

Abstract
The newly identified shieldin complex, composed of SHLD1, SHLD2, SHLD3, and REV7, lies downstream of 53BP1 and acts to inhibit DNA resection and promote NHEJ. Here, we show that Shld2 −/− mice have defective class switch recombination (CSR) and that loss of SHLD2 can suppress the embryonic lethality of a Brca1 Δ11 mutation, highlighting its role as a key effector of 53BP1. Lymphocyte development and RAG1/2‐mediated recombination were unaffected by SHLD2 deficiency. Interestingly, a significant fraction of Shld2 −/− primary B‐cells and 53BP1‐ and shieldin‐deficient CH12F3‐2 B‐cells permanently lose expression of immunoglobulin upon induction of CSR; this population of Ig‐negative cells is also seen in other NHEJ‐deficient cells and to a much lesser extent in WT cells. This loss of Ig is due to recombination coupled with overactive resection and loss of coding exons in the downstream acceptor constant region. Collectively, these data show that SHLD2 is the key effector of 53BP1 and critical for CSR in vivo by suppressing large deletions within the Igh locus.
Keywords: B cell, BRCA1, class switch recombination, shieldin, Shld2−/− mouse
Subject Categories: DNA Replication, Repair & Recombination; Immunology
The recently described Shieldin complex inhibits DNA resection. This study shows that Shieldin deficiency rescues BrcaΔ11 embryonic lethality and impairs class switch recombination in primary B‐cells through loss of immunoglobulin coding sequence as a result of enhanced DNA resection.
Introduction
Across multiple phyla, adaptive immunity requires programmed DNA damage and concomitant repair to generate antigen receptor diversity and fine‐tune the immune response. These phenomena are also particularly well suited for studying the various DNA repair pathways responsible for maintaining genome integrity. In particular, the humoral response mediated by B‐cells requires the non‐homologous end‐joining (NHEJ) repair pathway for V(D)J recombination of the antigen‐binding immunoglobulin (Ig) variable region, as well as class switch recombination (CSR) of the Ig constant regions (Kenter, 2005). Activation‐induced cytidine deaminase (AID) plays a central role in humoral immunity by inducing somatic hypermutation (SHM) and CSR that function to increase the antibody affinity and alter the antibody isotype, respectively (Muramatsu et al, 2000). AID accomplishes these processes by deaminating dC to produce dU within the immunoglobulin DNA encoding the variable region and the non‐coding switch regions (Martin et al, 2002; Petersen‐Mahrt et al, 2002; Bransteitter et al, 2003). At this point, both processes diverge: In the context of SHM, damage produced by AID at the variable region is engaged by base excision repair and mismatch repair pathways leading to small mutations that may potentially increase the affinity of the antibody to antigen (Cascalho et al, 1998; Wiesendanger et al, 2000; Di Noia & Neuberger, 2002; Martin et al, 2003; Rada et al, 2004; Wilson et al, 2005). By contrast in CSR, damage produced by AID at the switch μ (Sμ) and downstream switch regions (Sγ, Sε, and Sα) is engaged by the same DNA repair pathways, with the distinction that these lesions are converted to double‐stranded DNA breaks (DSBs), followed by synapsis and ligation of distant ends through NHEJ or the poorly defined alternative end joining (Pan et al, 2002; Kenter, 2005; Masani et al, 2016). The process of CSR allows for the expression of a novel downstream Ig constant region, with differing effector functions, with the pre‐existing variable region.
The joining reaction during CSR requires the NHEJ pathway. One member, 53BP1, plays a key role in CSR (Manis et al, 2004) by inhibiting DNA end resection (Bothmer et al, 2010; Bunting et al, 2010), thereby shuttling the repair of DSBs toward NHEJ instead of the homologous recombination pathway. Downstream of 53BP1 lies RIF1 that further facilitates DNA end protection and is necessary for CSR (Chapman et al, 2013; Di Virgilio et al, 2013; Escribano‐Diaz et al, 2013; Zimmermann et al, 2013). The recently identified shieldin complex, that lies immediately downstream of the 53BP1‐RIF1 axis, is necessary for CSR, NHEJ, and telomere protection (Dev et al, 2018; Findlay et al, 2018; Ghezraoui et al, 2018; Gupta et al, 2018; Noordermeer et al, 2018; Tomida et al, 2018). Shieldin is composed of REV7, SHLD1, SHLD2, and SHLD3. SHLD2 has three OB‐fold domains that bind to single‐stranded DNA (Noordermeer et al, 2018; Setiaputra & Durocher, 2019), much like RPA1 and POT1 proteins, and appears to be the factor proximally responsible for the end protection role ascribed to the 53BP1 pathway (Dev et al, 2018; Findlay et al, 2018; Gupta et al, 2018; Noordermeer et al, 2018; Tomida et al, 2018). Some shieldin components have been demonstrated to promote CSR in B‐cell lines (Gupta et al, 2018; Noordermeer et al, 2018), and 53BP1/RIF1/REV7‐deficient mice have also been observed to have profoundly impaired CSR (Manis et al, 2004; Chapman et al, 2013; Di Virgilio et al, 2013; Ghezraoui et al, 2018). By contrast, the importance of SHLD1, SHLD2, and SHLD3 on CSR in mice has not been tested. In this report, we show that SHLD2 deficiency impairs CSR in mice but has no impact on early B‐cell development and V(D)J recombination. We further show that Shld2 −/− primary B‐cells, as well as 53BP1‐, shieldin‐, and other NHEJ‐deficient CH12F3‐2 B‐cells (hereafter referred to as CH12 cells), exhibit a significant population with low‐to‐no expression of Ig upon induction of CSR. This effect is permanent as cells do not recover Ig expression. Our analysis shows that a large proportion of these Iglo CH12 cells have undergone CSR to IgA, however with major deletions of the Ighm and Igha loci that lead to loss of Cα constant region exons. These data suggest that 53BP1 or shieldin deficiency does not lead to reduced recombination at the DNA level per se, but rather to a relative increase in non‐productive CSR that inactivates the Igh locus. These results also support a role of the shieldin complex in preventing resection of DNA.
Results
Lymphocyte development and B‐cell populations largely unaffected by Shld2 deficiency
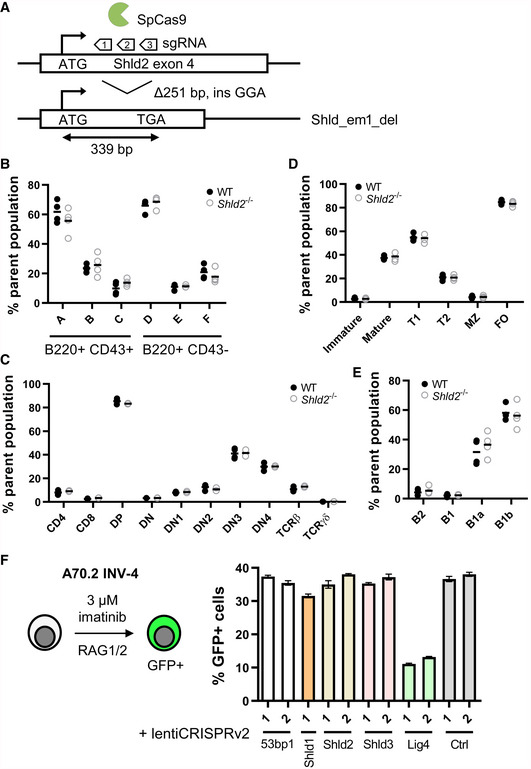
Shld2 knockout mice (Shld2 em_del/em_del, referred hereafter as Shld2 −/−) were generated by microinjection of Cas9 ribonucleoprotein complexes targeting exon 4 in the C57BL/6 background, and a founder mouse with a large out‐of‐frame deletion (248 bp) was generated (Fig 1A). Histopathology examination of various organs and tissues reported no significant pathologies in the Shld2 −/− mice relative to controls (see Materials and Methods).
Figure 1. SHLD2 does not affect lymphocyte development or V(D)J recombination.
-
ASchematic of the Shld2 exon 4 showing the 3 sgRNAs used to produce a 251 base pair deletion in one founder line that leads to a frameshift and usage of a premature stop codon.
- B
-
CCharacterization of the indicated thymic T‐cell populations in the bone marrow of 4 wild‐type and 4 Shld2 −/− littermate controls. Gating strategy for measuring these populations is shown in Fig EV1D.
-
DCharacterization of the indicated immature and mature splenic B‐cell populations in the bone marrow of 4 wild‐type and 4 Shld2 −/− littermate controls. Gating strategy for measuring these populations is shown in Fig EV1B.
-
ECharacterization of the indicated peritoneal B‐cell populations in the bone marrow of 4 wild‐type and 4 Shld2 −/− littermate controls. Gating strategy for measuring these populations is shown in Fig EV1C.
-
FLeft panel: Schematic of the A70.2 INV‐4 cell line strategy to induce RAG1/2‐mediated recombination using imatinib. Right panel: A70.2 INV‐4 cell lines were transduced with lentiviruses encoding the lentiCRISPRv2 expressing sgRNAs against the 53bp1, Shld1, Shld2, Shld3, and Lig4 genes. Guide RNA targeting chicken AID was used as a negative control (Ctrl). Cells were selected with puromycin and treated with 3 μM imatinib for 4 days after which GFP frequency was measured (mean ± SD of 3 biological replicates). The insertion–deletion (indel) penetrance as measured by TIDE analysis (Brinkman et al, 2014) of sequence for each of these sgRNA constructs is shown in Fig EV1E, and the baseline GFP frequency prior to imatinib stimulation is shown in Fig EV1F. sgRNA sequences used are shown in Table EV2.
One of the most remarkable genetic interaction observed among DNA repair‐coding genes is the ability of 53BP1 mutations to suppress the embryonic lethality and cell lethality associated with BRCA1 loss‐of‐function alleles (Cao et al, 2009; Bouwman et al, 2010; Bunting et al, 2010). There is currently some debate as to which effector of 53BP1 is responsible to enforce lethality in BRCA1 mutants since the loss PTIP‐interacting 53BP1‐S25 residue in mice results in viable Brca1 Δ11/Δ11 animals suggesting that PTIP is a critical mediator of the embryonic lethality of BRCA1‐deficient animals. However, Brca1 Δ11/Δ11; 53bp1 S25A/S25A mice display phenotypes such as premature aging, and mouse embryo fibroblasts derived from these mice remain profoundly HR‐deficient due to the presence of the RIF1‐shieldin axis (Callen et al, 2019). To test the impact of shieldin depletion on the viability of the Brca1 Δ11 allele (exon 11 deleted), we combined the Shld2‐null allele with the Brca1 Δ11 mutation by Shld2 −/+; Brca1 Δ11/+ x Shld2 −/+; Brca1 Δ11/+ intercrosses. We monitored the genotype of 96 live births and found that introduction of the Shld2 mutation suppressed the embryonic lethality of the Brca1 Δ11 allele (Xu et al, 2001) (Table 1; χ2 = 33.208, P = 5.649 × 10−5). At the time of submission, all double‐mutant mice were alive and appeared normal, with the oldest mouse being 27.5 weeks old. These results indicate that SHLD2 is a key effector of 53BP1 with respect to the lethality of BRCA1 loss.
Table 1.
Genotypes of the Shld2 +/−; Brca1 Δ11/+ x Shld2 +/−; Brca1 Δ11/+ intercross
| Genotype | Total mice | Observed frequency | Expected frequency |
|---|---|---|---|
| Shld2 +/+; Brca1 +/+ | 8 | 0.0833 | 0.0625 |
| Shld2 +/−; Brca1 +/+ | 6 | 0.0625 | 0.125 |
| Shld2 −/−; Brca1 +/+ | 10 | 0.1042 | 0.0625 |
| Shld2 +/+; Brca1 Δ11/+ | 12 | 0.1250 | 0.125 |
| Shld2 +/−; Brca1 Δ11/+ | 37 | 0.3854 | 0.25 |
| Shld2 −/−; Brca1 Δ11/+ | 14 | 0.1458 | 0.125 |
| Shld2 +/+; Brca1 Δ11/Δ11 | 0 | – | 0.0625 |
| Shld2 +/−; Brca1 Δ11/Δ11 | 0 | – | 0.125 |
| Shld2 −/−; Brca1 Δ11/Δ11 | 9 | 0.0938 | 0.0625 |
| Total | 96 |
χ2 = 33.208, P = 5.649 × 10−5.
In Shld2 −/− mice, there was no apparent block in B‐ and T‐cell development in the bone marrow and thymus, respectively, particularly at the Hardy fraction C or DN3 populations corresponding to the pre‐BCR and pre‐TCR selection stages (Figs 1B and C, and EV1A and D). Moreover, marginal zone and follicular B‐cells in the spleen, as well as B1 cells in the peritoneal cavity, were unaffected by SHLD2 deficiency (Figs 1D and E, and EV1B and C). This apparent lack of a defect in lymphocyte developmental suggests that V(D)J recombination mediated by the RAG1/2 recombinase is unaffected by SHLD2 deficiency. To test this notion, we transduced A70.2 INV‐4 cell line with CRISPR lentivirus targeting the 53bp1, Shld1, Shld2, Shld3, and Lig4 genes. In this cell line, imatinib‐induced RAG‐mediated recombination of a genomically integrated artificial substrate results in GFP expression (Bredemeyer et al, 2006). In the bulk‐edited A70.2 cells, despite similar indel penetrance using the various CRISPR constructs and low baseline frequency of GFP expression (Fig EV1E and F), the only defect in GFP expression was in cells transduced with Lig4‐targeting sgRNA (Fig 1F). Hence, SHLD2 deficiency does not impact V(D)J recombination, consistent with what was observed in the Mb1 cre/+ Rev7 fl/fl mice (Ghezraoui et al, 2018).
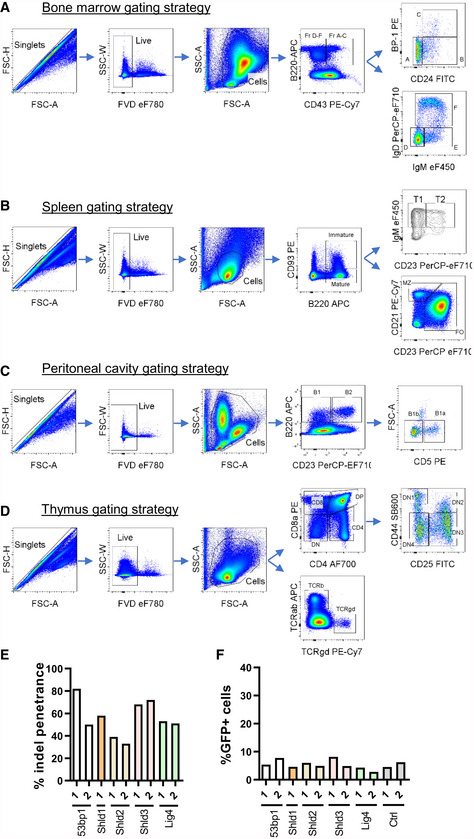
Figure EV1. Gating strategies for the assessment of B‐ and T‐cell populations in WT and Shld2 −/− mice.
-
AGating strategies used to quantitate Hardy fractions A‐F of progenitor B‐cells in bone marrows of WT and Shld2 −/− mice.
-
BGating strategies used to quantitate immature, mature, T1, T2, MZ, and FO B‐cell populations in the spleen of WT and Shld2 −/− mice.
-
CGating strategies used to quantitate B1, B2, B1a, and B1b populations in the peritoneal cavity of WT and Shld2 −/− mice.
-
DGating strategies used to quantitate various thymocyte populations in WT and Shld2 −/− mice.
-
EInsertion–deletion (indel) penetrance was measured by TIDE sequencing for the lentiCRISPRv2 constructs expressing the indicated sgRNAs targeting the 53bp1, Shld1, Shld2, Shld3, and Lig4 genes.
-
FBaseline GFP frequency of bulk gene‐edited A70.2 cells prior to imatinib stimulation (Fig 1F).
SHLD2 is necessary for class switch recombination
To determine whether SHLD2 functions in CSR in vivo, we first examined the steady‐state levels of serum Ig isotypes. We found that the levels of IgM in the serum of unimmunized Shld2 −/− mice were normal, but IgG2b and IgG3 isotypes had reduced concentrations relative to wild type (Fig 2A). Interestingly, IgA levels seemed to be elevated in Shld2 −/− animals, perhaps pointing to a reduced dependence of IgA CSR on NHEJ, as previously reported (Li et al, 2018). To further test the role of SHLD2 in CSR, we purified splenic B‐cells and induced them to switch to various isotypes using different stimulation cocktails. We found that ex vivo CSR to all tested Ig isotypes shows a clear impairment of Shld2 −/− B‐cells relative to WT (Figs 2B and EV2A), and this impairment was not due to a decrease in AID protein or sterile transcript expression (Fig EV2B and C). In addition, using an experimental system in which class switch is mediated by Cas9 (Ling et al, 2018), we found that neither 53BP1 nor SHLD2 deficiency affected this pseudo‐CSR (Fig EV2D). This finding is similar to previous results involving 53BP1 deficiency and I‐SceI‐mediated CSR (Bothmer et al, 2010), pointing to a unique relationship between 53BP1/shieldin function and AID‐mediated DNA damage.
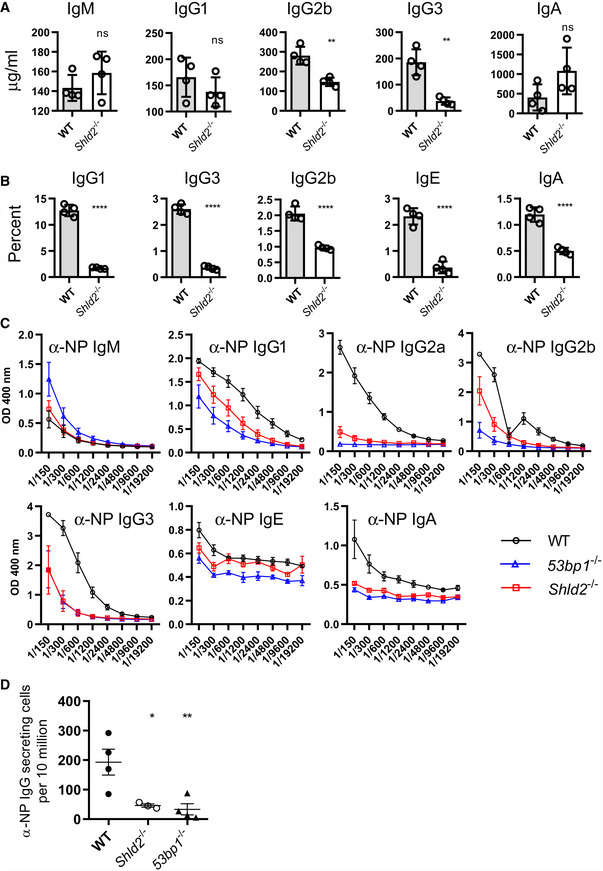
Figure 2. SHLD2‐deficient mice have defects in class switch recombination.
-
AConcentration of the various indicated isotypes in the serum of 6‐ to 8‐week‐old unimmunized WT and Shld2 −/− mice. Values are mean concentration ± SD of 4 biological replicates; **P ≤ 0.01, unpaired two‐tailed t‐test.
-
BB‐cells were purified from spleens from WT and Shld2 −/− mice, and stimulated to undergo CSR to the various indicated isotypes using different stimulation cocktails (Li et al, 2018). Cells were then analyzed by flow cytometry for expression of the various indicated isotypes, and the percent expression of each isotype is reported. Values are mean frequency ± SD of 4 biological replicates; ****P ≤ 0.0001, unpaired two‐tailed t‐test.
-
CWT, 53bp1 −/−, and Shld2 −/− mice were immunized with NP‐CGG, the serum was withdrawn 2 weeks post‐immunization, and serial dilutions were subjected to ELISA analysis for NP‐specific antibodies of the indicated isotypes. Values are mean absorbance ± SD of 4 biological replicates.
-
DWT, 53bp1 −/−, and Shld2 −/− mice were immunized with NP‐CGG, spleens were isolated, and anti‐IgG‐secreting cells were enumerated by the ELISPOT assay. Values are mean frequency ± SD of 4 biological replicates, except for Shld2 –/– (n = 3); *P ≤ 0.05, **P ≤ 0.01, unpaired two‐tailed t‐test.
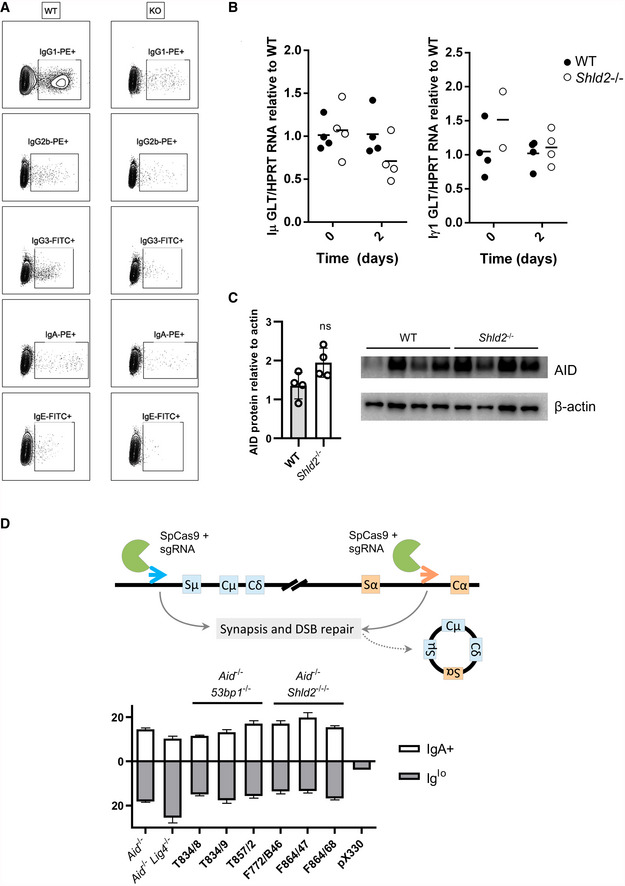
Figure EV2. Sterile transcript or AID protein levels were unaffected in Shld2 −/− B‐cells.
-
ARepresentative flow plots of switched ex vivo B‐cells in Fig 2B.
-
BPurified splenic B‐cells from WT and Shld2 −/− mice were unstimulated or stimulated for 2 days with LPS + IL4, and germline (sterile) transcripts for Iμ (left panel) and Iγ1 (right panel) were quantitated by qPCR and compared to HPRT mRNA levels. Data are shown relative to WT, which is set at 1.
-
CLysates of purified splenic B‐cells from WT and Shld2 −/− mice that were stimulated for 3 days with LPS + IL4 and subjected to Western blot analyses for AID and the internal control β‐actin.
-
DCas9‐induced switching was carried out on Aid −/−, Aid −/− Lig4 −/−, Aid −/− 53bp1 −/−, and Aid −/− Shld2 −/−/− CH12 clones and switching to IgA was measured 3 days post‐transfection. The percent of Iglo cells is also reported. Transfection with the empty vector pX330 served as negative control. Values are mean frequency ± SD of 3 biological replicates; *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001, two‐way ANOVA with post hoc Dunnett's test.
To examine the effect of SHLD2 on antigen‐specific CSR, mice were immunized with 4‐Hydroxy‐3‐nitrophenylacetyl Chicken Gamma Globulin (NP‐CGG). Shld2 −/− mice had reduced NP‐specific serum of the IgG1, IgG2a, IgG2b, and IgG3 classes relative to WT, as well as a trend to increased NP‐specific IgM (Fig 2C). In some cases, the NP‐specific serum Ig defect in Shld2 −/− mice was intermediate between WT and 53bp1 −/− animals. In addition, Shld2 −/− splenic NP‐specific IgG1‐secreting cells were ~7‐fold reduced compared to WT and congruent with 53bp1 −/− results (Fig 2D). Moreover, there was no apparent difference in splenic germinal center B‐cell frequency before or after NP‐CGG immunization, suggesting that the Shld2 −/− CSR defect is on the molecular level of end joining (Fig EV3). All together, these data show a critical function of SHLD2 in CSR in mice, supporting previous findings in the CH12 B‐cell line (Noordermeer et al, 2018).
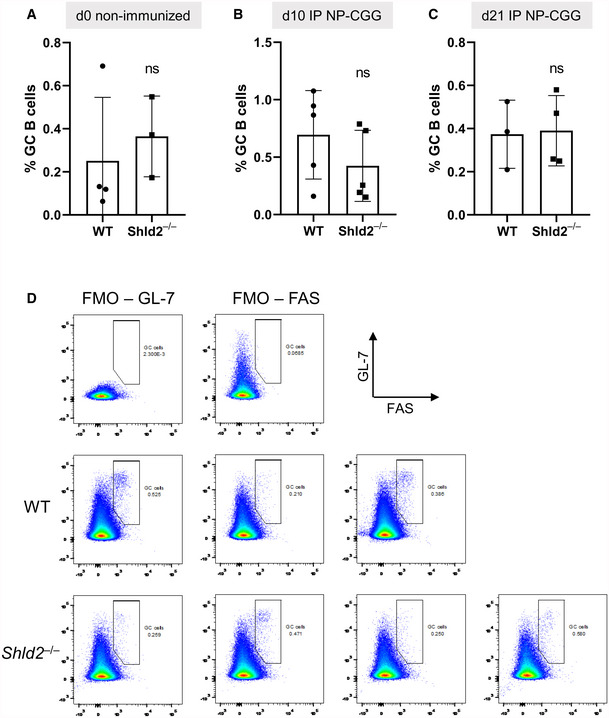
Figure EV3. Germinal center B‐cell frequency is not affected by SHLD2 deficiency.
-
AGerminal B‐cell (GL‐7+ Fas+) frequency relative to all B‐cells (B220+) in the spleen in unimmunized mice; mean ± SD of 3 or 4 biological replicates, ns P ≥ 0.05, unpaired two‐tailed t‐test.
-
BAs in A, but at day 10 post‐NP‐CGG immunization; mean ± SD of 4 or 5 biological replicates, ns P ≥ 0.05, unpaired two‐tailed t‐test.
-
CAs in A, but at day 21 post‐NP‐CGG immunization; mean ± SD of 3 or 4 biological replicates, ns P ≥ 0.05, unpaired two‐tailed t‐test.
-
DRepresentative flow plots of fluorescence minus one (FMO) controls and day 21 data points.
Deficiency in the shieldin complex and other NHEJ factors exhibit a permanent Iglo population upon CSR
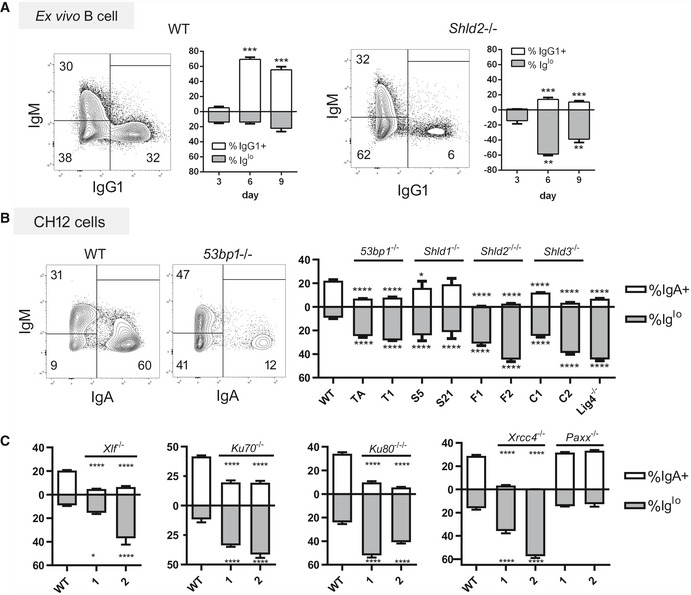
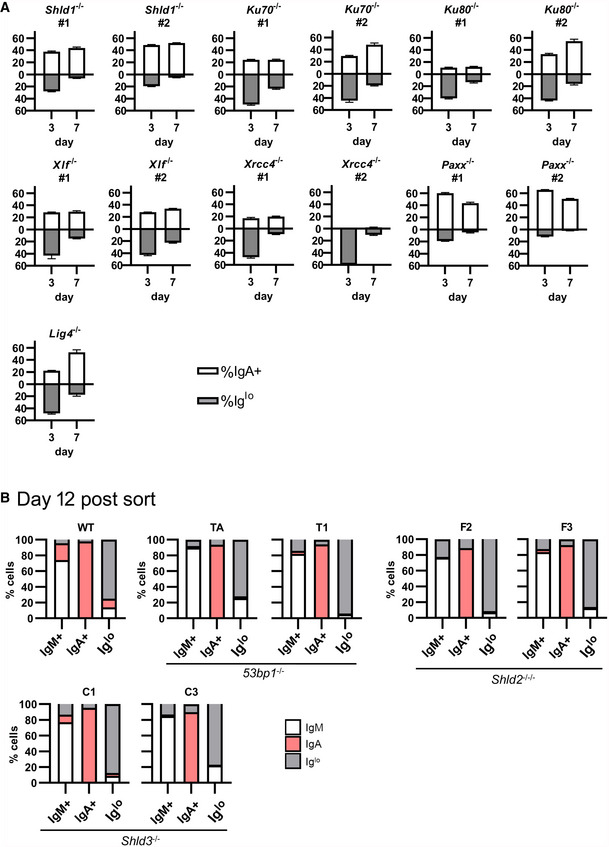
In carrying out the ex vivo CSR of WT and Shld2 −/− B‐cells, we observed that Shld2 −/− B‐cells stimulated with LPS and IL4 showed an increased proportion of IgMlo IgG1lo cells relative to WT cells at day 6 post‐stimulation (Fig 3A). This phenomenon was also recapitulated in 53bp1 −/−, Shld1 −/−, Shld2 −/−/−, and Shld3 −/− CH12 B‐cells at 3 days after stimulating with the CIT cocktail (anti‐CD40, IL4, TGFβ) that induces CSR to IgA (Fig 3B). We also found that CH12 cells deficient in other NHEJ factors such as KU70, KU80, DNA‐PKcs, XLF, XRCC4, and LIG4 (but not PAXX) (Appendix Fig S1) have reduced CSR to IgA and an increased Iglo population (Fig 3B and C). The presence of this Iglo population was temporary and was reduced by 9 days post‐stimulation in ex vivo B‐cells (Fig 3A) and 7 days post‐CIT stimulation in 53bp1 −/−, Shld2 −/−/−, and Shld3 −/− CH12F3‐2 B‐cells (Fig 4A), as well as in CH12 cells deficient in Shld1, KU70, KU80, XLF, XRCC4, and LIG4 (Fig EV4A).
Figure 3. SHLD2‐deficient B‐cells and B‐cells deficient in other NHEJ factors exhibit an Iglo population upon CSR induction.
-
AWT and Shld2 −/− B‐cells were purified from spleens and stimulated with LPS + IL4 and examined for IgM and IgG1 expression 3, 6, and 9 days post‐stimulation by flow cytometry. Representative plots are shown for both WT and Shld2 −/− B‐cells 6 days post‐stimulation. The graph plots show proportion of IgG1+ and Iglo cells, mean ± SD from 6 biological replicates; **P ≤ 0.01, ***P ≤ 0.001, two‐way ANOVA with post hoc Dunnett's test.
-
BWT CH12 cells, as well as two each of 53BP1‐, SHLD1‐, SHLD2‐, and SHLD3‐deficient clones generated previously (Noordermeer et al, 2018), as well as a LIG4‐deficient CH12 clone, were subjected to CSR induction with the CIT cocktail and measured for both IgM and IgA expression by flow cytometry. Representative flow plots for WT and 53bp1 −/− CH12 cells are shown at day 3 post‐CIT stimulation. The graph plots show proportion of IgA+ and Iglo CH12 cells, mean ± SD from 3 biological replicates; *P ≤ 0.05, ****P ≤ 0.0001, two‐way ANOVA with post hoc Dunnett's test. The letters below the x‐axis represent the clone codes for the 53BP1‐, SHLD1‐, SHLD2‐, and SHLD3‐deficient CH12 clones. One LIG4‐deficient CH12 clone was also used.
-
CThe Xlf, Ku70, Ku80, Xrcc4, and Paxx genes were knocked out in CH12 cells by CRISPR, and two independent clones each were analyzed as in Fig 3B with mean ± 3 biological replicates; *P ≤ 0.05, ****P ≤ 0.0001, two‐way ANOVA with post hoc Dunnett's test.
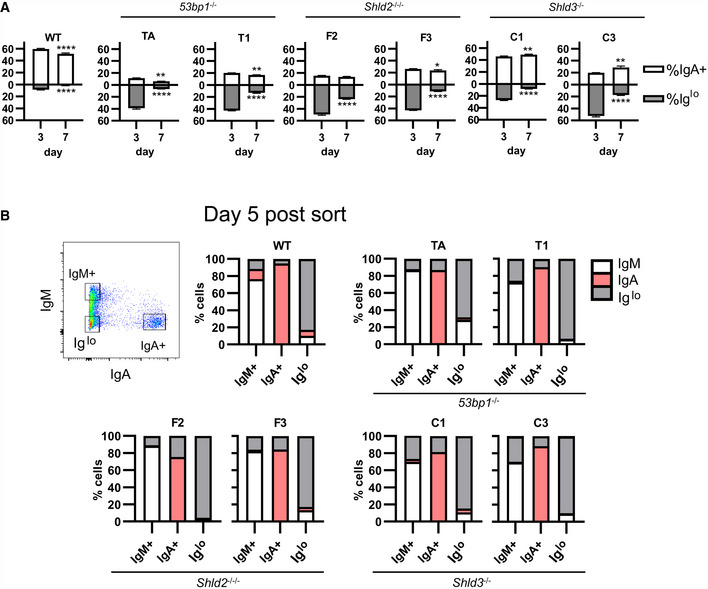
Figure 4. Reduced Ig expression in 53BP1‐, SHLD2‐, and SHLD3‐deficient CH12 cells is permanent and dependent on CSR.
-
AWT and two independent clones each of 53bp1 −/−, Shld2 −/−/−, and Shld3 −/− CH12 cells were stimulated with CIT and analyzed by flow cytometry for IgM and IgA expression. The Iglo population was reduced after 7 days in culture. Values are mean ± SD from 3 biological replicates; *P ≤ 0.05, **P ≤ 0.01, ****P ≤ 0.0001, two‐way ANOVA with post hoc Dunnett's test.
-
BWT and two independent clones each of 53bp1 −/−, Shld2 −/−/−, and Shld3 −/− CH12 clones were stimulated with CIT for 3 days. The IgM+, IgA+, and Iglo populations were sorted and reanalyzed for expression of IgM and IgA 5 days post‐sort as well as 12 days post‐sort (see Fig EV4B). Shown on bar graphs are sorted IgM+, IgA+, and Iglo populations (each column, 1 technical replicate) from WT and mutant CH12 clones, and the percent of cells expressing IgM, IgA, or low for both isotypes (Iglo) after 5 days of culture post‐sort. A representative flow plot of 53bp1 −/− CH12 3 days post‐CIT treatment is shown.
Figure EV4. CSR induces a permanent loss of Ig expression in CH12 cells.
-
AThe indicated NHEJ‐mutant CH12 clones were stimulated with CIT and analyzed by flow cytometry for IgM and IgA expression at days 3 and 7; mean ± SD of 3 biological replicates.
-
BWT, 53bp1 −/−, Shld2 −/−/−, and Shld3 −/− CH12 clones were stimulated with CIT for 3 days. The IgM+, IgA+, and Iglo populations were sorted and reanalyzed for expression of IgM and IgA 12 days post‐sort. Shown on bar graphs are sorted IgM+, IgA+, and Iglo populations (each column, 1 technical replicate) from WT and mutant CH12 clones, and the percent of cells expressing IgM, IgA, or low for both isotypes (Iglo) after 12 days of culture post‐sort.
DSB formation has been shown to transiently reduce expression of a gene, a process mediated by ATM (Harding et al, 2015). Hence, cells deficient in NHEJ factors might have Igh DSBs that persist longer than in WT cells, and thus impose a longer‐lasting decrease in Ig expression. To test whether this Iglo population was exhibiting a transient decrease in Ig expression or was being diluted from the population due to increased death and/or decreased proliferation, WT and mutant CH12 cells were stimulated with CIT for 3 days, and IgM+ IgA−, IgM− IgA+, and IgMlo IgAlo populations from WT, 53bp1 −/−, Shld2 −/−/−, and Shld3 −/− CH12 cells were sorted out and re‐cultured for 5 or 12 days. Notably, all three populations from all CH12 genotypes largely maintained their Ig expression phenotype at the point of sorting (Figs 4B and EV4B). These data suggest that CSR induces a permanent reduced Ig expression in 53bp1 −/− and Shld null CH12 cells.
CSR in 53bp1 −/− and Shld2 −/−/− CH12 cells leads to aberrant recombination involving deletions in the acceptor constant region
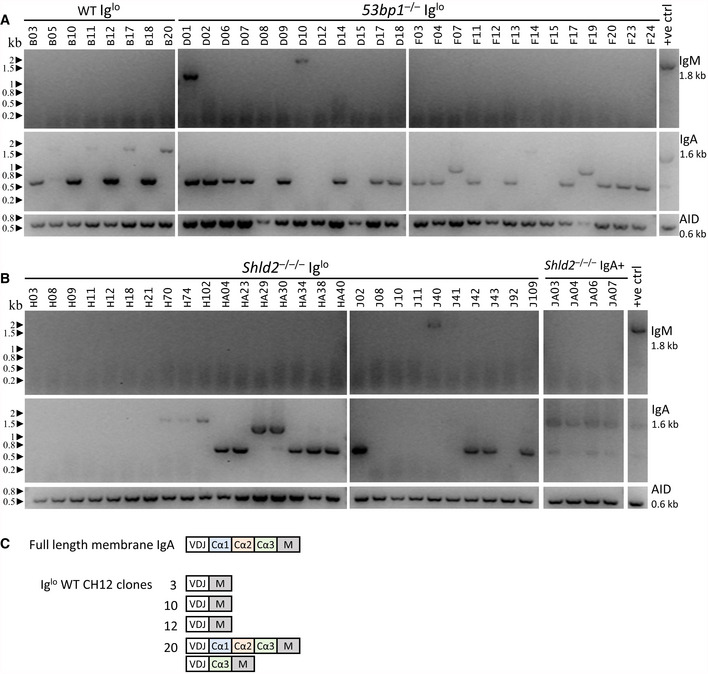
The finding that CSR induction generates a persistent Iglo population in 53bp1 −/− and Shld null CH12 cells suggests that the Igh locus has been inactivated. One possible explanation for this phenomenon is that 53bp1 −/− and other NHEJ‐deficient CH12 cells exhibited increased DNA resection during CSR, leading to loss of sequences that allow for surface expression of IgH. To test this notion, Iglo cells from WT, 53bp1 −/−, and Shld2 −/−/− CH12 cells were sorted, and subcloned, and expression for IgM and IgA was re‐assessed by flow cytometry to confirm that Ig expression remained negative or low (Appendix Fig S2). To test whether expression of IgM or IgA was affected at the mRNA level, we used primers that amplified the entire coding region of the IgM and IgA transcript from the leader sequence to stop codon. For the WT Iglo subclones, we found that most expressed IgA at the mRNA level; however, 50% (4/8) of tested subclones only expressed “truncated” (~0.7 kb) IgA cDNAs relative to the full‐length IgA mRNA (~1.6 kb); the positive control also has a truncated IgA cDNA band likely corresponding to a mis‐spliced product (Fig 5A). For the 53bp1 −/− Iglo subclones, 72% (18/25) of tested subclones only expressed truncated IgA cDNAs (~0.7 or 1 kb); a small fraction of subclones expressed neither IgM or IgA cDNA, and at least one clone (D01) appeared oligoclonal by expressing both IgM and IgA transcripts (Fig 5A). Likewise, Shld2 −/−/− Iglo subclones also expressed truncated or no IgA transcripts, as compared with Shld2 −/−/− IgA+ subclones which expressed the full‐length IgA transcript (Fig 5B).
Figure 5. Loss of Ig cell‐surface expression in CH12 cells is accompanied by aberrant IgA transcripts.
-
AmRNA expression analysis of IgM and IgA was carried out by RT–PCR for Iglo subclones from WT and two independent 53bp1 −/− CH12 (TA and T1) clones. The Iglo subclones derived from the 53bp1 −/− CH12 TA clone are listed as D01‐D18, while the Iglo subclones derived from the 53bp1 −/− CH12 T1 clone are listed as F03‐F24. Gels show the RT–PCR analysis for IgM cDNA (top), IgA cDNA (middle), and AID cDNA (bottom) as control. Appendix Fig S2 shows the expression of IgA and IgM by flow cytometry for each of these subclones.
-
BmRNA expression analysis as in A of Iglo subclones of two independent Shld2 –/–/– (F2 and F3) clones and IgA+ subclones from Shld2 –/–/– clone F3. The Iglo subclones derived from the Shld2 –/–/– CH12 F2 clone are listed as H03‐H40, while the Iglo subclones derived from the Shld2 –/–/–CH12 F3 clone are listed as J02‐J109.
-
CRepresentative IgA cDNA sequence analysis from WT Iglo clones.
To determine the nature of these truncated IgA cDNAs, we sequenced the IgA amplicons for select WT Iglo CH12 subclones. The analysis showed that these subclones expressed IgA mRNAs that were variably missing exons Cα1, Cα2, and Cα3 (Fig 5C). These data suggest that many of the Iglo clones in WT, 53bp1 −/−, and Shld2 −/−/− CH12 cells had developed deletions during CSR that either deleted or affected splicing of IgA constant region Cα exons to the variable region VDJ exon.
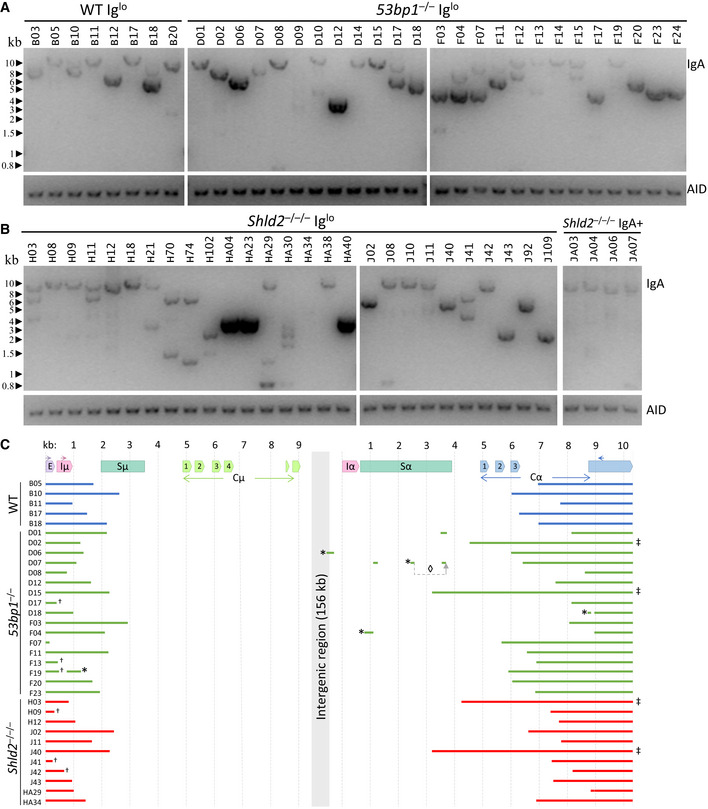
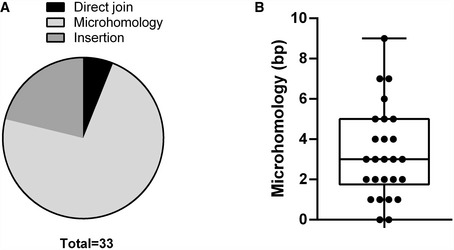
To confirm these deletions at the DNA level, we extracted DNA from these subclones and assayed for Ighm and Igha deletions on the assumption that most of the Iglo subclones had undergone recombination. Using a long‐range PCR assay, we attempted to amplify a region between the Ighm Iμ exon and the Igha M exon downstream of Cα3, with the maximal size of the amplicon expected to be at ~12 kb (Fig 6A). Almost all the subclones, both Iglo and IgA+, yielded long‐range amplicons and confirmed that Iglo cells have undergone recombination (Fig 6B and C). In many cases, there was more than one amplicon from each subclone, which is likely due to recombination of both the productive and non‐productive (without the rearranged VDJ exon) Igh alleles; alternatively but not mutually exclusively, some of the subclones may have been or become oligoclonal at the point of or after subcloning. Moreover, many of the amplicons from 53bp1 −/− and Shld2 –/–/– Iglo subclones were shorter than amplicons from Shld2 –/–/– IgA+ subclones, suggesting that the Iglo amplicons had large deletions within the IgH region (Fig 6B and C). To assess the extent of these deletions, some of these long‐range amplicons were sequenced (Fig 6D). Most of the sequenced amplicons had deletions of Cα exons, although 12% had no deletion of coding sequence, which may be due to the capture of both productive and non‐productive Igh alleles. Interestingly, only 6% of the Ighm‐Igha junctions were direct joins, with the other junctions either incorporating microhomology or sequence insertion (Fig EV5), suggesting that many of these junctions are the product of alternative end joining. All together, these data show that 53BP1 and SHLD2 deficiency leads to aberrant CSR that involves loss of coding sequence in the acceptor constant region. These data thereby provide an explanation for the increased loss of Ig expression in NHEJ‐deficient B‐cells undergoing CSR, which supports the role of the 53BP1‐Rif1‐shieldin axis in inhibiting DNA resection.
Figure 6. Loss of Ig expression in CH12 cells is accompanied by large deletions within the IgA constant region.
-
ALong‐range PCR of Iglo subclones from WT and two independent 53bp1 –/– CH12 (TA and T1) clones. The Iglo subclones derived from the 53bp1 −/− CH12 TA clone are listed as D01‐D18, while the Iglo subclones derived from the 53bp1 −/− CH12 T1 clone are listed as F03‐F24. Gels show long‐range amplicons (top) and AID genomic DNA PCR control (bottom).
-
BLong‐range PCR as in B of Iglo subclones of two independent Shld2 –/–/– (F2 and F3) clones (left 2 panels) and IgA+ subclones from Shld2 –/–/– clone F3.
-
CSequence analysis of a representative subset of long‐range PCR amplicons. Symbol annotations: (*) inverted fragment, (◊) rearranged fragment order, (†) amplified with the EμF primer rather than IμF, and (‡) undamaged Cα coding sequence.
Figure EV5. Repair junctions in Iglo cells have characteristics of alternative end joining.
-
ACategorization of Ighm‐Igha junctions from sequences in Fig 6D, separated into junctions incorporating DNA insertion, microhomology, or neither in the form of direct joins.
-
BMicrohomology distribution of Ighm‐Igha junctions from sequences in Fig 6D, except for those with insertions (26 independent sequences from one experiment). Median and lower/upper quartiles defined by box, and whiskers represent minimum and maximum values.
Discussion
The recently characterized shieldin complex composed of SHLD1/2/3, and REV7 exhibits increased end resection during DSB repair leading to defective NHEJ and CSR. This work has been accomplished in cell lines, with the exception of REV7 (Ghezraoui et al, 2018). In this report, we generated Shld2 −/− mice and report that SHLD2 deficiency largely recapitulated 53BP1 deficiency (Manis et al, 2004) in terms of a defect in CSR and largely normal lymphocyte development and V(D)J recombination. This work establishes the shieldin complex as an essential factor in CSR in vivo. Furthermore, this work further shows that SHLD2 is the main effector mediating the epistatic antagonism of 53BP1 on BRCA1. The observation that Brca1 Δ11/Δ11; Shld2 −/−/− mice are viable with no apparent pathologies at > 27 weeks of age suggests that the loss of shieldin allows for the rescue of HR.
We observed that murine Shld2 −/− B‐cells, in addition to CH12 cells deficient in 53BP1, SHLD2, and other NHEJ repair factors, exhibited an increase in an Iglo population during CSR induction. Two initial possibilities struck us as potential explanatory mechanisms: First, these cells may be temporarily decreasing Ig expression due to unrepaired DNA damage in the switch regions, a process that might be regulated by ATM (Harding et al, 2015). Alternatively, CSR induced an increase in non‐productive recombination events (Dong et al, 2015) in SHLD2‐ and other NHEJ‐deficient B‐cells. Indeed, we observed that most Iglo clones from 53BP1‐deficient CH12 cells were the product of non‐functional CSR into the Igha locus, leading to the production of transcripts that were missing IgA constant region exons. These recombination events also occur in WT CH12 cells, but because the frequency of Iglo cells is reduced in these cells, the frequency of such non‐productive CSR events is minimal in WT B‐cells. Interestingly, the aggregate frequency IgA+ and Iglo populations together in WT, SHLD2‐deficient, and other NHEJ‐deficient B‐cells is strikingly similar (Fig 3). This observation suggests that recombination at the DNA level per se is not necessarily reduced in 53BP1‐ and SHLD2‐deficient cells, but a larger proportion of such recombination events are non‐functional in repair‐deficient cells due to excessive resectioning and loss of coding sequence. Additionally, these non‐functional recombination events may be biased toward alternative end joining as a last‐ditch repair attempt (Fig EV5).
Inversional recombination has previously been observed to be increased in 53bp1 −/− B‐cells during CSR (Dong et al, 2015). Inversional recombination during CSR is predicted to lead to loss of Ig expression as constant region coding sequences downstream of variable region VDJ exon would be inverted and incapable of being translated. Curiously, however, we find that Iglo clones from 53BP1‐deficient CH12 cells are largely due to loss of IgA constant region sequences during CSR and not due to inversional recombination (Fig 5). These data suggest that inversional recombination may occur at frequencies lower than previously reported (Dong et al, 2015). Nevertheless, we conclude that the shieldin complex promotes CSR by inhibiting excessive DNA end resection, and the presence of a significant Iglo population during CSR may be a useful proxy indicator for increased DNA resection in the Igh locus.
Materials and Methods
Mouse generation and husbandry
Shld2 −/− mice were generated by injecting Cas9 ribonucleoprotein complexes and single guide RNA(s) with spacer sequences of GTCCACTAGTCATATCACTC, TCTTTGGAAGTTCCGAACGC, and TCACGATGTCCTGTCGGCTC targeting ENSMUSE00000618046 into C57Bl/6 embryos, resulting in a 251‐bp del Chr14:34268371 to 34268621, as well as an insertion of GGA. These mice were backcrossed for several generations to the C57Bl/6 background. Shld2 +/− mice were bred with Shld2 +/− to generate Shld2 −/− and WT littermates for experimental use (sex‐matched; ~6–8 weeks old). 53bp1 +/− mice were bred with 53bp1 +/− to generate 53bp1 −/− and WT littermates for experimental use (sex‐matched; ~ 8 weeks old). All mice were maintained under pathogen‐free conditions. The experimental procedures were approved by the Animal Care Committee of University of Toronto. Histopathology examination carried out by Dr. S. Camilleri at the Centre for Phenogenomics (Toronto) included the following organs and tissues: calvarium, brain, eyes, ears, tongue, Harderian gland, zymbal gland, salivary glands, nasal sinuses, teeth, trachea, lungs, heart, thymus, parathyroid gland, spleen, liver, gall bladder, exocrine pancreas, endocrine pancreas, esophagus, stomach, large intestine, small intestine, urinary organs and tract, testis, epididymis, vas deference, seminal vesicle, prostate, penis, adrenal gland, lymph nodes, spinal cord, bone marrow, skeletal muscles, brown fat, and skin.
Spleen, bone marrow, thymus, and peritoneal cavity profiling
Single‐cell suspensions of spleen, bone marrow, thymus, and peritoneal cavity were prepared from Shld2 −/− or WT littermate mice (6–8 weeks old). The cells were resuspended in staining buffer (PBS + 2% FBS) and incubated with mouse Fc blocker (2.4G2 mAb). As previously described (Li et al, 2018), splenocytes were stained with rat anti‐CD45R/B220 APC (RA3–6B2; Southern Biotech; 1/150 dilution), rat anti‐IgM eFluor450 (II/41; eBioscience; 1/150 dilution), rat anti‐CD93 PE (AA4.1; eBioscience; 1/50 dilution), rat anti‐CD23 PerCP‐eFluor710 (B3B4; eBioscience; 1/100 dilution), and rat anti‐CD21/CD35 PE‐Cy7 (8D9; eBioscience; 1/100 dilution); germinal center B‐cells in the spleen were stained with anti‐CD45R/B220 eFluor 450 (eBioscience; 1/100 dilution), GL‐7 Alexa Fluor 488 (GL7; eBioscience; 1/100 dilution), and FAS/CD95 APC (15A7; eBioscience; 1/100 dilution); BM cells were stained with anti‐CD45R/B220 APC, anti‐IgM eFluor450, rat anti‐IgD PerCP‐eFluor710 (11‐26c; eBioscience; 1/100 dilution), rat anti‐CD43 PE‐Cy7 (S7; BD Pharmingen; 1/150 dilution), rat anti‐CD24 FITC (30‐F1; eBioscience; 1/100 dilution), and rat ant‐ BP‐1 PE (6C3; BioLegend; 1/33 dilution); peritoneal cells were stained with anti‐CD45R/B220 APC, anti‐CD23 PerCP‐eFluor710, and rat anti‐CD5 PE (53‐7.3; eBioscience; 1/100 dilution); and thymocytes were stained with anti‐CD25 FITC, anti‐CD44 Super Bright 600, anti‐CD4 Alexa Fluor 700, anti‐CD8α PE, anti‐TCRβ APC, and anti‐TCRγδ. Stained cells were acquired on an LSR II (BD Biosciences) and analyzed by FlowJo software.
Ex vivo CSR
Splenic B‐cells were purified from Shld2 −/− mice (6–8 weeks old), and age‐ and sex‐matched littermate controls, using the EasySep Mouse B‐cell Isolation Kit (Stemcell Technologies) and stimulated in vitro as described previously (Li et al, 2018). The stained cells were acquired on a LSR II (BD Biosciences) and analyzed by FlowJo software.
NP‐specific ELISA and ELISPOT assay
Shld2 −/− or WT littermates (6–8 weeks old) and 53bp1 −/− mice (10–12 weeks) were intraperitoneally immunized with 100 μg of 1 mg/ml NP20‐CGG in PBS (Biosearch) precipitated with an equal volume of Imject Alum (Thermo) according to the manufacturer instructions. At day 21 post‐immunization, sera and spleens were harvested and subjected to ELISA and ELISPOT as previously described (Li et al, 2018).
In vitro RAG1/2 and Cas9 induced switching
2 × 105 puromycin‐resistant A70.2 INV‐4 cells transduced with lentiCRISPRv2 (Addgene #52961) were stimulated as previously described for 4 days (Bredemeyer et al, 2006). Aid −/− CH12 cells were electroporated with sgRNA/Cas9 vectors targeting upstream of Sμ and Sα as previously described (Ling et al, 2018).
Cell culture and CRISPR/Cas9 editing
Gene targeting and CH12 cells were performed as described previously (Ramachandran et al, 2016). The CH12 parental clone I (referred to above as WT CH12) was used to delete the following genes using CRISPR: Lig4, Xlf, Ku70, Ku80, XRCC4, and Paxx using sgRNA sequences listed in Table EV2. Verification of gene targeting was accomplished by sequencing. The 53BP1‐, SHLD1‐, SHLD2‐, and SHLD3‐deficient CH12 clones were previously generated (Noordermeer et al, 2018).
PCR and qPCR
RNA was isolated with TRIzol (Thermo Fisher) and cDNA was synthesized with Maxima H Minus reverse transcriptase (Thermo Fisher) according to the manufacturer's instructions. Genomic DNA was isolated by proteinase K digest and phenol–chloroform extraction. Quantitative PCR was performed with qPCRBIO SyGreen Blue Mix (PCR Biosystems) and CFX384 Real‐Time PCR Detection System (Bio‐Rad) according to the manufacturers’ instructions. Reverse‐transcription PCR of IgM and IgA cDNA performed with Ighv_F and either IgM_R or IgA_R. Long‐range PCR of the Ighm‐Igha amplicon was accomplished using Platinum SuperFi II polymerase (Invitrogen) using the manufacturer's instructions and an 8 min polymerase extension step, using either EμF or IμF and CαR primers. The primers used for all reactions are listed in Table EV1.
Statistics
All analyses were performed on GraphPad Prism. For Student's t‐tests, and two‐way analysis of variance (ANOVA), P values of 0.05 or less were considered significant: *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. All error bars represent standard deviations.
Author contributions
AKL, DD, and AM conceived the study. AKL and AM wrote the manuscript. AKL, MM, NC, CL, MB, and BW performed all of the experiments under the supervision of DD and AM.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Review Process File
Acknowledgements
We would like to thank the Martin and Durocher laboratories for helpful discussions, and Marc Shulman for providing feedback on the manuscript. The authors would like to acknowledge Dr. S. Camilleri and the Pathology Core at The Centre for Phenogenomics for their technical services as well as Lauryl Nutter at The Centre for Phenogenomics for generation of the Shld2 −/− mutant mouse. We thank Kefei Yu for AID, LIG4, and KU80‐mutant CH12 cells. This research is supported by grants from the Canadian Institutes of Health Research to D.D. (FDN143343) and to A.M. (Grant PJT‐153307).
EMBO Reports (2020) 21: e49823
References
- Bothmer A, Robbiani DF, Feldhahn N, Gazumyan A, Nussenzweig A, Nussenzweig MC (2010) 53BP1 regulates DNA resection and the choice between classical and alternative end joining during class switch recombination. J Exp Med 207: 855–865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouwman P, Aly A, Escandell JM, Pieterse M, Bartkova J, van der Gulden H, Hiddingh S, Thanasoula M, Kulkarni A, Yang Q et al (2010) 53BP1 loss rescues BRCA1 deficiency and is associated with triple‐negative and BRCA‐mutated breast cancers. Nat Struct Mol Biol 17: 688–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bransteitter R, Pham P, Scharff MD, Goodman MF (2003) Activation‐induced cytidine deaminase deaminates deoxycytidine on single‐stranded DNA but requires the action of RNase. Proc Natl Acad Sci USA 100: 4102–4107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredemeyer AL, Sharma GG, Huang CY, Helmink BA, Walker LM, Khor KC, Nuskey B, Sullivan KE, Pandita TK, Bassing CH et al (2006) ATM stabilizes DNA double‐strand‐break complexes during V(D)J recombination. Nature 442: 466–470 [DOI] [PubMed] [Google Scholar]
- Brinkman EK, Chen T, Amendola M, van Steensel B (2014) Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res 42: e168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunting SF, Callen E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez‐Capetillo O, Cao L et al (2010) 53BP1 inhibits homologous recombination in Brca1‐deficient cells by blocking resection of DNA breaks. Cell 141: 243–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callen E, Zong D, Wu W, Wong N, Stanlie A, Ishikawa M, Pavani R, Dumitrache LC, Byrum AK, Mendez‐Dorantes C et al (2019) 53BP1 enforces distinct pre‐ and post‐resection blocks on homologous recombination. Mol Cell 77: 26–38.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L, Xu X, Bunting SF, Liu J, Wang RH, Cao LL, Wu JJ, Peng TN, Chen J, Nussenzweig A et al (2009) A selective requirement for 53BP1 in the biological response to genomic instability induced by Brca1 deficiency. Mol Cell 35: 534–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascalho M, Wong J, Steinberg C, Wabl M (1998) Mismatch repair co‐opted by hypermutation. Science 279: 1207–1210 [DOI] [PubMed] [Google Scholar]
- Chapman JR, Barral P, Vannier JB, Borel V, Steger M, Tomas‐Loba A, Sartori AA, Adams IR, Batista FD, Boulton SJ (2013) RIF1 is essential for 53BP1‐dependent nonhomologous end joining and suppression of DNA double‐strand break resection. Mol Cell 49: 858–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dev H, Chiang TW, Lescale C, de Krijger I, Martin AG, Pilger D, Coates J, Sczaniecka‐Clift M, Wei W, Ostermaier M et al (2018) Shieldin complex promotes DNA end‐joining and counters homologous recombination in BRCA1‐null cells. Nat Cell Biol 20: 954–965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Noia J, Neuberger MS (2002) Altering the pathway of immunoglobulin hypermutation by inhibiting uracil‐DNA glycosylase. Nature 419: 43–48 [DOI] [PubMed] [Google Scholar]
- Di Virgilio M, Callen E, Yamane A, Zhang W, Jankovic M, Gitlin AD, Feldhahn N, Resch W, Oliveira TY, Chait BT et al (2013) Rif1 prevents resection of DNA breaks and promotes immunoglobulin class switching. Science 339: 711–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J, Panchakshari RA, Zhang T, Zhang Y, Hu J, Volpi SA, Meyers RM, Ho YJ, Du Z, Robbiani DF et al (2015) Orientation‐specific joining of AID‐initiated DNA breaks promotes antibody class switching. Nature 525: 134–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escribano‐Diaz C, Orthwein A, Fradet‐Turcotte A, Xing M, Young JT, Tkac J, Cook MA, Rosebrock AP, Munro M, Canny MD et al (2013) A cell cycle‐dependent regulatory circuit composed of 53BP1‐RIF1 and BRCA1‐CtIP controls DNA repair pathway choice. Mol Cell 49: 872–883 [DOI] [PubMed] [Google Scholar]
- Findlay S, Heath J, Luo VM, Malina A, Morin T, Coulombe Y, Djerir B, Li Z, Samiei A, Simo‐Cheyou E et al (2018) SHLD2/FAM35A co‐operates with REV7 to coordinate DNA double‐strand break repair pathway choice. EMBO J 37:e100158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghezraoui H, Oliveira C, Becker JR, Bilham K, Moralli D, Anzilotti C, Fischer R, Deobagkar‐Lele M, Sanchiz‐Calvo M, Fueyo‐Marcos E et al (2018) 53BP1 cooperation with the REV7‐shieldin complex underpins DNA structure‐specific NHEJ. Nature 560: 122–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta R, Somyajit K, Narita T, Maskey E, Stanlie A, Kremer M, Typas D, Lammers M, Mailand N, Nussenzweig A et al (2018) DNA repair network analysis reveals shieldin as a key regulator of NHEJ and PARP inhibitor sensitivity. Cell 173: 972–988 e923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding SM, Boiarsky JA, Greenberg RA (2015) ATM dependent silencing links nucleolar chromatin reorganization to DNA damage recognition. Cell Rep 13: 251–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy RR, Carmack CE, Shinton SA, Kemp JD, Hayakawa K (1991) Resolution and characterization of pro‐B and pre‐pro‐B cell stages in normal mouse bone marrow. J Exp Med 173: 1213–1225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenter AL (2005) Class switch recombination: an emerging mechanism. Curr Top Microbiol Immunol 290: 171–199 [DOI] [PubMed] [Google Scholar]
- Li C, Irrazabal T, So CC, Berru M, Du L, Lam E, Ling AK, Gommerman JL, Pan‐Hammarstrom Q, Martin A (2018) The H2B deubiquitinase Usp22 promotes antibody class switch recombination by facilitating non‐homologous end joining. Nat Commun 9: 1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling AK, So CC, Le MX, Chen AY, Hung L, Martin A (2018) Double‐stranded DNA break polarity skews repair pathway choice during intrachromosomal and interchromosomal recombination. Proc Natl Acad Sci USA 115: 2800–2805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manis JP, Morales JC, Xia Z, Kutok JL, Alt FW, Carpenter PB (2004) 53BP1 links DNA damage‐response pathways to immunoglobulin heavy chain class‐switch recombination. Nat Immunol 5: 481–487 [DOI] [PubMed] [Google Scholar]
- Martin A, Bardwell PD, Woo CJ, Fan M, Shulman MJ, Scharff MD (2002) Activation‐induced cytidine deaminase turns on somatic hypermutation in hybridomas. Nature 415: 802–806 [DOI] [PubMed] [Google Scholar]
- Martin A, Li Z, Lin DP, Bardwell PD, Iglesias‐Ussel MD, Edelmann W, Scharff MD (2003) Msh2 ATPase activity is essential for somatic hypermutation at a‐T basepairs and for efficient class switch recombination. J Exp Med 198: 1171–1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masani S, Han L, Meek K, Yu K (2016) Redundant function of DNA ligase 1 and 3 in alternative end‐joining during immunoglobulin class switch recombination. Proc Natl Acad Sci USA 113: 1261–1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T (2000) Class switch recombination and hypermutation require activation‐induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell 102: 553–563 [DOI] [PubMed] [Google Scholar]
- Noordermeer SM, Adam S, Setiaputra D, Barazas M, Pettitt SJ, Ling AK, Olivieri M, Alvarez‐Quilon A, Moatti N, Zimmermann M et al (2018) The shieldin complex mediates 53BP1‐dependent DNA repair. Nature 560: 117–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Q, Petit‐Frere C, Lahdesmaki A, Gregorek H, Chrzanowska KH, Hammarstrom L (2002) Alternative end joining during switch recombination in patients with ataxia‐telangiectasia. Eur J Immunol 32: 1300–1308 [DOI] [PubMed] [Google Scholar]
- Petersen‐Mahrt SK, Harris RS, Neuberger MS (2002) AID mutates E. coli suggesting a DNA deamination mechanism for antibody diversification. Nature 418: 99–103 [DOI] [PubMed] [Google Scholar]
- Rada C, Di Noia JM, Neuberger MS (2004) Mismatch recognition and uracil excision provide complementary paths to both Ig switching and the A/T‐focused phase of somatic mutation. Mol Cell 16: 163–171 [DOI] [PubMed] [Google Scholar]
- Ramachandran S, Haddad D, Li C, Le MX, Ling AK, So CC, Nepal RM, Gommerman JL, Yu K, Ketela T et al (2016) The SAGA deubiquitination module promotes DNA repair and class switch recombination through ATM and DNAPK‐Mediated gammaH2AX formation. Cell Rep 15: 1554–1565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setiaputra D, Durocher D (2019) Shieldin ‐ the protector of DNA ends. EMBO Rep 20: e47560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomida J, Takata KI, Bhetawal S, Person MD, Chao HP, Tang DG, Wood RD (2018) FAM35A associates with REV7 and modulates DNA damage responses of normal and BRCA1‐defective cells. EMBO J 37: e99543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesendanger M, Kneitz B, Edelmann W, Scharff MD (2000) Somatic hypermutation in MutS homologue (MSH)3‐, MSH6‐, and MSH3/MSH6‐deficient mice reveals a role for the MSH2‐MSH6 heterodimer in modulating the base substitution pattern. J Exp Med 191: 579–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson TM, Vaisman A, Martomo SA, Sullivan P, Lan L, Hanaoka F, Yasui A, Woodgate R, Gearhart PJ (2005) MSH2‐MSH6 stimulates DNA polymerase eta, suggesting a role for A: T mutations in antibody genes. J Exp Med 201: 637–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Qiao W, Linke SP, Cao L, Li WM, Furth PA, Harris CC, Deng CX (2001) Genetic interactions between tumor suppressors Brca1 and p53 in apoptosis, cell cycle and tumorigenesis. Nat Genet 28: 266–271 [DOI] [PubMed] [Google Scholar]
- Zimmermann M, Lottersberger F, Buonomo SB, Sfeir A, de Lange T (2013) 53BP1 regulates DSB repair using Rif1 to control 5′ end resection. Science 339: 700–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Review Process File