

Abstract
Transmembrane domain (TMD) mutations of ERBB2 have previously been reported in lung cancer patients in addition to well‐studied kinase domain (KD) mutations, which may stabilize ERBB2 heterodimerization with other EGFR family members and favor a kinase active conformation. However, the frequency and clinical significance of ERBB2 TMD mutations in Chinese population is unknown. We prospectively analyzed the next‐generation sequencing data of 34 368 Chinese lung cancer patients with different sample types, including tumor tissue, plasma, cerebrospinal fluid, and pleural effusion. Patients' clinical characteristics and treatment history were retrieved from the database for further evaluation. Our findings show that ERBB2 V659/G660 mutations were detected at a frequency of 0.13% (45/34 368), of which the most frequent was V659D/E (88.9%), with a trend in nonsmokers and male. Moreover, 18% of patients (8/45) showed EGFR and/or ERBB2 amplification, whereas nine patients presented EGFR L858R or exon19 deletion. Interestingly, novel ERBB3 TMD mutation I646R was found coexisting in three patients with ERBB2 V659D and one patient with ERBB2 G660D, which might influence its heterodimerization with ERBB2 and further activate ERBB2. Four ERBB2 TMD mutation‐positive patients received afatinib monotherapy or combination therapy, but showed variable responses. One patient with V659E responded well to ERBB2 inhibitor lapatinib plus capecitabine as well as subsequent afatinib treatment upon progression. Our study provides valuable insights into the distribution of ERBB2 TMD mutations by employing the largest Asian lung cancer cohort thus far. Patients with ERBB2 TMD mutations who received afatinib, a pan‐ERBB inhibitor, demonstrated mixed responses, posing the urgent need to develop more effective therapeutic strategy for patients who carry ERBB2 TMD mutations.
Keywords: comprehensive genomic profiling, ERBB2, NSCLC, TMD, transmembrane domain mutation
Transmembrane domain (TMD) mutations of ERBB2 were identified in 0.13% of a large Chinese lung cancer cohort with a ratio of 0.17% in lung adenocarcinomas. Novel ERBB3 TMD mutation I646R was found co‐existing in four ERBB2 TMD mutation carriers, which might affect ERBB2‐ERBB3 heterodimerization. ERBB2 TMD carriers demonstrated mixed response to afatinib, posing the need for more effective therapeutic strategy.

1. Introduction
Since the discovery of actionable activating epidermal growth factor receptor (EGFR) mutation and anaplastic lymphoma kinase (ALK) rearrangement in lung cancer, thoracic oncologists have led the way in comprehensive genomic profiling (CGP) of solid malignancies for precision medicine. Indeed, multiple additional actionable driver alterations have been identified including some of these mutations are at < 1% incidence in non‐small‐cell lung cancer (NSCLC) such as neurotrophic tropomyosin‐related kinase rearrangement (Gatalica et al., 2019) and NRG1 fusions (Drilon et al., 2018). Another rare but potentially actionable driver mutation is located in the transmembrane domain (TMD) at valine amino acid residue 659 and glycine at amino acid residue 660 in Erb‐b2 receptor tyrosine kinase 29 (ERBB2) (Bargmann et al., 1986; Pahuja et al., 2018). Clinically, it was initially described in the index case of a family with familial lung cancer (Yamamoto et al., 2014). TMD mutations are typically mutually exclusive from HER2 kinase domain (KD) mutations and other oncogenic driver mutations. TMD mutations are located within the glycine zipper motif at the N‐terminal portion of TMD which is critically important to the dimerization of ERBB2 to other EGFR family members (Bocharov et al., 2008; Mineev et al., 2010). NMR studies of the protein revealed that TMD mutations stabilize the N‐terminal dimerization and favor a kinase active conformation (Ou et al., 2017). In vitro experiments confirmed that ERBB2 TMD mutations can be inhibited by afatinib, a second‐generation EGFR/ERBB2 tyrosine kinase inhibitor (TKI), but not by gefitinib, a first‐generation EGFR TKI (Yamamoto et al., 2018). A subsequently larger survey of 8551 lung adenocarcinoma patients from a US‐based genomic profiling database revealed that ERBB2 TMD mutations are very rare at a frequency of 0.18%. The study further suggested that ERBB2 TMD mutant patients may respond to afatinib (Ou et al., 2017). In this study, considering the unique mutation spectrum of Chinese lung cancer patients compared to the western population, we investigated the frequency and mutation spectrum of ERBB2 TMD mutations in Chinese lung cancer population, which would further our insights for treating ERBB2 TMD mutant patients.
2. Material and methods
2.1. Patient cohort, sample preparation, and next‐generation sequencing
From January 2014 to July 2019, 34 368 unique cases of lung cancer were analyzed using CGP in a Clinical Laboratory Improvement Amendments‐certified, College of American Pathologists accredited laboratory (Nanjing Geneseeq Technology Inc., Nanjing, China), as previously described (Fang et al., 2019; Shu et al., 2017). Written consent was obtained from each patient. The study methodologies conformed to the standards set by the Declaration of Helsinki and were approved by the local ethics committee.
The pathologic diagnosis of each case was confirmed on routine hematoxylin and eosin (H&E) stained slides and all tumor samples forwarded for DNA extraction contained a minimum of 20% tumor nuclear area. DNA from formalin‐fixed paraffin‐embedded or fresh tumor samples, and/or cell‐free DNA (cfDNA) from plasma, cerebrospinal fluid (CSF), or pleural effusion (PE) were extracted as previously described (Jiang et al., 2017; Yang et al., 2018). The extracted DNA samples were assayed by next‐generation sequencing (NGS)‐based CGP through hybrid capture targeting all coding exons of solid tumor‐related genes and introns involved in cancer‐related gene fusions, including all EGFR family members. Captured libraries were sequenced using Illumina HiSeq platform (Illumina, San Diego, CA, USA) to a coverage depth of > 3000×, > 500×, > 100× for ctDNA, tissue and whole blood control samples, respectively. The resultant sequences were analyzed for base substitutions, insertions, deletions, copy number alterations, and gene fusions as previously described (Yang et al., 2018).
2.2. In silico modeling of ERBB2‐ERBB3 TMD dimerization structure
The hetero‐dimerization structural model of ERBB2–Erb‐b2 receptor tyrosine kinase 3 (ERBB3) TMDs was built based on the NMR structure of EGFR‐TMD homo‐dimer (PDB ID: 2M20), which provided the structural basis for EGFR activation (Endres et al., 2013). The NMR structures of ERBB2‐TMD (PDB ID: 2N2A) and ERBB3‐TMD (PDB ID: 2L9U) were used individually in the superimpose. The built ERBB2–ERBB3 TMDs model was further optimized in the structure to eliminate any existing steric clashes with ucsf PLOP6.0 software (Jacobson et al., 2002).
3. Results
3.1. ERBB2 TMD mutations are rare in lung cancer, and majority are lung adenocarcinoma
Among the 34 368 unique clinical lung cancer cases evaluated with CGP in this study, 23 247 were NSCLC (71.3%), including 21 762 cases of adenocarcinoma (ADC, 63.3% of total), 2389 cases of squamous cell carcinoma (SCC, 6.9%), 260 cases of mixed ADC and SCC (mixed ADC & SCC, 0.8%), 97 cases of large cell carcinoma (0.3%), 681 cases of small cell carcinoma (SCLC, 2.0%), and 9179 cases of lung cancer not otherwise specified (Unknown; 26.7%; Fig. 1A).
Fig. 1.
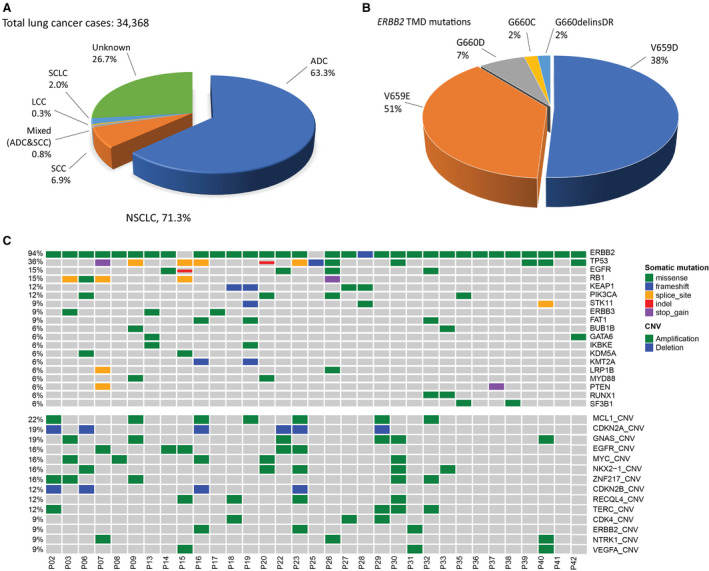
Genomic landscape of lung cancer patients with ERBB2 TMD mutation. (A) Distribution of the histological subtypes of lung cancer cases analyzed. LCC, large‐cell carcinomas. (B) Distribution of ERBB2/HER2 TMD mutations in a cohort of 45 patients with lung ADCs. (C) Mutation profile of lung cancer patients with ERBB2/HER2 TMD mutations in tumor tissue. Each column represents one patient. Total number of mutation and amplification in each patient were shown in the top bar graph. Distributions of individual gene mutations (middle panel) and amplification (bottom panel) detected at least in two patients were shown.
The most common ERBB2 mutations were the KD mutations at 2.3% (802 out of 34 368), followed by extracellular domain mutations (1.0%, 340 out of 34 368), which were identified in multiple subtypes of lung cancer. ERBB2 TMD mutations were extremely rare at 0.13% (45 out of 34 368). Summary of the clinical characteristics of patients with ERBB2 TMD mutation was shown in Table 1. Except six patients with unknown histology and one patient with mixed ADC and SCC, all patients with ERBB2 TMD mutations were ADCs at a frequency of 0.17% (38 out of 21 762), consistent with the previous report in western lung cancer patients (Ou et al., 2017). There was slightly more male (51%) than female (45%) in patients with ERBB2 TMD mutation. The median age of these patients was 59.5 ranging from 39 to 76. Of the thirteen patients with a known smoking history, nine were never‐smokers.
Table 1.
Summary of clinical characteristics of ERBB2/HER2 TMD mutant patients.
| Characteristics | All patients (N = 45) |
|---|---|
| Median age (range) | 59.5 (39–76) |
| Gender—No. (%) | |
| Male | 23 (51) |
| Female | 20 (45) |
| Unknown | 2 (4) |
| Smoking—No. (%) | |
| Smokers | 4 (9) |
| Never smokers | 9 (20) |
| Unknown | 32 (71) |
| Histology—No.(%) | |
| ADC | 38 (85) |
| Mixed ADC and SCC | 1 (2) |
| Unknown | 6 (13) |
| Stage—No. (%) | |
| I | 3 (7) |
| II | 0 (0) |
| III | 4 (9) |
| IV | 16 (35) |
| Unknown | 22 (49) |
| ERBB2 TMD mutation—No. (%) | |
| V659 | 40 (89) |
| G660 | 5 (11) |
A comparison of ERBB2 TMD mutation in a Western cohort and this Asian cohort was shown in Table 2, and distribution of different ERBB2 TMD mutation was shown in Fig. 1B. 40 cases were found to harbor ERBB2 TMD mutations at amino acid residue 659, including 23 cases of V659E and 17 cases of V659D. Only five cases with G660 TMD mutation were found in our cohort, including three cases of G660D mutation, one case of D660C and one case of G660delinsDR. Seven patients with ERBB2 TMD mutation were found to be heterozygous for BIM deletion polymorphism.
Table 2.
Comparison of ERBB2/HER2 TMD mutations between US and Chinese NSCLC patients.
| USA | Chinese | |
|---|---|---|
| Cohort size | 15 | 45 |
| Median age | 54 | 59.5 |
| Female gender | 73.3% | 45% |
| Specific mutation (%) | ||
| V659E | 9 (60.0) | 23 (51.1) |
| V659D | 3 (20.0) | 17 (37.8) |
| G660D | 2 (13.3) | 3 (6.7) |
| G660C | 0 (0.0) | 1 (2.2) |
| G660R | 1 (6.7) | 0 (0.0) |
| G660insDR | 0 (0.0) | 1 (2.2) |
| V659_600VE | 1 (6.7) | 0 (0.0) |
| Concurrent alterations (%) | ||
| ERBB2 amplification | 2 (13.3) | 4 (8.9) |
| EGFR amplification | 5 (11.1) | |
| BIM deletion polymorphism | ||
| Heterozygous | 7 (15.6) | |
3.2. Concomitant EGFR family gene amplifications or activating mutations exist in ERBB2 TMD mutant patients
In this cohort, patients had multiple sample types for NGS analysis including plasma (15), tumor tissue (34), and CSF (four). Two tumor samples were excluded from further analysis because they were sequenced with a different target gene panel (Fig. S1). Mutational landscape of patients with ERBB2 TMD mutation in tumor tissue was shown in Fig. 1C. Identified gene alterations in plasma and CSF were shown in Table S1. Apart from the ERBB2 mutation (94%, 30 out of 32), TP53 alterations showed the highest ratio (38%, 12 out of 32) among 32 cases with ERBB2 TMD mutation (Fig. 1C). In P15 and P25, ERBB2 TMD mutation was identified in CSF and/or plasma but not in tumor tissue.
EGFR mutations were identified and accounted for 20% (nine out of 45) of all cases (Fig. 1C and Table S1). The activating EGFR mutation L858R was identified in tumor tissue of two patients with either ERBB2 G660C (P22) or G660D (P26). In plasma and CSF, EGFR mutation L858R was identified in four patients (two ERBB2 V659D and two ERBB2 G660D) including P26 (Table S1). Patient 14 had two tumor tissue from the right upper and lower lobe of the lung, respectively. One lobe harbored EGFR amplification, while the other lobe was identified with ERBB2 V596D and EGFR sensitizing mutation G719A of which occurs at ~ 2–3% in EGFR‐mutated lung tumors (Mitsudomi and Yatabe, 2010). Patient 15 with ERBB2 TMD V659D detected both in plasma and in CSF had concurrent EGFR amplification and EGFR exon19 deletion. Patient 32 carried V659E TMD mutation and was also detected to carry the extracellular EGFR missense mutation A289V which has been previously reported as an oncogenic mutation in glioblastoma (Dai et al., 2018; Thorne et al., 2017). Patient 42 with ERBB2 V659D had a ERBB2 extracellular domain mutation S310F.
Myeloid leukemia cell differentiation protein 1 (22%) was the most frequent amplification found among ERBB2 TMD carriers. EGFR amplification was identified in five patients with ERBB2 TMD mutations, and ERBB2 amplification was in four patients (Fig. 1C and Table S1). P23 with EGFR and ERBB2 double amplification was also identified with fourteen gene amplifications which may be due to the chromosome instability (Fig. 1C).
3.3. Recurrent ERBB3 TMD mutation in patients with ERBB2 TMD mutation may affect heterodimerization between ERBB2 and ERBB3
Concurrent ERBB3 mutation was also identified among ERBB2 TMD carriers. A novel ERBB3 TMD mutation I649R was found in tumor tissue of three patients with ERBB2 V659D (P03, P13, and P17). P24 with ERBB2 G66D had ERBB3 I649R detected in CSF (Table S1). Meanwhile, no other known driver mutation was detected in the four patients. Especially in P17 and P24, apart from ERBB2 and ERBB3 TMD mutations, no other gene alteration was identified in all the samples.
Activation mechanism underlying the hetero‐dimerization of ERBB2/ERBB3 TMDs was still unknown despite the well‐established hetero‐dimerization activation mechanism of KD. Comparison of in silico model of ERBB2‐ERBB3 TMD hetero‐dimer and experimental structure of EGFR TMD homo‐dimer showed that ERBB3‐Ile649 was at the same location to EGFR‐Val651 in the TMD region (Fig. 2), whose polar mutation was predicted to enhance TMD dimerization and activating the receptor. In addition, I649R in ERBB3 greatly increased the hydrophilic surface of the N‐terminal dimerization motif as well as its polarity, which had the potential to facilitate transition of receptor dimer to an active state independently of ligand binding (Notsuda et al., 2017). It was highly likely that I649 on ERBB3 could enhance TMD heterodimerization and activating ERBB2.
Fig. 2.
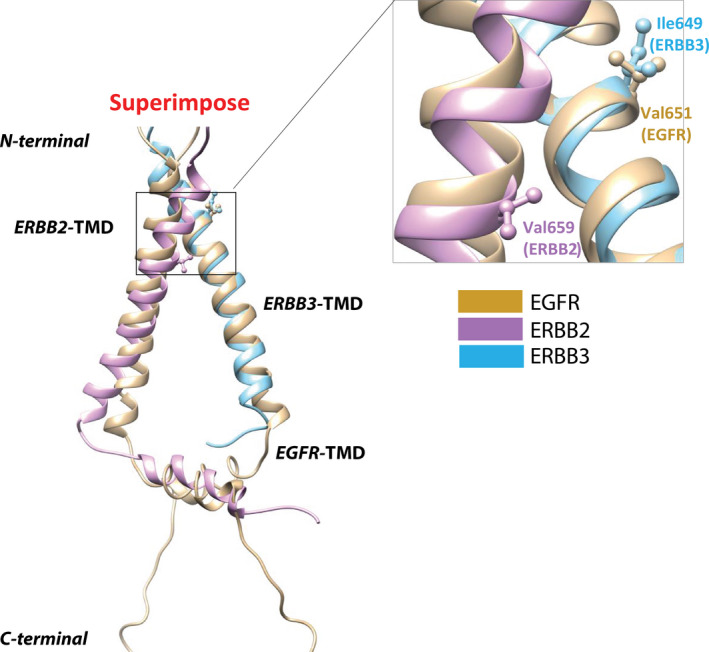
Structural analysis of influence of concurrent ERBB2–ERBB3 TMD mutations on hetero‐dimerization. Superimpose of EGFR‐TMD homo‐dimer structure (PDB ID: 2M20, tan color) and modeled ERBB2–ERBB3 TMD hetero‐dimer structure (HER2‐TMD PDB ID: 2N2A, magenta color; ERBB3‐TMD PDB ID: 2L9U, cyan color). The highlighted residues including Val659 in ERBB2, Ile649 in ERBB3, and Val651 in EGFR are shown in stick‐and‐ball. Left panel: general view of the whole structure of TMD. Right panel: focused view of the interested region bearing somatic mutations.
3.4. Treatment response of patients with ERBB2 TMD mutation
The treatment history and clinical outcomes of target therapy were available for a total of eight patients (Fig. 3). Target therapy with tyrosine kinase inhibitor (TKI) alone or combination therapy was administrated with variable responses. Two patients (P01 and P44) were administrated with icotinib, a first‐generation EGFR TKI. Patient 1 (V659E) reached a PFS of 6 months, while patient 44 (G660D) was still on treatment after 9 months. No EGFR alteration was detected in P01. P44 was identified with EGFR L858R in the plasma.
Fig. 3.
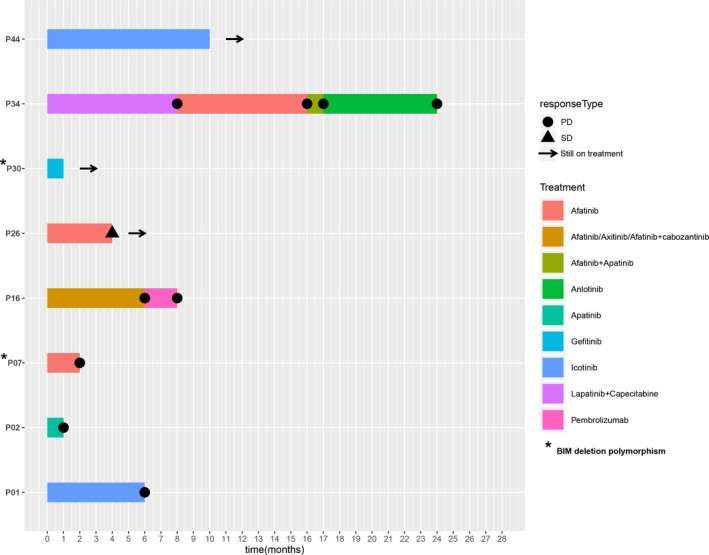
Treatment outcomes of different target therapy in patients with ERBB2 TMD mutations. The horizontal axis shows the duration (months) since initiation of TKI treatment. The drugs used in each treatment were indicated by different colors. /: sequential treatment; +: combined treatment.
Four patients were administrated with afatinib which is TKI target both EGFR and ERBB2. Patient 7 had ERBB2 V659E mutation as well as EGFR amplification. He was treated with afatinib with rapid disease progression within 2 months. The patient in case 16 (V659E) was a 39‐year‐old female diagnosed as lung ADC. She achieved no response to the 6‐month sequential treatment of afatinib, axitinib, and afatinib plus cabozantinib. Then, she underwent immunotherapy with pembrolizumab, a PD‐1 inhibitor, and had progressive disease (PD) after 2 months of treatment. Patient 26 carrying both ERBB2 G660D and EGFR L858R mutation received afatinib treatment and achieved stable disease (SD) for only 4 months, which may have been due to a concurrent PIK3CA E545K mutation that can influence the efficacy of TKI treatment. Indeed, a follow‐up NGS testing at disease progression revealed a dramatic increase in the mutant allele frequencies of all three mutations. Interestingly, patient 34 harboring ERBB2 V659E was given ERBB2 TKI lapatinib plus capecitabine with a progression‐free survival (PFS) of 8 months. He then switched to afatinib treatment and further achieved a PFS of 8 months, although the subsequent afatinib plus apatinib treatment and anlotinib therapy were not effective upon disease progression.
4. Discussion
In this study, we reported on the clinical and molecular characteristics of ERBB2 TMD in Asian lung cancer patients. Our analysis revealed that ERBB2 TMD mutation accounted for 0.13% of all lung cancer cases, which was extremely rare and majority were restricted to lung ADC. This observation also suggested a similar frequency of TMD mutation ratio in ADC (0.17%, 38 out of 21 762). Besides the previously reported V659D/E and G660D mutations, we also observed novel ERBB2 TMD mutation G660C and G660delinsDR in our cohort.
Here, the incidence of ERBB2 TMD mutation was analyzed with different sample type including plasma, tissue, and CSF. Given this was a retrospective analysis of a large Asian NSCLC cohort, the inconsistency in sample type was quite common in this real‐world situation. Tumor tissues could only be obtained when patients underwent surgery. And in some patients, only the non‐invasive liquid biopsies were performed. Generally, positive rate of NGS detection in plasma, tissue, and CSF is 75%, 93%, and 82%, respectively, calculating with all NGS data from 34 368 Chinese lung cancer patients. This difference in sample type and sensitivity of NGS detection might have impact on the interpretation on ERBB2 TMD mutation prevalence and concurrent genomic alterations, which could be considered as a major limitation of this study.
Afatinib has demonstrated response in patients with ERBB2 TMD mutation in other study (Ou et al., 2017). In this cohort, two patients achieved PD and one patient achieved SD on afatinib treatment. Despite coexistence of other driver event which may impact the efficacy of afatinib, another reason for the inferior response to afatinib may be the BIM deletion polymorphism. BIM is a proapoptotic member of the B‐cell CLL/lymphoma 2 (BCL2) family of proteins, and its upregulation is required for TKIs to induce apoptosis in kinase‐driven cancers. It has been reported that EGFR‐mutated NSCLC patients with BIM polymorphism experienced significantly worse responses to TKIs than patients without this polymorphism (Ng et al., 2012). Further investigation needs to be carried out on the association of BIM polymorphism and clinical outcomes of ERBB2‐mutated NSCLC patients under TKI treatment.
Regarding concurrent ERBB3 TMD mutation at I649, previous reports have shown that ERBB3 mutations alone are not sufficient to result in oncogenic transformation (Jaiswal et al., 2013). Notably, recurrent of ERBB3–ERBB2 TMD double mutations in ERBB2 TMD carriers and structure analysis suggested that ERBB3 I649R may be a aid in driving the oncogenicity of ERBB2. Further study is warranted to test this hypothesis. Similar to V659/G660 of ERBB2, V651/G652 of EGFR was predicted to act in the same fashion by enhancing TMD dimerization in certain conformation (Ou et al., 2017). Interestingly, we also identified seven patients with the EGFR TMD mutation (three with EGFR TMD V651M, two with EGFR TMD V651L, one with EGFR TMD G652D, and one with EGFR TMD G652W; Table S2). The four patients with EGFR TMD mutation at V651 carried concurrent EGFR classic oncogenic mutation L858R or exon19 deletion which suggested that these EGFR TMD mutations may not necessarily be the driver mutation in these patients.
5. Conclusion
Our study provided valuable insights into the distribution of ERBB2 TMD mutations by employing the largest Asian lung cancer cohort thus far. Our findings showed that ERBB2 V659/G660 mutations were detected in 0.17% of lung ADC cases and 0.4% (one out of 260) of cases with mixed ADC and SCC. Patients with ERBB2 TMD mutations who received afatinib—a pan‐ERBB inhibitor demonstrated mixed responses, posing the urgent need to develop more effective therapeutic strategy for patients who carry ERBB2 TMD mutations.
6. Implication for practice
ERBB2 TMD alterations are presented in 0.17% of Chinese lung ADC patients.
Eighteen percent of the patients who were identified with ERBB2 TMD mutations had concurrent EGFR mutations or EGFR/ERBB2 amplification.
A novel ERBB3 TMD mutation I646R was found coexisting in four patients with ERBB2 TMD mutation, which might affect its ERBB2‐ERBB3 heterodimerization and activate ERBB2.
Patients with ERBB2 TMD mutations who received afatinib—a pan‐ERBB inhibitor—demonstrated mixed responses. More effective treatment options are in urgent need for patients who carry ERBB2 TMD mutations.
Conflict of interest
RY, XW, RC, XC, and QO are the employees of Geneseeq Technology Inc., Canada. YWS is an employee and shareholder of Nanjing Geneseeq Technology Inc., Nanjing, Jiangsu, China. MN received honorarium from Astra Zeneca and Tempus. SHIO has received speaking/advisory honorarium from Pfizer, Merck, Roche/Genentech, Takea/ARIAD, and AstraZeneca. SHIO is a stock owner and former member of the scientific advisory board of Turning Point Therapeutics, Inc. The remaining authors have no conflict of interest to declare.
Author contributions
YF and JQ involved in conception and design. YF, JQ, and JZ carried out provision of study material or patients. RY, XW, and QO involved in collection and/or assembly of data. RY, XW, QO, YWS, XC, and RC analyzed and interpreted the data. SHIO, JZ, and MN wrote the manuscript. SHIO involved in final approval of manuscript.
[Correction added on 11 July 2020, after first online publication: the author name Xiaoxi Chen(XC) has been included in Conflict of interest and Author contributions sections]
Supporting information
Fig. S1. Consort diagram of sample inclusion criteria.
{kind=link}
Table S1. Gene alterations identified in plasma and CSF of HER2 TMD mutant patients.
Table S2. Clinicopathologic features of EGFR TMD mutations in lung cancer patients.
Acknowledgements
We would like to thank the patients and their families for giving consent for publication. We also thank all research staff and coinvestigators involved in this study.
Yun Fan and Jinrong Qiu contributed equally to the work
Contributor Information
Jiexia Zhang, Email: drzjxcn@126.com, Email: siou@uci.edu.
Sai‐Hong Ignatius Ou, Email: drzjxcn@126.com, Email: siou@uci.edu.
References
- Bargmann CI, Hung MC and Weinberg RA (1986) Multiple independent activations of the neu oncogene by a point mutation altering the transmembrane domain of p185. Cell 45, 649–657. [DOI] [PubMed] [Google Scholar]
- Bocharov EV, Mineev KS, Volynsky PE, Ermolyuk YS, Tkach EN, Sobol AG, Chupin VV, Kirpichnikov MP, Efremov RG and Arseniev AS (2008) Spatial structure of the dimeric transmembrane domain of the growth factor receptor ErbB2 presumably corresponding to the receptor active state. J Biol Chem 283, 6950–6956. [DOI] [PubMed] [Google Scholar]
- Dai L, Su X, Lu L and Lv D (2018) Nonsmall cell lung cancer with rare exon 7 p. A289V mutation in the EGFR gene responds to Icotinib treatment: a case report. Medicine 97, e13809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drilon A, Somwar R, Mangatt BP, Edgren H, Desmeules P, Ruusulehto A, Smith RS, Delasos L, Vojnic M, Plodkowski AJ et al (2018) Response to ERBB3‐directed targeted therapy in NRG1‐rearranged cancers. Cancer Discov 8, 686–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endres NF, Das R, Smith AW, Arkhipov A, Kovacs E, Huang Y, Pelton JG, Shan Y, Shaw DE, Wemmer DE et al (2013) Conformational coupling across the plasma membrane in activation of the EGF receptor. Cell 152, 543–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang W, Ma Y, Yin JC, Hong S, Zhou H, Wang A, Wang F, Bao H, Wu X, Yang Y et al (2019) Comprehensive genomic profiling identifies novel genetic predictors of response to anti‐PD‐(L)1 therapies in non‐small cell lung cancer. Clin Cancer Res 25, 5015–5026. [DOI] [PubMed] [Google Scholar]
- Gatalica Z, Xiu J, Swensen J and Vranic S (2019) Molecular characterization of cancers with NTRK gene fusions. Mod Pathol 32, 147–153. [DOI] [PubMed] [Google Scholar]
- Jacobson MP, Friesner RA, Xiang Z and Honig B (2002) On the role of the crystal environment in determining protein side‐chain conformations. J Mol Biol 320, 597–608. [DOI] [PubMed] [Google Scholar]
- Jaiswal BS, Kljavin NM, Stawiski EW, Chan E, Parikh C, Durinck S, Chaudhuri S, Pujara K, Guillory J, Edgar KA et al (2013) Oncogenic ERBB3 mutations in human cancers. Cancer Cell 23, 603–617. [DOI] [PubMed] [Google Scholar]
- Jiang BY, Li YS, Guo WB, Zhang XC, Chen ZH, Su J, Zhong WZ, Yang XN, Yang JJ, Shao Y et al (2017) Detection of driver and resistance mutations in leptomeningeal metastases of NSCLC by next‐generation sequencing of cerebrospinal fluid circulating tumor cells. Clin Cancer Res 23, 5480–5488. [DOI] [PubMed] [Google Scholar]
- Mineev KS, Bocharov EV, Pustovalova YE, Bocharova OV, Chupin VV and Arseniev AS (2010) Spatial structure of the transmembrane domain heterodimer of ErbB1 and ErbB2 receptor tyrosine kinases. J Mol Biol 400, 231–243. [DOI] [PubMed] [Google Scholar]
- Mitsudomi T and Yatabe Y (2010) Epidermal growth factor receptor in relation to tumor development: EGFR gene and cancer. FEBS J 277, 301–308. [DOI] [PubMed] [Google Scholar]
- Ng KP, Hillmer AM, Chuah CT, Juan WC, Ko TK, Teo AS, Ariyaratne PN, Takahashi N, Sawada K, Fei Y et al (2012) A common BIM deletion polymorphism mediates intrinsic resistance and inferior responses to tyrosine kinase inhibitors in cancer. Nat Med 18, 521–528. [DOI] [PubMed] [Google Scholar]
- Notsuda H, Bradbury PA and Tsao MS (2017) HER2 transmembrane domain mutations: rare new target for non‐small cell lung cancer therapy. J Thorac Oncol 12, 422–424. [DOI] [PubMed] [Google Scholar]
- Ou SI, Schrock AB, Bocharov EV, Klempner SJ, Haddad CK, Steinecker G, Johnson M, Gitlitz BJ, Chung J, Campregher PV et al (2017) HER2 transmembrane domain (TMD) mutations (V659/G660) that stabilize homo‐ and heterodimerization are rare oncogenic drivers in lung adenocarcinoma that respond to afatinib. J Thorac Oncol 12, 446–457. [DOI] [PubMed] [Google Scholar]
- Pahuja KB, Nguyen TT, Jaiswal BS, Prabhash K, Thaker TM, Senger K, Chaudhuri S, Kljavin NM, Antony A, Phalke S et al (2018) Actionable activating oncogenic ERBB2/HER2 transmembrane and juxtamembrane domain mutations. Cancer Cell 34, 792–806.e795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu Y, Wu X, Tong X, Wang X, Chang Z, Mao Y, Chen X, Sun J, Wang Z, Hong Z et al (2017) Circulating tumor DNA mutation profiling by targeted next generation sequencing provides guidance for personalized treatments in multiple cancer types. Sci Rep 7, 583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorne AH, Binder ZA, Bakas S, Wileyto EP, Morrissette JJD, Akbari H, Rathore S, Scott A, Davatzikos C, O'Rourke D et al (2017) EXTH‐56. EGFR extracellular domain point mutant A289V: a therapeutically targetable driver of glioblastoma invasion. Neuro‐oncology 19, vi85. [Google Scholar]
- Yamamoto H, Higasa K, Sakaguchi M, Shien K, Soh J, Ichimura K, Furukawa M, Hashida S, Tsukuda K, Takigawa N et al (2014) Novel germline mutation in the transmembrane domain of HER2 in familial lung adenocarcinomas. J Natl Cancer Inst 106, djt338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto H, Toyooka S, Ninomiya T, Matsumoto S, Kanai M, Tomida S, Kiura K, Muto M, Suzawa K, Desmeules P et al (2018) Therapeutic potential of afatinib for cancers with ERBB2 (HER2) transmembrane domain mutations G660D and V659E. Oncologist 23, 150–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Yang N, Ou Q, Xiang Y, Jiang T, Wu X, Bao H, Tong X, Wang X, Shao YW et al (2018) Investigating novel resistance mechanisms to third‐generation EGFR tyrosine kinase inhibitor osimertinib in non‐small cell lung cancer patients. Clin Cancer Res 24, 3097–3107. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Consort diagram of sample inclusion criteria.
Table S1. Gene alterations identified in plasma and CSF of HER2 TMD mutant patients.
Table S2. Clinicopathologic features of EGFR TMD mutations in lung cancer patients.