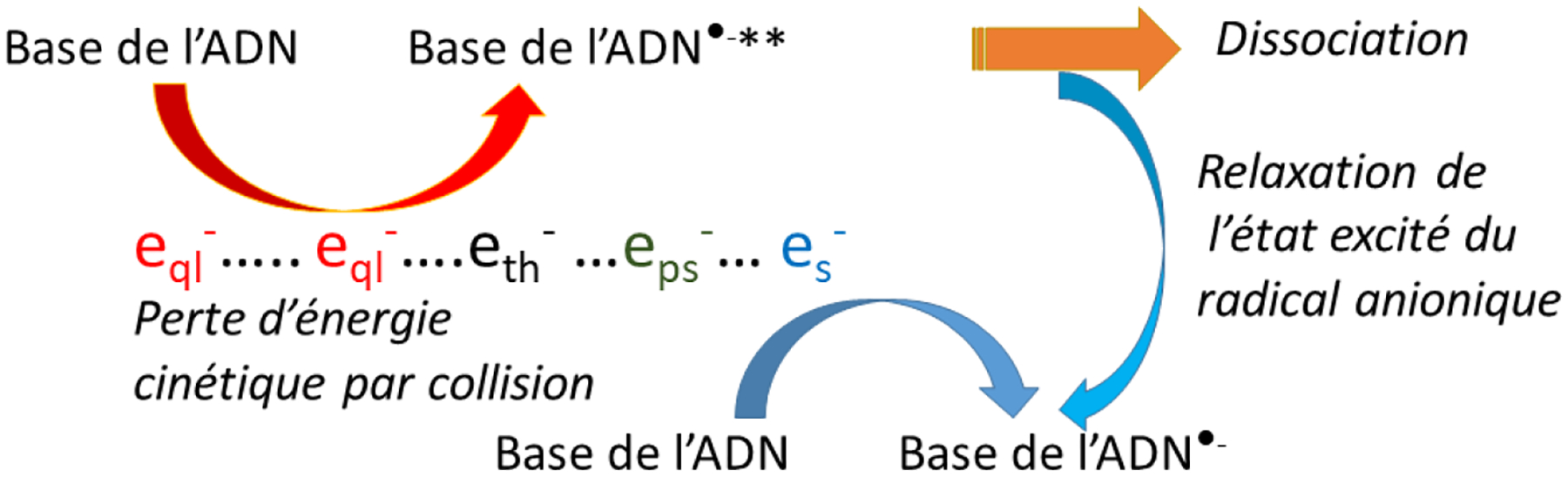
Schéma représentant la réactivité de eql− et de es− avec une base de l’ADN et les deux voies possibles du déclin du radical anionique excité (Base d’ADN•-*) : relaxation ou dissociation. Dans l’eau la dissociation de l’anion excité n’est pas observée.
Au contraire le même soluté Ag(CN)2− réagit avec l’électron hydraté sans processus de dissociation :
AgCN•- est observé comme le produit de la réaction seulement dans le cas de la réaction d’un électron quasi-libre et non dans le cas de l’électron hydraté [23]. Par rapport aux systèmes biologiques étudiés plus haut, Ag(CN)2− possède peu de degrés de vibration et on peut se demander si le transfert d’énergie via de nombreuses modes de vibration entre le soluté et le solvant est le facteur déterminant. En effet, si l’état excité de l’anion perd rapidement son énergie par couplages vibrationnels, on peut penser que cette voie de la dissipation d’énergie l’emporte sur celle de la dissociation.
Attachement électronique dissociatif de la ribothymidine dans le diéthylène glycol
Nous avons mené des études similaires sur un système biologique dans un autre solvant, le diéthylène glycol (DEG). La raison du choix de ce solvant est triple. Tout d’abord, le temps de solvatation des électrons dans DEG, comme pour les autres alcools, est relativement long et la solvatation se produit à l’échelle de quelques dizaines de picosecondes [23]. Ainsi nous pouvons, avec la résolution temporelle de notre montage (5 ps), observer directement non seulement es− mais aussi eps− et ainsi pouvoir distinguer la réactivité de eps− et celle de eql−. Deuxièmement, la valeur de la constante diélectrique de DEG (εr = 31,69) est bien plus faible que celle de l’eau libre mais plus proche de celle d’un cytosol ou d’un noyau très concentré où l’eau est très liée aux molécules biologiques et riche en sels [24]. De plus, dans DEG, l’ADN peut conserver la même structure native double brin que dans l’eau [25]. Finalement, nous avons trouvé que la ribothymidine (rT), un nucléoside de l’ARN choisi comme modèle, peut être suffisamment dissoute dans DEG (jusqu’à 0,5 M) pour favoriser la capture des électrons quasi-libres dans un temps ultracourt. Ainsi le choix de DEG nous permet d’étudier les réactions élémentaires plus pertinentes pour le système biologique. Nous avons donc réalisé des études similaires sur des solutions de rT dans DEG à différentes concentrations (0 – 0,5 M) à la température ambiante, pour rechercher s’il existe un processus d’attachement électronique dissociatif avec rT [26] (figure 4).
Figure 4 -.
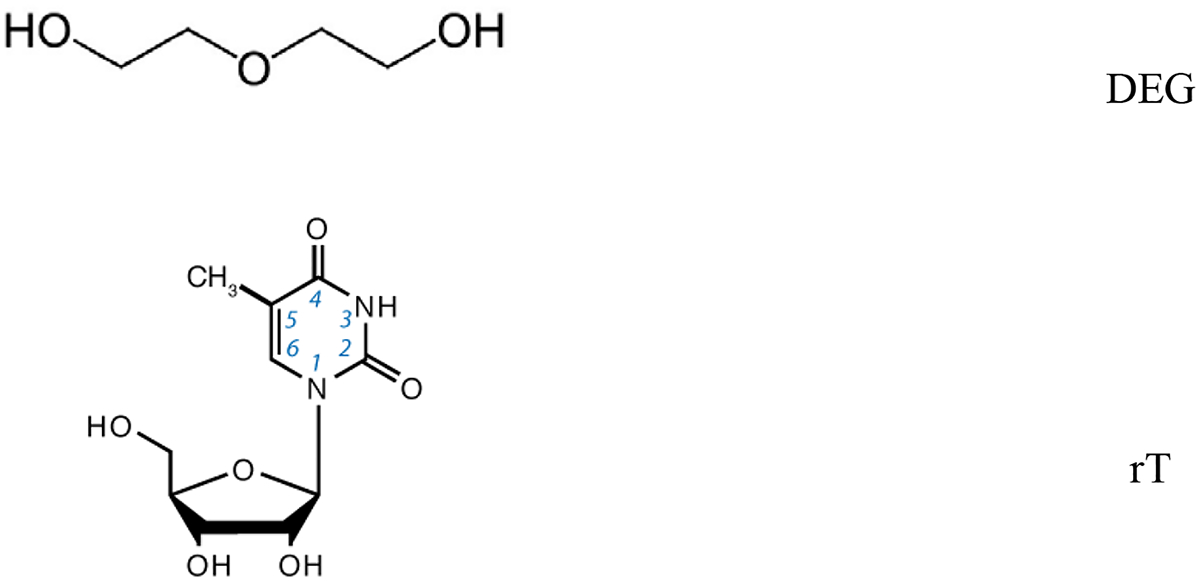
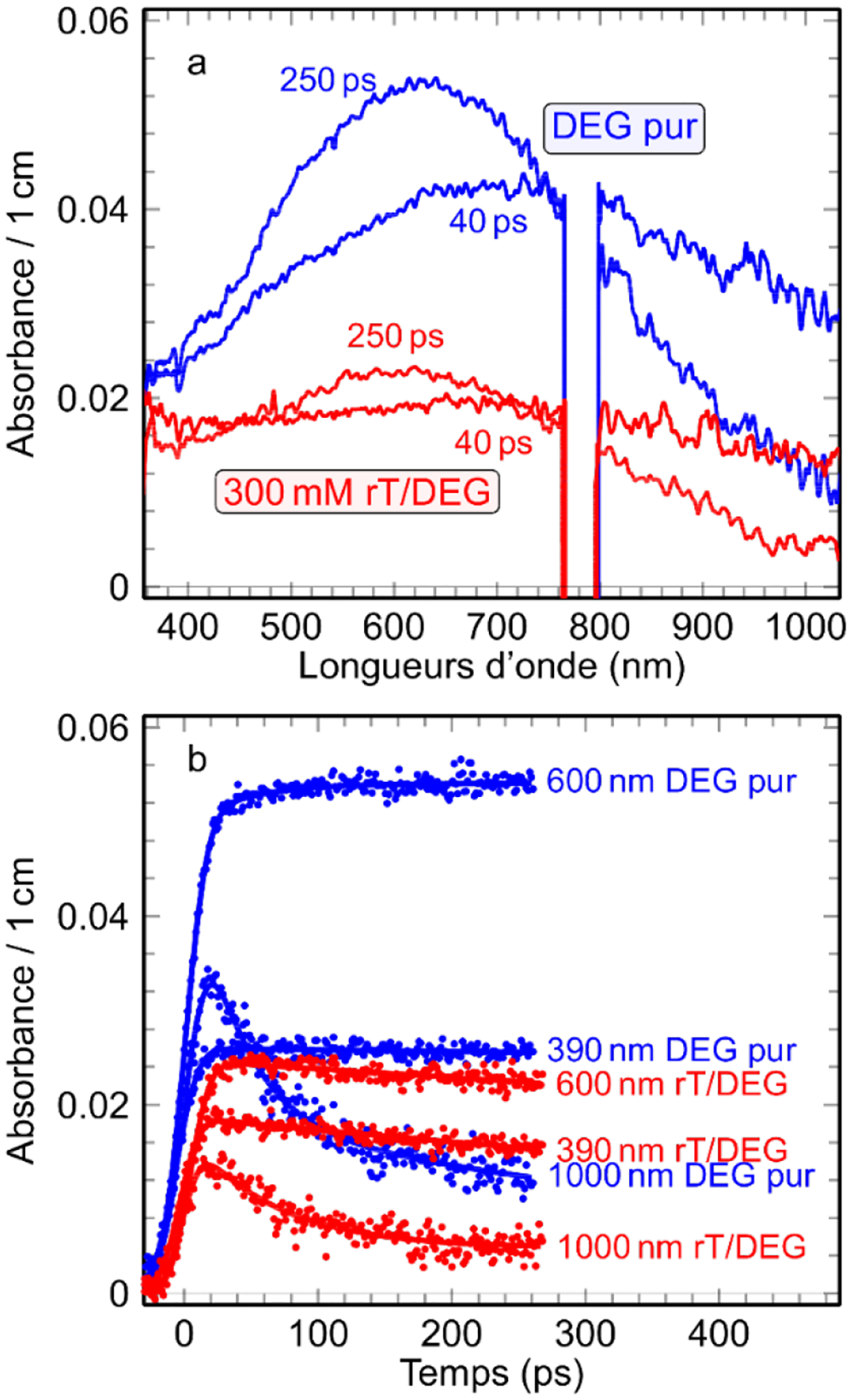
En haut : formules chimiques du diéthylène glycol (DEG) et de la ribothymidine (rT). (a) Exemples de spectres d’absorption transitoire dans le diéthylène glycol (DEG) pur et de solutions de ribothymidine (rT) 0,3 M dans DEG obtenus par radiolyse impulsionnelle. Le signal du laser empêche des mesures autour de la longueur d’onde de 800 nm. (b) Cinétiques enregistrées à différentes longueurs d’onde. La durée de l’impulsion électronique est d’environ 5 ps. La dose par impulsion électronique était de 25 Gy [26].
Les spectres d’absorption transitoire de l’électron en excès dans DEG pur sont illustrés sur la figure 5. Une fraction significative des électrons formés dans DEG immédiatement après l’impulsion subit progressivement le processus de la solvatation. L’absorbance au-dessus de 900 nm diminue rapidement, et un spectre d’absorption optique large et intense avec un maximum à environ 750 nm, due aux électrons pré-solvatés dans DEG, subit un décalage continu d’environ 120 nm vers le bleu, accompagné d’une amplitude croissante sur environ 250 ps. A la fin de cette évolution spectrale (figure 5a), on observe une bande d’absorption large avec un maximum à 630 nm. Sur la base d’études antérieures portant sur divers alcools [27], cette bande est attribuée au spectre de l’électron solvaté dans DEG, eDEG−.
Figure 5 –
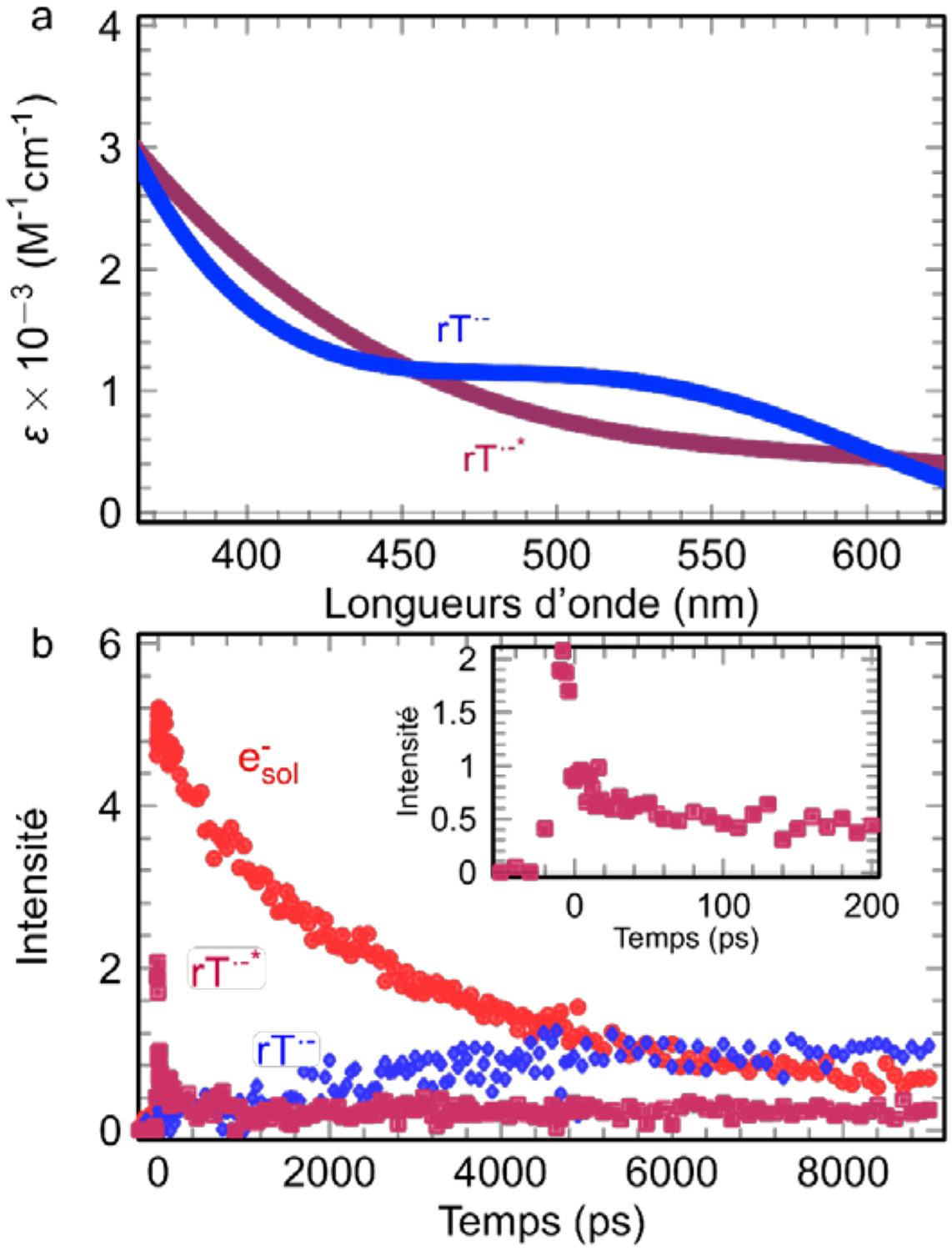
(a) : Coefficients d’extinction molaire de l’état excité et de l’état fondamental des radicaux anioniques, rT•¯* (obtenu en solution très concentrée) et rT•¯ (obtenus en solution diluée ou bien en solution concentrée mais à temps long corrélé avec le déclin de es¯). (b) : Cinétiques obtenues par la déconvolution des données expérimentales (figure 1). Encart : Cinétique de rT•¯* jusqu’à 200 ps [26]. (Reproduit avec la permission [Ref. 26] Copyright 2019 Nature Springer.
Pour étudier l’attachement des électrons à la ribothymidine, rT et la formation consécutive de rT•-*, des études de radiolyse par impulsions picosecondes ont été réalisées dans des solutions de 50 à 500 mM en rT. Dans ces solutions, l’ionisation directe ou l’excitation du soluté lui-même n’est pas significative. Comme exemple, les spectres transitoires et les cinétiques à différentes longueurs d’onde sont présentés pour la concentration de 0,3 M sur les figures 8a et 8b. La capture rapide d’électrons par rT génère un nouveau signal transitoire qui est observé immédiatement après l’impulsion dans les régions visibles - UV (figure 5). Contrairement à une légère augmentation du signal observé dans DEG pur, la cinétique transitoire dans tout le domaine de longueur d’onde 370–600 nm des solutions de rT montre un déclin marqué (figure 5). Une diminution substantielle de l’absorbance initiale dans le proche infrarouge en corrélation avec l’augmentation de la concentration en rT montre clairement que rT a capturé une fraction importante des électrons libres, eql−, avant qu’ils ne soient solvatés dans DEG. Par exemple, selon la concentration, environ 12 % à 75 % des électrons sont capturés par des molécules rT aux concentrations de 0,05 et 0,5 M, respectivement.
De plus, dans la région infrarouge de 720 à 1000 nm, où les absorbances ne sont associées qu’à l’électron pré-solvaté, epre¯, quelle que soit la concentration de rT, elles déclinent avec une vitesse pratiquement identique à celles observées dans DEG pur. Par conséquent, ces résultats établissent que la présence de rT n’affecte pas de manière significative le processus de la solvatation des électrons dans DEG. Cela signifie aussi que rT ne réagit pas avec epre¯ sur l’échelle de temps de centaines de ps. En nous basant sur nos attributions des états eql− (sans absorption dans le visible ou proche IR), epre¯(absorbant dans le proche IR) et eDEG¯ (absorbant dans le visible), nous concluons que les électrons capturés par rT pendant la durée de l’impulsion (≤ 5 ps) sont les électrons quasi-libres qui ont une certaine énergie cinétique. La signature de cette réaction est d’une part la baisse de la quantité de epre¯ et par conséquent celle de eDEG¯ observée en fin d’impulsion, et d’autre part la formation de rT•¯* observée simultanément. La déconvolution des données spectroscopiques à différentes concentrations nous permet d’établir le spectre d’absorption distinct de rT•¯* et de rT•¯ et de leurs cinétiques respectives (figure 5). Le déclin très rapide de rT•¯* sans donner rT•¯ correspond à une dissociation de la liaison C-N qui a été identifiée par ailleurs par spectrométrie de masse dans des solutions irradiées par rayonnement gamma. Une telle dissociation a été observée pour des réactions d’oxydation par des radicaux •OH.28,29 On peut illustrer cette similitude par le schéma ci-dessous (figure 6) montrant la comparaison entre la réaction de eql− et rT et une des réactions du radical •OH avec le thymidine. Dans les deux cas : la liaison glycosidique N-C est coupée et par conséquent la base thymine est relâchée et un radical de ribose est formé. Notons que la nature des deux radicaux riboses n’est pas identique et que ces deux radicaux vont subir d’autres réaction dans le milieu.
Figure 6-.
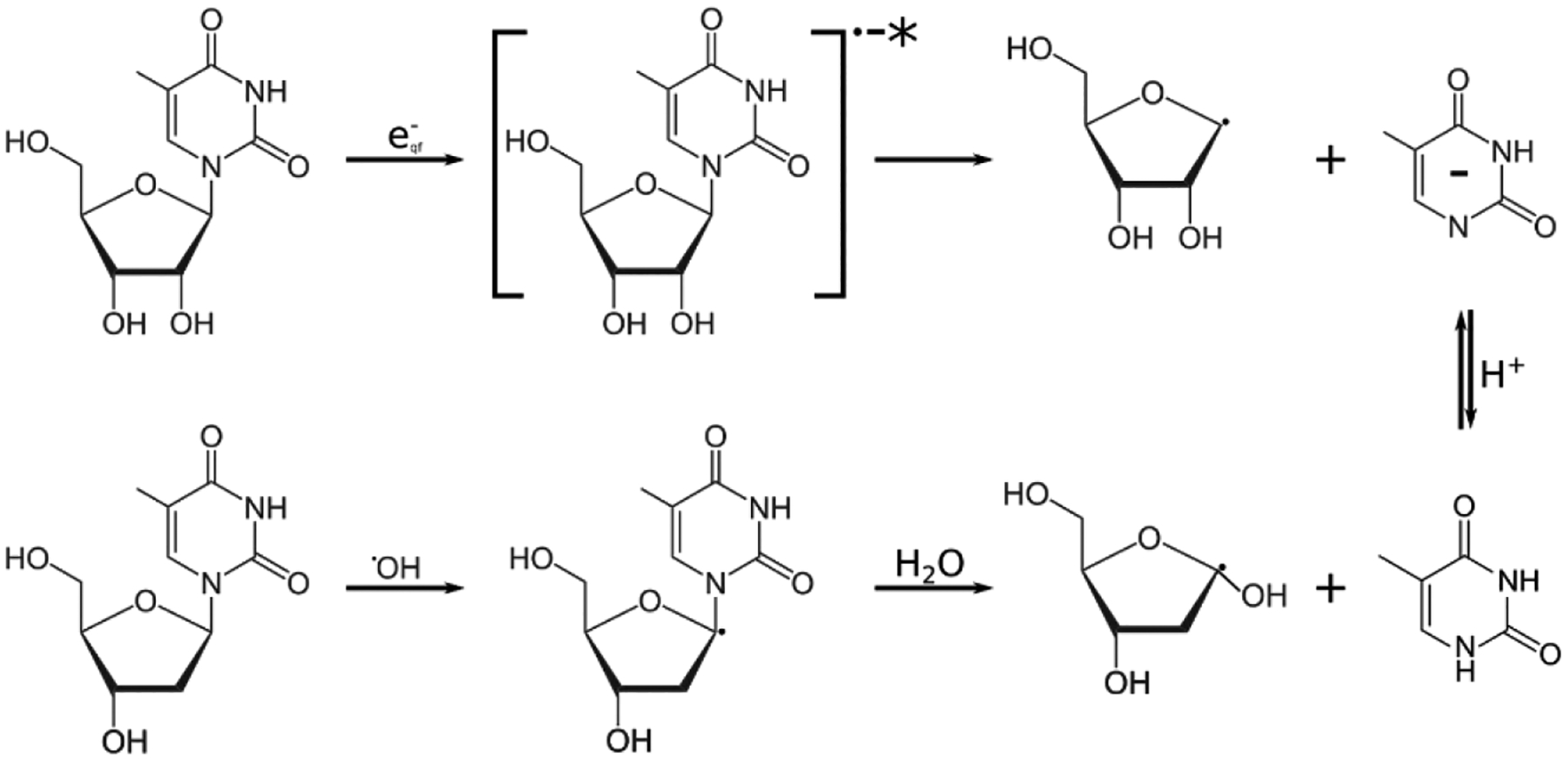
Réaction entre eql− et rT et celle du radical •OH et Thymidine.
Les expériences dans DEG ont donc permis, grâce au ralentissement des évènements, d’observer un attachement dissociatif de eql−. Les phénomènes dans l’eau aux concentrations plus limitées, que nous avons pu étudier, n’ont pas permis de conclure à cette dissociation. Néanmoins, dans les conditions biologiques réelles où la fraction de l’ADN est très élevée, on peut penser que de telles dissociations induites par eql− peuvent aussi avoir lieu.
Conclusion
Nos études en radiolyse impulsionnelle picoseconde ont clairement montré que des réactions ultrarapides peuvent prendre place quand un système biologique est soumis à l’irradiation [30]. Dans cette étude nous nous sommes focalisés sur la capture de l’électron quasi libre en solution à la température ambiante. Nous avons constaté que, dans l’eau, eql− réagit efficacement avec les bases d’ADN en formant des anions stables. En revanche, les mesures dans le diéthyl-glycol démontrent que eql− peut réagir avec la ribothymidine (rT), permettent d’identifier le spectre d’absorption du radical anionique excité, rT•¯*. Mais le principal résultat est que cet attachement électronique est dissociatif. Cette démonstration a pu se réaliser grâce à des mesures précises de l’absorbance des produits de réactions, juste quelques picosecondes après le dépôt d’énergie dans les solutions. Cette démonstration est une première importante puisqu’il signifie que non seulement la voie oxydative peut causer des dommages irréversibles de coupures de liaison, mais paradoxalement qu’un électron, connu pour provoquer des réactions de réduction en formant un anion stable, peut aussi, quand il a de l’énergie cinétique, casser des liaisons glycosidiques en solution en formant un état excité dissociatif et ainsi oxyder l’ADN (figure 6). L’étude des réactions de eql− en solution avec les systèmes biologiques est un enjeu important pour mieux connaitre les premières étapes du mécanisme de dommage à l’ADN par les rayonnements ionisants. Nos données permettent aussi d’améliorer les codes de calculs utilisés pour simuler les dommages à l’ADN.
Encadré : Radiolyse impulsionnelle.
Les phénomènes cinétiques ultrarapides sont observés grâce à l’installation de radiolyse pulsée picoseconde ELYSE du Laboratoire de Chimie Physique [9]. Elle est basée sur un accélérateur d’électrons qui sont générés à partir d’une photocathode par un laser femtoseconde. Le montage synchronise le faisceau d’électrons pour l’ionisation avec une fraction du faisceau du laser pour sonder les espèces créées par l’impulsion d’électrons.
A gauche :
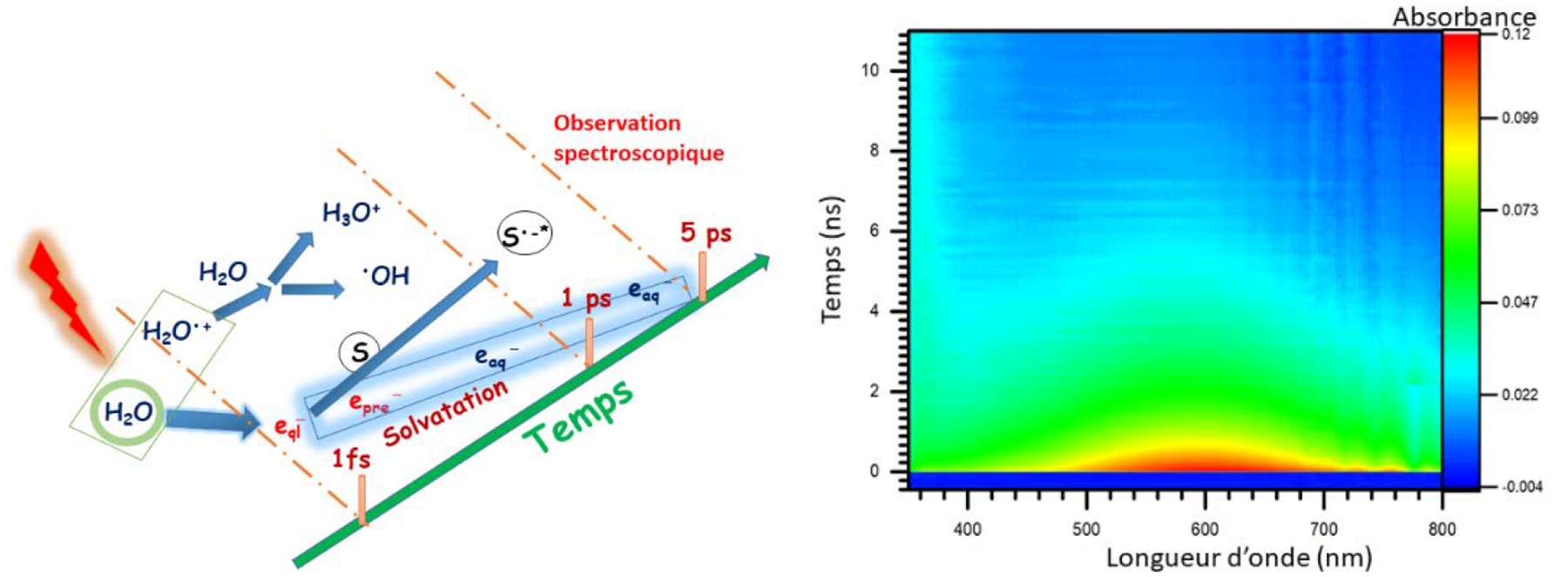
Une impulsion ionise l’eau et eql− est émis avec une certaine énergie cinétique. Cet électron perd son énergie et se thermalise par des collisions et finalement s’hydrate en eaq−. Mais en présence d’un soluté S, eql− peut réagir avec ce dernier et former un anion radical à l’état excité S•-*. L’observation de cet anion à l’état excité, ou de son produit de fragmentation, est effectuée par spectroscopie optique après l’impulsion de 5 ps. A droite : Exemple de données expérimentales enregistrées dans une solution de 0.3 M ribothymidine dans DEG obtenues par la radiolyse impulsionnelle. La ligne de retard permet de suivre la cinétique jusqu’ 11 ns avec des pas de 5 ps (voir figure 5). L’échelle des absorbances est croissante du bleu au rouge.
Remerciement :
J. M. remercie la Fondation nationale chinoise des sciences naturelles (subvention n° 11975122 et subvention n° 21906083) et la Fondation scientifique pour la jeunesse de la province du Jiangsu (subvention n° BK2019030384) de leur soutien. A. A. remercie le National Cancer Institute of the National Institutes of Health (Grant RO1CA045424) et la National Science Foundation sous le numéro de subvention CHE-1920110. De plus, A. A. remercie également R.E.F. et C.B.R. à l’Université d’Oakland pour leur soutien.
Contributor Information
Jun Ma, Professeur à l’Université de Nanjin en Chine (Aeronautics et Astronautics). Il a effectué sa thèse et une partie de son post-doctorat au Laboratoire de Chimie Physique..
Sergey Denisov, Chargé de recherche au CNRS au Laboratoire de Chimie Physique (UMR 8000) à Orsay, développant les méthodes spectroscopiques sur ELYSE..
Amitava Adhikary, Research Assistant Professor à l’Université d’Oakland aux Etats-Unis. Expert des dommages à l’ADN par le rayonnement ionisant..
Mehran Mostafavi, Professeur à l’Université Paris-Sud, responsable de l’équipe « Actes chimiques élémentaires en phase condensée » du Laboratoire de Chimie Physique (UMR 8000) et directeur adjoint scientifique au CNRS en charge des grands instruments de recherche à l’institut de Chimie (INC). Distingué par le Prix Chercheur Confirmé 2019, de la Division de Chimie Physique de la SCF..
References
- [1].Belloni J, Mostafavi M, Douki D, Spotheim-Maurizot M La chimie sous rayonnement : des aspects fondamentaux aux applications dans les sciences des matériaux et les sciences de la vie, Act. Chim 2008, n° 316 - février. [Google Scholar]
- [2].von Sonntag C Free-Radical-Induced DNA Damage and Its Repair: A Chemical Perspective; Springer: Berlin, 2006; pp 211–504. [Google Scholar]
- [3].Cadet J, Douki T, Gasparutto D, Ravanat JL, Wagner JR In Radical and Radical Ion Reactivity in Nucleic Acid Chemistry; Greenberg M, Ed.; John Wiley & Sons: Hoboken, NJ, 2010; pp 69–98. [Google Scholar]
- [4].Ma J, Denisov SA, Marignier JL, Pernot P, Adhikary A, Seki S Mostafavi M Ultrafast Electron Attachment and Hole Transfer Following Ionizing Radiation of Aqueous Uridine Monophosphate J. Phys. Chem. Lett 2018; 9; pp 5105–5109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Boudaiffa B, Cloutier P, Hunting D, Huels MA Sanche L Resonant formation of DNA strand breaks by low-energy (3 to 20 eV) electrons. Science 2000, 287, 1658–1660. [DOI] [PubMed] [Google Scholar]
- [6].Martin F, et al. DNA strand breaks induced by 0–4 eV electrons: the role of shape resonances. Phys. Rev. Lett 2004, 93, 068101. [DOI] [PubMed] [Google Scholar]
- [7].Ptasinska S, Denifl S, Scheier P, Illenberger E Märk TD Bond- and site-selective loss of H atoms from nucleobases by very-low-energy electrons (<3 eV). Angew. Chem. Int. Ed. Engl 2005, 44, 6941–6943. [DOI] [PubMed] [Google Scholar]
- [8].Zheng Y, Cloutier P, Hunting DJ, Wagner JR, Sanche L Glycosidic bond cleavage of thymidine by low-energy electrons. J. Am. Chem. Soc 2004, 126, 1002–1003. [DOI] [PubMed] [Google Scholar]
- [9].Belloni J, Monard H, Gobert F, Larbre J-P, Demarque A, De Waele V, Lampre I, Marignier J-L, Mostafavi M, Bourdon JC, Bernard M, Borie H, Garvey T, Jacquemard B, Leblond B, Lepercq P, Omeichb M, Roch M, Rodier J, Roux R, ELYSE – A picosecond electron accelerator for pulse radiolysis research. Nucl. Instruments Methods Phys. Res. Sect. A Accel. Spectrometers, Detect. Assoc. Equip 2005, 539, 527–539. [Google Scholar]
- [10].Ma J, Schmidhammer U, Pernot P, Mostafavi M Reactivity of the strongest oxidizing species in aqueous solutions: The short-lived radical cation H2O•+. J. Phys. Chem. Lett 2014, 5, 258–261. [DOI] [PubMed] [Google Scholar]
- [11].Mostafavi M, H2O•+, l’espèce la plus fugace et oxydante L’Act. Chim 2018, 429. [Google Scholar]
- [12].Ma J, Schmidhammer U, Mostafavi M, Picosecond pulse radiolysis of highly concentrated phosphoric acid solutions: mechanism of phosphate radical formation. J. Phys. Chem. B 2014, 119, 7180–7185. [DOI] [PubMed] [Google Scholar]
- [13].Wang F, Schmidhammer U, Larbre JP, Zong ZZ, Marignier JL, Mostafavi M Time-dependent yield of the hydrated electron and the hydroxyl radical in D2O: a picosecond pulse radiolysis study. Phys. Chem. Chem. Phys 2018, 20, 15671–15679 [DOI] [PubMed] [Google Scholar]
- [14].Migus A, Gauduel Y, Martin JL, Antonetti A Excess electrons in liquid water: first evidence of a prehydrated state with femtosecond lifetime. Phys. Rev. Lett 1987, 58, 1559–1562. [DOI] [PubMed] [Google Scholar]
- [15].Kumar A, Sevilla MD in Handbook of Computational Chemistry, Shukla MK, Leszczynski J, Eds. Springer-Verlag, Berlin, 2012, pp. 1215–1256. [Google Scholar]
- [16].Gu J, Leszczynski J, Schaefer HF, Interactions of electrons with bare and hydrated biomolecules: From nucleic acid bases to DNA segments. Chem. Rev 2012, 112, 5603–5640. [DOI] [PubMed] [Google Scholar]
- [17].Sevilla MD, Becker D, Kumar A, Adhikary A Gamma and ion-beam irradiation of DNA: Free radical mechanisms, electron effects, and radiation chemical track structure. Radiat. Phys. Chem 2016, 128, 60–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ma J, Furong W, Sergey DA, Adhikary A, Mostafavi M Reactivity of prehydrated electrons toward nucleobases and nucleotides in aqueous solution. Science Advances 2017, 3:e1701669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Bernhard WA Sites of electron trapping in DNA as determined by ESR of one-electron-reduced oligonucleotides. J. Phys. Chem 1989, 93, 2187–2189. [Google Scholar]
- [20].Wang W, Sevilla MD Protonation of nucleobase anions in gamma-irradiated DNA and model systems. Which DNA base is the ultimate sink for the electron? Radiat. Res 1994, 138, 9–17. [PubMed] [Google Scholar]
- [21].Steenken S, Jovanovic SV How easily oxidizable is DNA? One-electron reduction potentials of adenosine and guanosine radicals in aqueous solution. J. Am. Chem. Soc 119, 617–618 (1997). [Google Scholar]
- [22].Wang F, Archirel P, Muroya Y, Yamashita S, Pernot P, Yin C, El Omar AK, Schmidhammer U, Teuler J, Mostafavi M Effect of the solvation state of electron in dissociative electron attachment reaction in aqueous solutions. Phys. Chem. Chem. Phys 2017, 19, 23068–23077. [DOI] [PubMed] [Google Scholar]
- [23].Bonin J, Lampre I, Pernot P et al. Solvation dynamics of electron produced by two-photon ionization of liquid polyols. III. Glycerol. J. Phys. Chem. A 2008, 112, 1880–1886. [DOI] [PubMed] [Google Scholar]
- [24].Goncharenko AV, Chang YC Effective dielectric properties of biological cells: generalization of the spectral density function approach. J. Phys. Chem. B, 2009, 113, 9924–9931). [DOI] [PubMed] [Google Scholar]
- [25].Eliasson R, Hammarsten E, Lindahl T, Björk I; Laurent TC The stability of deoxyribonucleic acid in glycol solution. Biochim. Biophys. Acta 1963, 68, 234–239. [Google Scholar]
- [26].Ma J, Kumar A, Muroya Y, Muroya Y, Yamashita Sh., Sakurai T, Denisov SA, Michael D. Sevilla, Amitava Adhikary, Shu Seki & Mehran Mostafavi et al. Observation of dissociative quasi-free electron attachment to nucleoside via excited anion radical in solution NATURE COMMUNICATIONS 2019, 10, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Jou FJ, Freeman GR Band resolution of optical spectra of solvated electrons in water, alcohols, and tetrahydrofuran. Can. J. Chem 1979, 57, 591–597. [Google Scholar]
- [28].Dizdaroglu M, Neuwald K, von Sontag C Model Compounds, IX* Carbohydrate Products in the y-Radiolysis of Thymidine in Aqueous Solution. The Radical-Induced Scission of the N-Glycosidic Bond. Z. Naturforsch 1976, 31, 227–233. [Google Scholar]
- [29].Sy D, Savoye C, Begusova M, Michalik V, Charlier M, Spotheim-Maurizot M Sequence-dependent variations of DNA structure modulate radiation-induced strand breakage Int. J. Radiat. Biol 1997, 72, 147–155. [DOI] [PubMed] [Google Scholar]
- [30].Ma J, Denisov SA, Adhikary A, Mostafavi M Ultrafast Processes Occurring in Radiolysis of Highly Concentrated Solutions of Nucleosides/Tides. Invited Review (special issue, “Radiation Damage to DNA” (J. Rak, M. Zdrowowicz (Eds.)), Int. J. Mol. Sci 2019, 20, article no. 4963. [DOI] [PMC free article] [PubMed] [Google Scholar]