
Figure 2. Atrial ectopy. Molecular pathways promoting Ca2+-mediated ectopy.
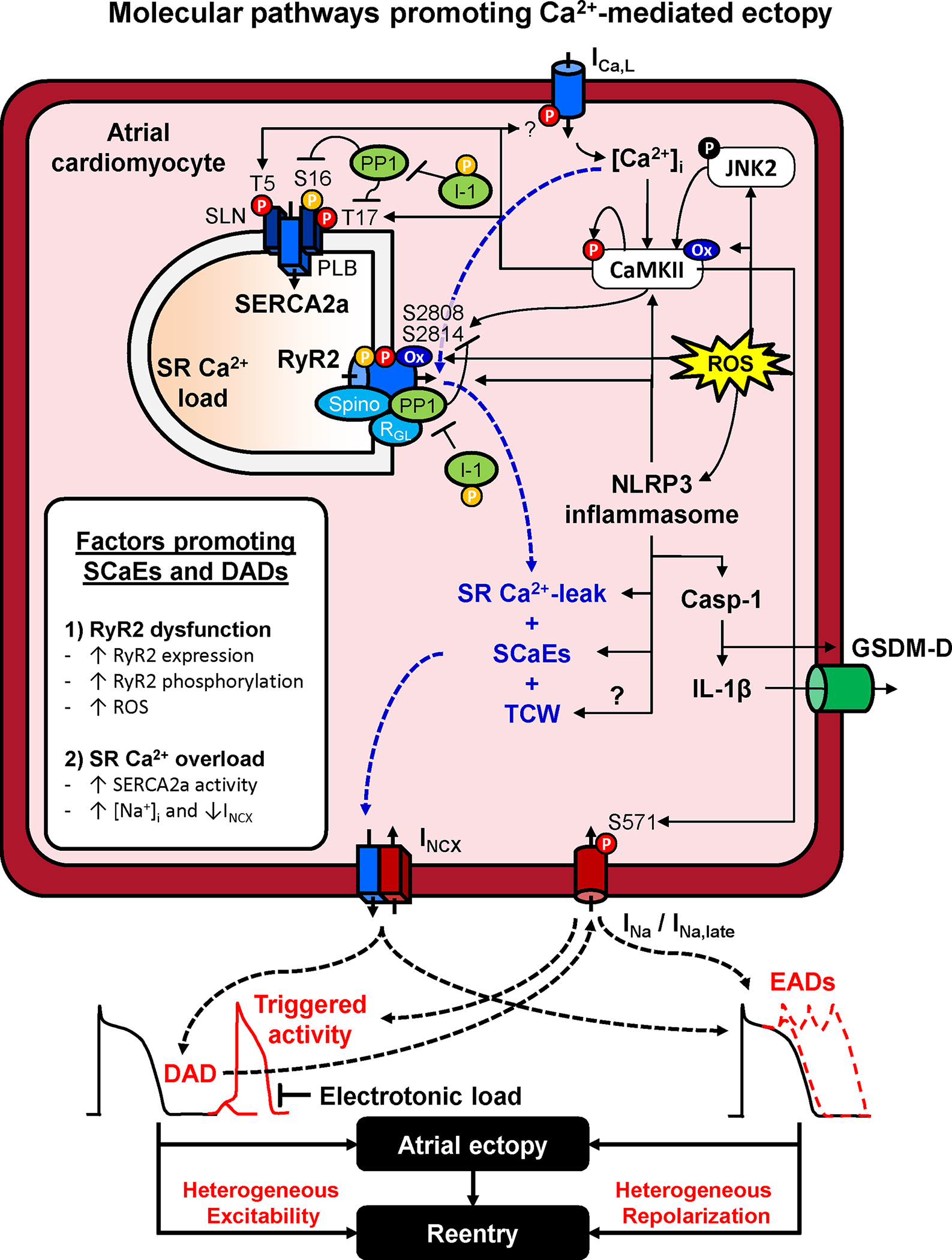
Increased sarcoplasmic reticulum (SR) Ca2+ leak and spontaneous SR Ca2+-release events (SCaEs) primarily result from dysfunction of the cardiac ryanodine receptor type-2 (RyR2) channel or SR Ca2+-overload. RyR2 dysfunction is promoted by increased RyR2 expression, hyperphosphorylation (e.g., due to increased Ca2+/calmodulin-dependent protein kinase-II, CaMKII, activity or improper targeting of protein phosphatase-1, PP1), or RyR2 oxidation due to increased reactive oxygen species (ROS). ROS mediated NLRP3 inflammasome activation amplifies the Ca2+-handling abnormalities and activates caspase-1 (Casp-1) which increases interleukin (IL)-1β generation and the formation of gasdermin-D-derived plasmamembrane-pores, allowing the release of IL-1β out of the cell, spreading inflammatory signaling. SR Ca2+-overload is promoted by increased activity of the SR Ca2+-ATPase-2a (SERCA2a) or elevated intracellular Na+, reducing Ca2+-extrusion via the Na+/Ca2+ exchanger type-1 (NCX1). SR Ca2+ overload also promotes L-type Ca2+-current (ICa,L)-dependent triggered Ca2+ waves (TCW). SCaEs and TCW activate a transient-inward current mediated by NCX (INCX) resulting in DADs or EADs, depending on their timing relative to the atrial action potential. DADs and EADs can promoted atrial ectopy, as well as reentry through increased heterogeneity of excitability and repolarization. Abbreviations: GSDM-D, N-terminal Gasdermin-D fragment; I-1, inhibitor-1 of PP1; JNK2, c-Jun N-terminal kinases-2; NLRP3, NACHT, LRR and PYD domains-containing protein 3; PLB, phospholamban; SLN, sarcolipin.