
Figure 6. Atrial fibrillation-promoting Ca2+/calmodulin-dependent protein kinase-II (CaMKII)- and NACHT, LRR and PYD domains-containing protein 3 (NLRP3)-inflammasome feed-forward signaling network.
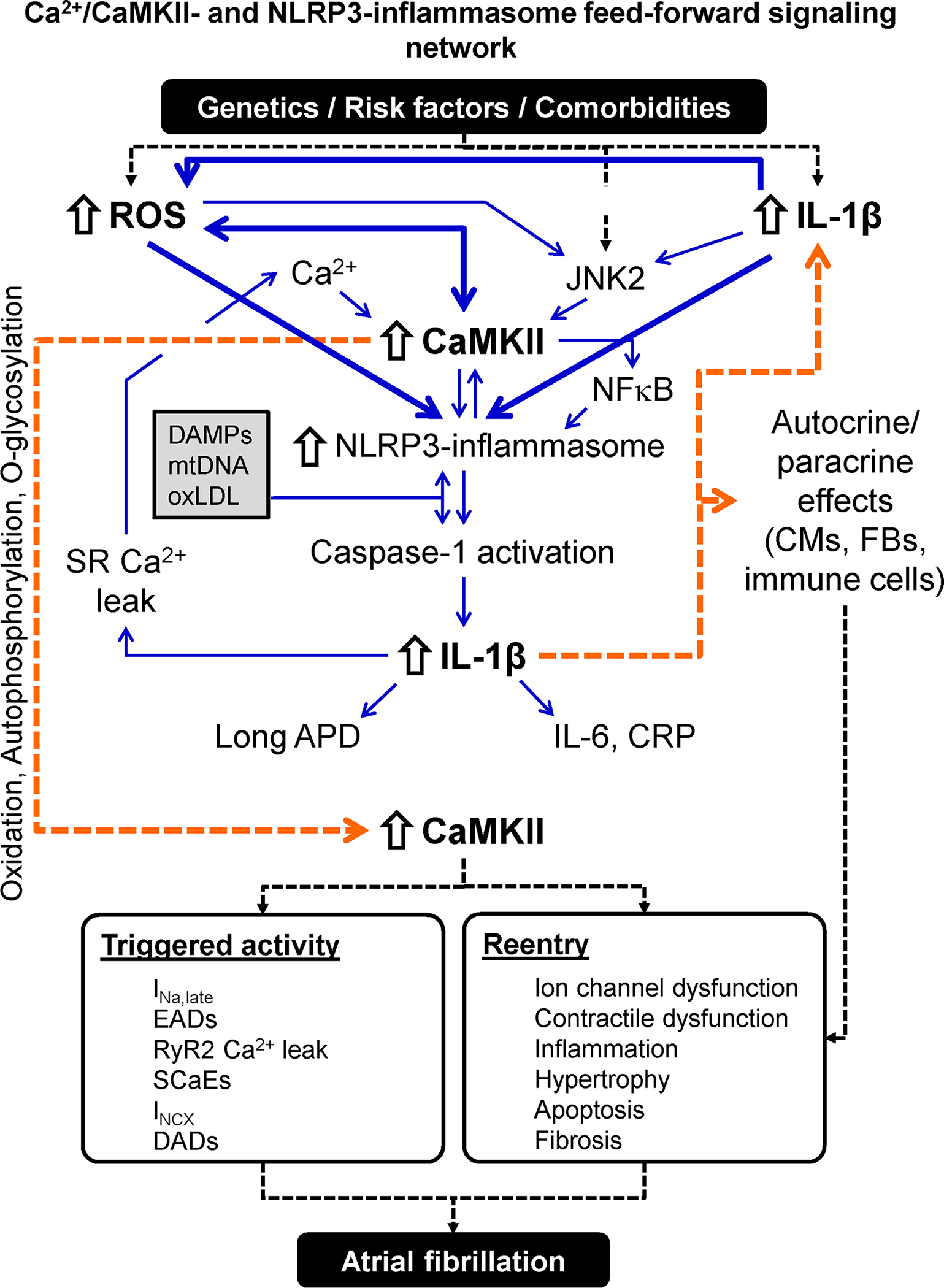
Risk factors and comorbidities create an environment in which danger-associated molecular patterns (DAMPs), mitochondrial DNA (mtDNA) and oxLDL activates the atrial NLRP3 inflammasome. Cardiac-restricted increases in reactive oxygen species (ROS) production and c-Jun N-terminal kinase-2 (JNK2) activity further stimulate the NLRP3 inflammasome via CaMKII-dependent and -independent pathways. The resulting stimulation of caspase-1 maturates interleukin (IL)-1β, which leaves the cell, thereby spreading the inflammatory signaling and increasing the synthesis of IL-6 and C-reactive protein (CRP). IL-1β amplifies the NLRP3 inflammatory signaling and promotes sarcoplasmic reticulum (SR) Ca2+ leak and action potential duration (APD) changes in cardiomyocytes (CMs), creating a feedforward signaling network. IL-1β also exerts paracrine effects on cardiac fibroblasts (CFs) and immune cells causing hypertrophy, apoptosis and fibrosis. Activation and perpetuation of this feedforward Ca2+/CaMKII/NLRP3-inflammasome signaling network promotes triggered activity and reentry and increases AF susceptibility. Abbreviations: DADs, delayed afterdepolarizations; EADs, early afterdepolarizations; INa,late, Persistent/late Na+ current; INCX, Na+-Ca2+-exchanger current; NFκB, nuclear factor kappa-light-chain-enhancer of activated B cells; RyR2, ryanodine receptor type-2; SCaEs, spontaneous SR Ca2+-release events.