

Abstract
At the neuromuscular junction (NMJ), lipoprotein‐related receptor 4 (LRP4) mediates agrin‐induced MuSK phosphorylation that leads to clustering of acetylcholine receptors (AChRs) in the postsynaptic region of the skeletal muscle. Additionally, the ectodomain of LRP4 is necessary for differentiation of the presynaptic nerve terminal. However, the molecules regulating LRP4 have not been fully elucidated yet. Here, we show that the CT domain of connective tissue growth factor (CTGF/CCN2) directly binds to the third beta‐propeller domain of LRP4. CTGF/CCN2 enhances the binding of LRP4 to MuSK and facilitates the localization of LRP4 on the plasma membrane. CTGF/CCN2 enhances agrin‐induced MuSK phosphorylation and AChR clustering in cultured myotubes. Ctgf‐deficient mouse embryos (Ctgf −/−) have small AChR clusters and abnormal dispersion of synaptic vesicles along the motor axon. Ultrastructurally, the presynaptic nerve terminals have reduced numbers of active zones and mitochondria. Functionally, Ctgf −/− embryos exhibit impaired NMJ signal transmission. These results indicate that CTGF/CCN2 interacts with LRP4 to facilitate clustering of AChRs at the motor endplate and the maturation of the nerve terminal.
Keywords: acetylcholine receptor, cellular communication network factor 2, connective tissue growth factor, low‐density lipoprotein‐related receptor 4, neuromuscular junction
Subject Categories: Development & Differentiation, Neuroscience
The connective tissue growth factor CTGF/CCN2 is required for the formation of neuromuscular junctions (NMJ). CTGF functions together with the lipoprotein‐related receptor 4 (LRP4) in pre‐ and post‐synaptic regions of NMJ.
Introduction
The neuromuscular junction (NMJ) is a synapse between the spinal motor neuron (SMN) and the skeletal muscle. At the NMJ, the skeletal muscle is controlled by the neurotransmitter acetylcholine (ACh) released from the motor nerve terminal (Hall & Sanes, 1993). To achieve efficient neuromuscular signal transmission, densely clustered acetylcholine receptors (AChRs) on the postsynaptic membrane of muscle fiber and appropriate differentiation and juxtaposition of the motor nerve terminals are critical (Wu et al, 2010). A combination of molecules secreted by these cells act in a concerted manner to regulate pre‐ and postsynaptic differentiation of the NMJ and AChR clustering (Fischbach & Rosen, 1997; Wu et al, 2010).
Low‐density lipoprotein receptor‐related protein 4 (LRP4), firstly identified as an inhibitory receptor for Wnt/β‐catenin signaling, plays a central role in pre‐ and postsynaptic differentiation of the NMJ. AChR clustering is primarily mediated by SMN‐derived agrin (Burden, 2002, 2011). At the postsynaptic region of the NMJ, an LRP4 dimer binds to a dimer of muscle‐specific receptor tyrosine kinase (MuSK), forming heterotetrameric receptor for agrin (Zong et al, 2012). Agrin binds to LRP4 and phosphorylates MuSK (Kim et al, 2008; Zhang et al, 2008), which leads to rapid tyrosine phosphorylation on the AChR β subunit (Borges et al, 2008) and facilitates AChR clustering on subsynaptic scaffolding proteins, including rapsyn (Gautam et al, 1995). At the presynaptic nerve terminal of SMN, a retrograde signal from postsynaptic LRP4 induces the clustering of synaptic vesicles and active zones (Wu et al, 2012; Yumoto et al, 2012). Although LRP4 plays a central role in inducing the pre‐ and postsynaptic structure of the NMJ, the molecule(s) regulating LRP4 functions at the NMJ are not fully elucidated.
Connective tissue growth factor/cellular communication network factor 2 (CTGF/CCN2), encoded by the Ccn2 gene, is an extracellular matrix‐associated protein and a member of the CCN family of proteins (Yeger & Perbal, 2016; Takigawa, 2017), which include Cyr61 (Ccn1), Nov (Ccn3), WISP1 (Ccn4), WISP2 (Ccn5), and WISP3 (Ccn6). CCN family proteins function in many biological processes, including cell adhesion, migration, proliferation, angiogenesis, skeletal development, neuronal development, and tissue wound repair, and are critically involved in various diseases such as fibrosis, inflammation, muscular dystrophies, neurodegenerative diseases, diabetes mellitus, retinopathy, and several types of malignancies (Jun & Lau, 2011; Krupska et al, 2015; Takigawa, 2017; Gonzalez & Brandan, 2019). Members of the CCN family of proteins, including CTGF/CCN2, have four conserved cysteine‐rich domains: insulin‐like growth factor‐binding protein (IGFBPs), von Willebrand factor type C (VWC), thrombospondin type‐1 (TSP1), and C‐terminal cystine knot‐like (CT) (Takigawa, 2017). CCN family proteins function by binding to various extracellular proteins including integrin receptors, heparin sulfate proteoglycans (HSPGs), TrkA, members of the TGF‐β superfamily, VEGF, perlecan, slit, and LRPs (Holbourn et al, 2008). Indeed, the secreted CTGF protein binds to LRP6, a protein in the LRP family, to inhibit Wnt signaling (Mercurio et al, 2004). Ctgf deficiency in mice results in lethality immediately after birth due to respiratory failure caused by thoracic skeletal defects (Ivkovic et al, 2003). CTGF is also important in diseases of neuronal tissues and skeletal muscles (Mercurio et al, 2004; Sun et al, 2008). CTGF is one of the extracellular matrix (ECM) proteins concentrated at the adult NMJ (Sun et al, 2008; Ohno et al, 2012); however, its physiological role in synaptogenesis remains unknown.
Here, we characterized the functions of CTGF/CCN2 in vitro, in cellulo, and in mouse embryonic NMJ. We show that CTGF physically interacts with LRP4 and enhances binding between LRP4 and MuSK. CTGF stabilizes LRP4 on the sarcoplasmic membrane and enhances agrin‐mediated MuSK phosphorylation in myotubes. Finally, Ctgf −/− mouse embryos exhibit reduced AChR clusters at the postsynaptic motor endplate, abnormal axonal dispersion of synaptic vesicles, fewer active zones in the presynaptic motor terminal, and impaired NMJ signal transmission. We therefore conclude that CTGF/CCN2 plays a critical role in the formation and function of the embryonic NMJ, in particular presynaptic differentiation.
Results
Interaction between CTGF and LRP4 enhances the binding of LRP4 to MuSK
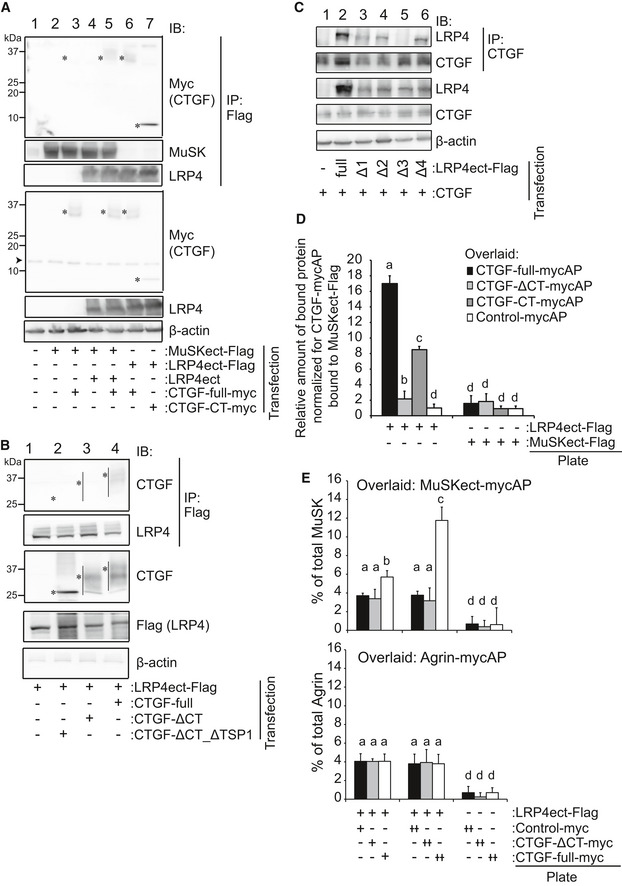
In order to determine the associations between CTGF and the agrin‐LRP4‐MuSK complex, we first examined the binding of CTGF to LRP4 and MuSK. An immunoprecipitation assay revealed that CTGF bound to the Flag‐tagged ectodomain of LRP4 (LRP4ect‐Flag) but not to that of MuSK (MuSKect‐Flag) (Fig 1A, lanes 3 and 6). We observed that the CT domain of CTGF was sufficient for binding to LRP4 (Fig 1A, lane 7) and that the deletion of the CT domain abrogated binding to LRP4 (Fig 1B). Conversely, deletion of each β‐propeller domain of LRP4 (LRP4‐Δ1, LRP4‐Δ2, LRP4‐Δ3, and LRP4‐Δ4) showed that the 3rd β‐propeller domain of LRP4 was required for binding to CTGF (Fig 1C). We also confirmed direct binding of CTGF and LRP4 by a cell‐free plate‐binding assay. Purified full‐length CTGF and the CT domain of CTGF that were tagged with myc and alkaline phosphatase (CTGF‐mycAP and CTGF‐CT‐mycAP, respectively) indeed bound to the plate‐coated ectodomain of LRP4 (LRP4ect‐Flag), but not to that of MuSK (MuSKect‐Flag) (Fig 1D). In contrast, purified CTGF lacking the CT domain (CTGF‐ΔCT‐mycAP) had weak binding compared to full‐length CTGF.
Figure 1. Interaction between the CT domain CTGF and the third β‐propeller domain of LRP4 enhances the binding of LRP4 to MuSK .
-
A–CRepresentative immunoprecipitation (IP) of HEK293 cells transfected with indicated cDNAs. (A) CTGF interacts with LRP4 but not with MuSK. Full‐length CTGF (CTGF‐full‐myc) was immunoprecipitated with the ectodomain of LRP4 (LRP4ect‐Flag, lane 6) but not with the ectodomain of MuSK (MuSKect‐Flag, lane 3). However, when CTGF‐full‐myc, LRP4ect, and MuSKect‐Flag were expressed together, CTGF‐full‐myc was immunoprecipitated with MuSKect‐Flag (lane 5). The CT domain of CTGF (CTGF‐CT‐myc) was sufficient to immunoprecipitate LRP4ect‐Flag (lane 7). (B) Deletion of the CT domain of human CTGF (CTGF‐ΔCT_ΔTSP1 and CTGF‐ΔCT) impairs the interaction between CTGF and LRP4ect‐Flag. (A, B) Predicted positions of full‐length and shortened CTGF proteins are indicated by bars and/or asterisks. (C) Anti‐CTGF antibody immunoprecipitated human LRP4 deleted with the 1st, 2nd, and 4th β‐propeller domains (Δ1, Δ2, and Δ4) but not with the 3rd β‐propeller domain (Δ3).
-
D, ECell‐free plate‐binding assays. A purified tagged protein was attached to a plastic plate, and a purified recombinant protein fused to alkaline phosphatase (AP) was overlaid. Bound protein was quantified by measuring AP activity. (D) LRP4ect‐Flag, but not MuSKect‐Flag, bound to overlaid CTGF‐full‐mycAP and CTGF‐CT‐mycAP. In contrast, LRP4ect‐Flag did not bind to CTGF‐ΔCT‐mycAP. (E, upper panel) LRP4ect‐Flag bound to overlaid MuSKect‐mycAP in a dose‐dependent manner of CTGF‐full‐myc, but not of CTGF‐ΔCT‐myc. (E, lower panel) LRP4ect‐Flag bound to overlaid agrin‐mycAP independent of CTGF‐full‐myc. (D, E) Rspo2‐mycAP and Rspo2‐myc were used as Control‐mycAP and control‐myc, respectively. Mean and SD (n = 9 wells; 3 wells in 3 independent experiments) are indicated. P‐value < 0.05 by two‐way repeated measures ANOVA for the three panels in (D) and (E). P < 0.05 by post hoc Tukey test is indicated by a single letter representing each group.
Source data are available online for this figure.
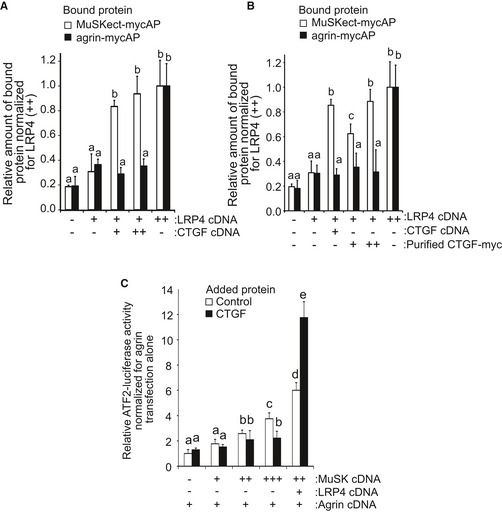
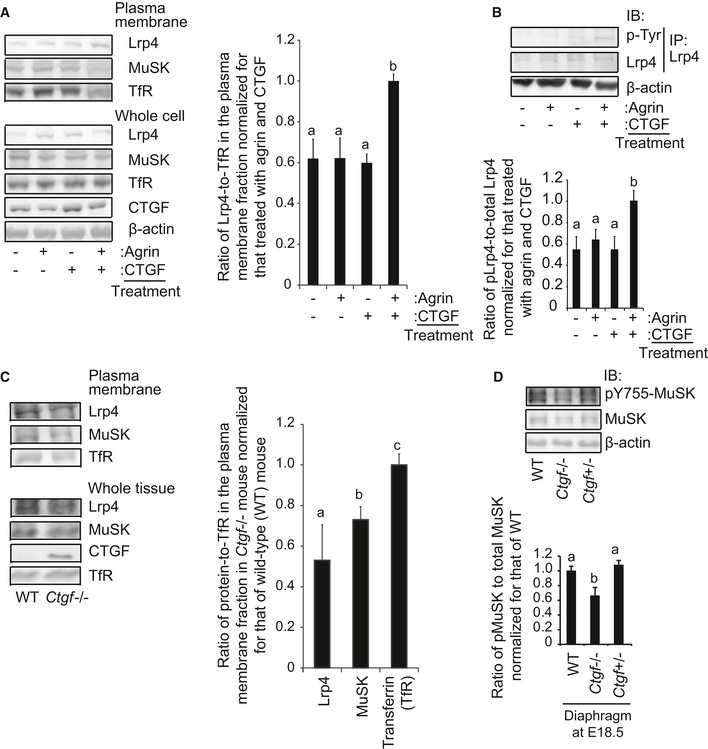
Cell‐free plate‐binding assay also revealed that purified full‐length CTGF‐myc on a plate had no binding to MuSK‐mycAP or partial agrin‐mycAP (Fig 1E). However, full‐length CTGF, but not CTGF‐ΔCT, enhanced the binding of LRP4 and MuSKect‐mycAP on the plate, while CTGF had no effect on the binding of LRP4 and agrin‐mycAP (Fig 1E). To examine whether CTGF enhances the binding of LRP4 and MuSK on the cell membrane, we performed a cell surface‐binding assay. Expression of CTGF (Fig EV1A), as well as addition of CTGF in the medium (Fig EV1B), enhanced the binding of MuSKect‐mycAP, but not of agrin‐mycAP, to LRP4 on the surface of HEK293 cells. We also observed in agrin‐treated C2C12 myotubes that CTGF increased the amount of Lrp4 on the plasma membrane, but not that in whole cells (Fig 2A). In addition, CTGF increased phosphorylation of Lrp4 in agrin‐treated C2C12 myotubes (Fig 2B). Similarly, the absence of Ctgf reduced the amount of Lrp4 on the plasma membrane in mouse embryonic muscles (Fig 2C). However, transgenic (lanes 2–4 in Fig EV1A) or exogenous (lanes 3–5 in Fig EV1B) CTGF failed to increase the binding of agrin to LRP4 on the surface of HEK293 cells. CTGF, thus, likely stabilizes LRP4 on the plasma membrane in the presence of NMJ‐specific proteins like agrin and MuSK.
Figure EV1. CTGF functions with LRP4.
-
A, BCell surface‐binding assays. Variable concentrations of LRP4 cDNA and CTGF cDNA were introduced to HEK293 cells. Purified MuSKect‐mycAP (30 nM) or purified agrin‐mycAP (30 nM), both of which carried alkaline phosphatase in fusion, was added to the cultured medium. In (B), 30 (+) or 60 (++) nM CTGF was also added as indicated. Bound protein was quantified by measuring alkaline phosphatase activity. CTGF enhanced binding of LRP4 to MuSKect‐mycAP but not to Agrn‐mycAP. Mean and SD (n = 9 wells; 3 wells in 3 independent experiments) are indicated. P‐value < 0.05 by two‐way repeated measures ANOVA for both (A and B). P < 0.05 by post hoc Tukey test is indicated by a single letter representing each group.
-
CATF‐2 luciferase assay. HEK293 cells were transfected with 1 (+), 2 (++), or 5 (+++) ng pExpress‐1‐Musk encoding full‐length MuSK, 5 ng phLRP4 encoding full‐length LRP4, 5 ng Agrin/pAPtag‐5 encoding partial agrin retaining AChR clustering activity and 5 ng ATF‐2 luciferase reporter to monitor MuSK phosphorylation. The cells were then treated with 250 pM BSA (Control) or CTGF for 24 h. Purified recombinant CTGF enhanced MuSK activation in the presence of LRP4. Mean and SD (n = 9 wells; 3 wells in 3 independent experiments) are indicated. P‐value < 0.05 by two‐way repeated measures ANOVA. P < 0.05 by post hoc Tukey test is indicated by a single letter representing each group.
Source data are available online for this figure.
Figure 2. CTGF stabilizes Lrp4 on the plasma membrane of C2C12 myotube and the embryonic muscle.
-
ARepresentative immunoblotting of Lrp4, MuSK, transferrin receptor (TfR), CTGF, and β‐actin in whole‐cell lysates and the plasma membrane fraction of C2C12 myotubes treated with 1 pM (1 ng/ml) agrin and/or 250 pM (100 ng/ml) CTGF for 24 h. CTGF increases Lrp4 on the plasma membrane in the presence of agrin.
-
BC2C12 myotubes were treated with 1 pM agrin and/or 250 pM CTGF for 6 h. Lrp4 was immunoprecipitated (IP) with an anti‐Lrp4 antibody, and phosphorylated Lrp4 was immunoblotted with an anti‐phosphotyrosine (p‐Tyr) antibody.
-
CRepresentative immunoblotting of Lrp4, MuSK, CTGF, and TfR in whole tissue lysates and the plasma membrane fraction of lower limb muscles at E18.5 of wild‐type (WT) and Ctgf −/− mice.
-
DRepresentative immunoblotting of Y755‐phosphorylated MuSK, total MuSK, and β‐actin in the diaphragm at E18.5 of wild‐type (WT), Ctgf −/−, and Ctgf +/− mice.
CTGF promotes MuSK phosphorylation and AChR clustering in C2C12 myotubes
We then analyzed the effects of CTGF on the agrin‐LRP4‐MuSK complex. We first analyzed the effect of CTGF on MuSK activation in vivo. Tyrosine (Y) 755 is located in an activation loop of the MuSK kinase domain and is self‐phosphorylated by MuSK to induce AChR clustering. In the mouse gastrocnemius, Y755‐phosphorylated MuSK is localized at the NMJ (Camurdanoglu et al, 2016). The absence of Ctgf reduced phosphorylation of MuSK Y755 in the mouse diaphragm at E18.5 (Fig 2D). We then examined the effects of CTGF on activation of the MuSK signaling pathway by expressing LRP4, MuSK, and a JNK‐responsive ATF‐2 luciferase reporter (ATF2‐luc) (van der Sanden et al, 2004; Ohkawara et al, 2014). In the presence of agrin, increased expressions of MuSK alone potentiated the ATF2‐luc activity, and addition of LRP4 further enhanced the ATF2‐luc activity (Fig EV1C). CTGF had no effect on the ATF2‐luc activity in the absence of LRP4, but increased the ATF2‐luc activity 2‐fold in the presence of LRP4. These results suggest that CTGF upregulate MuSK signaling in an LRP4‐dependent manner.
In C2C12 myotubes, addition of full‐length CTGF, but not CTGF‐ΔCT, to the culture medium enhanced agrin‐induced MuSK phosphorylation (Fig 3A), and its downstream AChR clustering (Fig 3B). We next knocked down endogenous Ctgf expression in C2C12 myotubes using shRNA‐expressing lentivirus (Figs 3D and EV2B). Knockdown of Ctgf reduced agrin‐induced MuSK phosphorylation (Fig 3E) and AChR clustering (Fig 3F) in C2C12 myotubes. In addition, the ectodomain of LRP4 restored shCTGF‐induced reduction of MuSK phosphorylation (Fig EV2C) and AChR clustering (Fig EV2D) in C2C12 myotubes. These results suggest that CTGF enhances MuSK phosphorylation and promotes AChR clustering via LRP4 in C2C12 myotubes.
Figure 3. CTGF enhances agrin‐mediated AChR clustering in C2C12 myotubes.
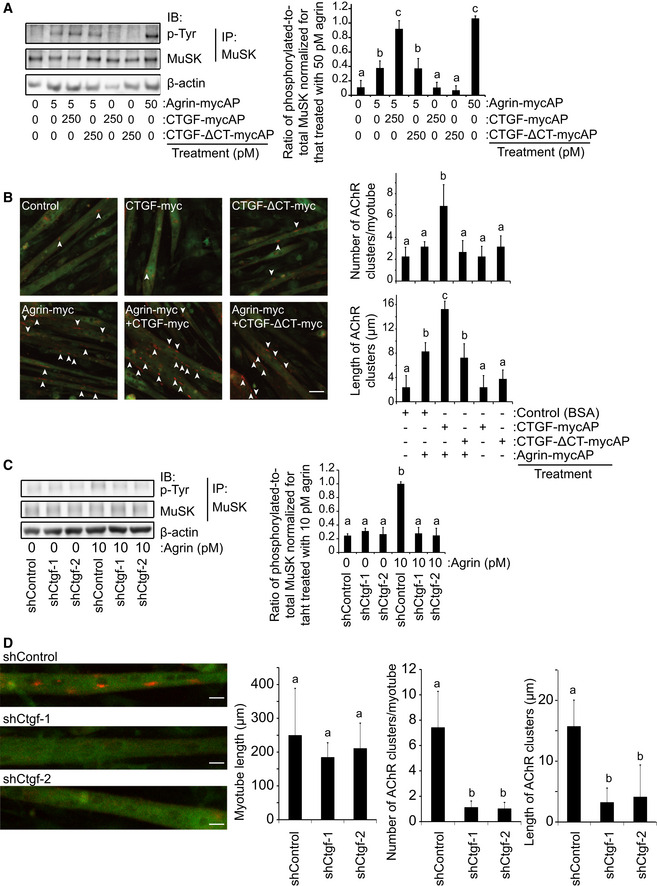
-
AC2C12 myotubes were treated with indicated concentrations of agrin‐mycAP, CTGF‐mycAP, and/or CTGF‐ΔCT‐mycAP for 3 h. Total MuSK was immunoprecipitated (IP) with an anti‐MuSK antibody, and phosphorylated MuSK was immunoblotted with an anti‐phosphotyrosine (p‐Tyr) antibody. CTGF‐mycAP, but not CTGF‐ΔCT‐mycAP, enhanced agrin‐mediated MuSK phosphorylation.
-
BC2C12 myotubes were treated with 5 pM agrin‐mycAP, 250 pM CTGF‐mycAP, and/or 250 pM CTGF‐ΔCT‐mycAP for 18 h. BSA was added to the control medium. AChRs and the plasma membrane were visualized with Alexa 594‐conjugated α‐bungarotoxin (red) and CellMask (green), respectively. Scale bar = 20 μm. Arrowheads point to the AChR clusters with an axis length of 4 μm or more. CTGF enhances agrin‐mediated AChR clustering.
-
CC2C12 myotubes were infected with lentivirus expressing shControl, shCtgf‐1, or shCtgf‐2. Doxycycline was added for 2 days to induce shRNA expression. BSA or agrin was added to the medium at 10 pM. Total MuSK was immunoprecipitated (IP) by an anti‐MuSK antibody, and phosphorylated MuSK was immunoblotted with an anti‐phosphotyrosine (p‐Tyr) antibody. Ctgf knockdown suppressed MuSK phosphorylation.
-
DTen pM agrin was added to the medium. AChRs were visualized with Alexa 594‐conjugated α‐bungarotoxin (red signals) in the infected myotube (green GFP signals). Ctgf knockdown decreased the number and length of AChR clusters. Scale bar = 10 μm.
Figure EV2. CTGF enhances agrin‐ and LRP4‐mediated AChR clustering in C2C12 myotubes.
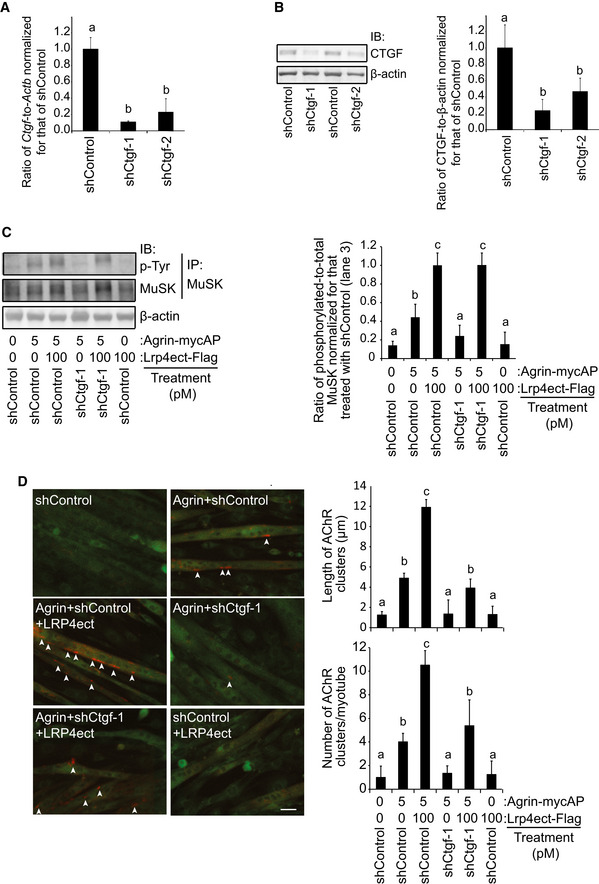
-
A, BC2C12 myotubes were infected with lentivirus expressing shControl, shCtgf‐1, or shCtgf‐2. Doxycycline was added for 2 days to induce shRNA expression. (A) Quantitative RT–PCR. (B) Immunoblotting. Mean and SD (n = 3 independent experiments) are indicated. P‐value < 0.05 by two‐way repeated measures ANOVA for both (A and B). P < 0.05 by post hoc Tukey test is indicated by a single letter representing each group.
-
C, DC2C12 myotubes were infected with GFP‐expressing lentivirus carrying shControl or shCtgf‐1. Doxycycline was added for 2 days to induce shRNA expression. C2C12 myotubes were treated with indicated concentrations of purified agrin‐mycAP and/or purified LRP4ect‐Flag for 3 h. (C) Total MuSK was immunoprecipitated (IP) with anti‐MuSK antibody, and phosphorylated MuSK was immunoblotted with anti‐phosphotyrosine (p‐Tyr) antibody. Quantification of immunoblots is shown in the right panel. Ctgf knockdown decreased MuSK phosphorylation, which was rescued by adding LRP4ect‐Flag. (D) AChRs were visualized with Alexa 594‐conjugated α‐bungarotoxin (red signals) in the infected myotube (green GFP signals). AChR cluster with an axis length of 4 μm or more (arrowheads) was recognized and measured by the MetaMorph software. Analysis was performed in a blinded manner. Ctgf knockdown decreased the number and the length of AChR clusters, which was partially rescued by adding LRP4ect‐Flag. Scale bar = 20 μm. Mean and SD are indicated in the right panels (n = 60–75 myotubes in 3 independent experiments). P‐value < 0.05 by one‐way ANOVA for the three panels in (C and D). P < 0.05 by post hoc Tukey test is indicated by a single letter representing each group.
Source data are available online for this figure.
Ctgf is detected at the NMJs during embryogenesis
We scrutinized molecules that are regulated by CTGF, as well as molecules that regulate CTGF expression. In C2C12 myotubes, exogenous CTGF induced the expression of endogenous Ctgf, but not of Chrne, MuSK, or Lrp4 (Fig 4A). We then examined the effects of neuregulin‐1, a secreted modulator of AChR clustering (Ngo et al, 2012), and agrin on Ctgf expression. Neuregulin‐1, but not agrin, induced Ctgf expression (Fig 4B). These results indicate that Ctgf expression is induced by neuregulin‐1 and CTGF itself in myotubes.
Figure 4. Ctgf is enriched at the neuromuscular junction.
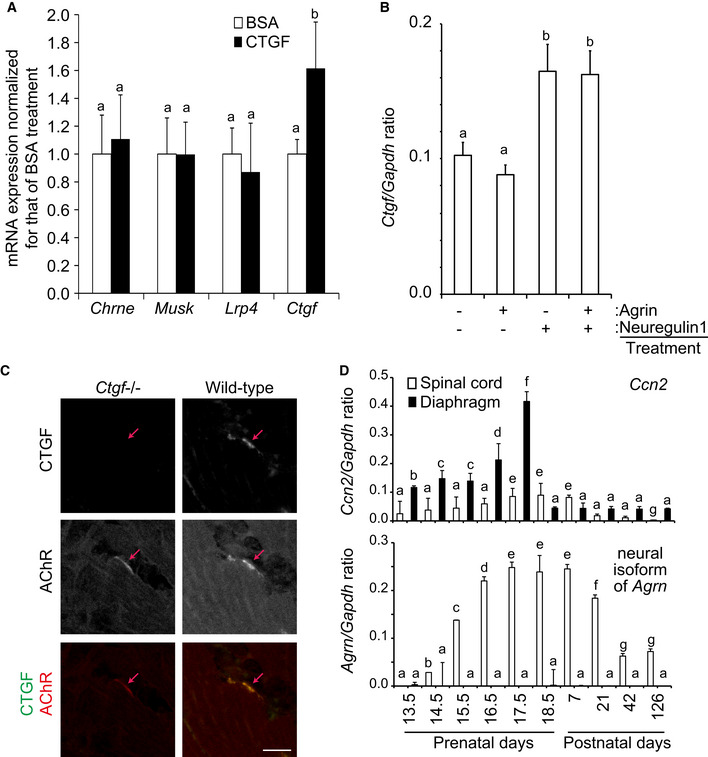
-
A, B(A) Quantitative RT–PCR of Chrne, Musk, Lrp4, and Ctgf in C2C12 myotubes treated with 10 ng/ml BSA or recombinant human CTGF. (B) Quantitative RT–PCR of Ctgf in C2C12 myotubes treated with indicated combinations of 10 ng/ml agrin and/or 10 ng/ml neuregulin‐1. Mean and SD (n = 3 mice) are indicated. P‐value < 0.05 by two‐way repeated measures ANOVA (A) and one‐way ANOVA (B). P < 0.05 by post hoc Tukey test is indicated by a single letter representing each group.
-
CLocalizations of CTGF and AChR in a representative cross section of the lower limb muscle at E18.5. Co‐localization of AChR (red) and CTGF (green) is indicated in the merged images. Red arrow points to an AChR cluster. Scale bar = 20 μm.
-
DQuantitative RT–PCR of Ctgf and neural Agrn in the spinal cord and the diaphragm in mouse. Mean and SD (n = 3 mice) are indicated. P‐value < 0.05 by two‐way repeated measures ANOVA. P < 0.05 by post hoc Tukey test is indicated by a single letter representing each group.
We then quantified Ctgf expression in embryonic skeletal muscle. First, we confirmed that CTGF protein was colocalized with AChR clusters in mouse NMJs of the lower limb muscle at embryonic day (E) 18.5 (Fig 4C). We analyzed temporal profiles of Ctgf and neural Agrin expressions in the diaphragm and the spinal cord during development. Expression of Ctgf was detected in the diaphragm from E13.5 and peaked at E17.5, when the Ctgf expression was 4 times higher than that in the spinal cord (Fig 4D). Expression of neural agrin was also detected in the spinal cord from E14.5 and peaked from E17.5 to postnatal day (P) 7. Thus, the temporal profile of the Ctgf expression in the diaphragm was correlated with embryonic NMJ formation (Burden, 2011). Although the diaphragm produced more Ctgf mRNA than the spinal cord, the origin of CTGF concentrated at the NMJs remains undetermined.
Loss of Ctgf compromises AChR clustering, NMJ ultrastructure, and NMJ signal transduction at embryonic day (E) 18.5
Ctgf deficiency resulted in lethality immediately after birth because of respiratory failure caused by thoracic skeletal defects (Ivkovic et al, 2003). We analyzed the NMJs of the diaphragm in Ctgf‐deficient (Ctgf −/−) mice at E18.5. The diaphragm of Ctgf −/− mouse was normal in size (Table EV1). The expressions of marker genes for myogenesis and NMJ formation were essentially unchanged in the Ctgf −/− diaphragm at E18.5 (Fig EV3A). The muscle contractile units, sarcomeres, had normal ultrastructure in Ctgf −/− mice (Fig EV3B). The band formed by AChR‐positive endplates (Fig EV3C–E) and branching of the phrenic nerve (Fig EV3C, D and F) were normal at low magnification in the Ctgf −/− diaphragm at E18.5. Almost all NMJ synapses were adjacent to S100β signals, a marker for Schwann cells (Fig EV4A–C). In electron microscopy, the presynaptic nerve terminal was also adjacent to terminal Schwann cell (Fig EV4D). These results suggest that Ctgf deficiency exhibits no gross defects in muscle differentiation and axonal guidance of motor neurons to muscle fibers.
Figure EV3. Ctgf −/− diaphragm shows no significant differences in myogenic gene expressions, myofibril ultrastructure, width of the AChR band, or axonal branches of the phrenic nerves.
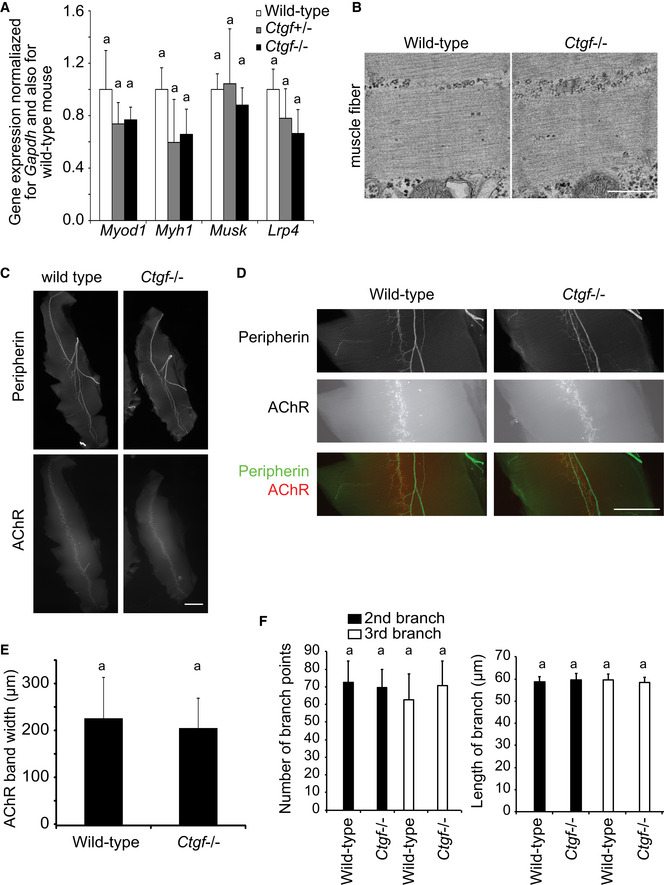
-
AQuantitative RT–PCR of myogenic genes (Myod and Myh1) and genes expressed at the NMJs (MuSK and Lrp4) in the mouse embryonic diaphragm. Mean and SD (n = 6 mice) are indicated. P‐value > 0.05 by two‐way repeated measures ANOVA. Lack of statistical difference is indicated by a single letter “a.”
-
BRepresentative electron micrographs of muscle fibers in the diaphragm of wild‐type and Ctgf −/− mice at embryonic day (E) 18.5. Scale bar = 1 μm.
-
C–FRepresentative low (C) and high (D) magnification images of the whole‐mount left diaphragms at E18.5 stained for peripherin (anti‐peripherin antibody, upper panels) and AChR (α‐bungarotoxin, lower panels). Scale bar = 1 mm. (E) Blinded morphometry of the endplate bandwidth of AChR clusters in (C). (F) Blinded morphometry of the 2nd and 3rd branches in (C). Mean and SD (n = 6 left diaphragms) are indicated. P‐value > 0.05 by one‐way ANOVA for the three panels in (E and F). Lack of statistical difference is indicated by a single letter “a.”
Figure EV4. Ctgf −/− diaphragm shows no significant differences in number of Schwann cells at the NMJ .
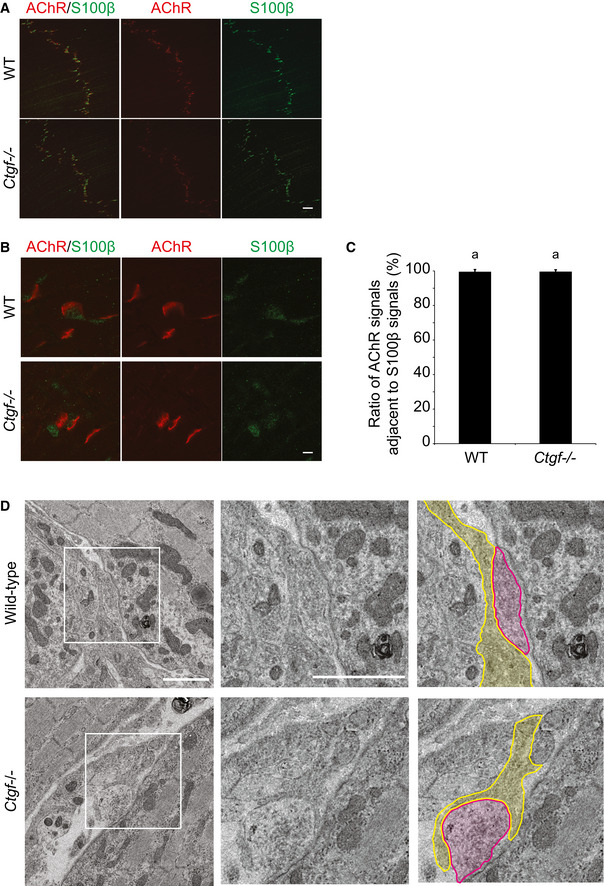
-
A–CRepresentative low (A) and high (B) magnification images of the whole‐mount left diaphragms at E18.5 stained for S100β (green) and AChR (α‐bungarotoxin, red). (C) Blinded morphometry to examine the ratio of AChR signals adjacent to S100β signals. Mean and SD (n = 30 NMJs in 5 left diaphragms) are indicated. Scale bar = 20 μm (A) and 2 μm (B). P‐value > 0.05 by Student's t‐test. Lack of statistical difference is indicated by a single letter “a.”
-
DRepresentative electron micrographs of the NMJs in the diaphragm of wild‐type and Ctgf −/− mice at embryonic day (E) 18.5. Nerve terminal (red) is juxtaposed with Schwann cells (yellow). Scale bar = 1 μm.
However, higher magnification images of the diaphragm NMJs showed that 10.0 ± 3.2% (mean and SD) of peripherin‐positive axons passed through the AChR‐positive areas in Ctgf −/− mice, whereas 2.0 ± 1.4% of them passed through the AChR‐positive areas in wild‐type mice (Fig 5A). In addition, synaptophysin‐positive areas were abnormally observed along motor axons in the Ctgf −/− diaphragm (Figs 5B and EV5A), which may represent the immaturity of the nerve terminal. Blinded morphometric analysis revealed that the area of AChR clusters was smaller in the Ctgf −/− diaphragm than that in the wild‐type diaphragm, while the numbers and intensities of AChR‐positive areas and of synaptophysin‐positive areas were unchanged (Fig 5C and D). Furthermore, the ratio of synaptophysin‐positive areas that were not colocalized with AChR signals was increased in Ctgf −/− mice (Fig 5E), which likely represents dispersion of synaptophysin along the motor axon (Fig 5B). The ultrastructure of NMJs of the Ctgf −/− diaphragm at E18.5 showed a lower density of synaptic vesicles, fewer active zones, and significantly fewer mitochondria in the presynaptic nerve terminals (Fig 5F and Table 1). These structural analyses suggest that CTGF is important for the maturation of the presynaptic nerve terminal.
Figure 5. Apposition of the nerve terminal and the motor endplate is compromised in the E18.5 diaphragm of Ctgf −/− mice.
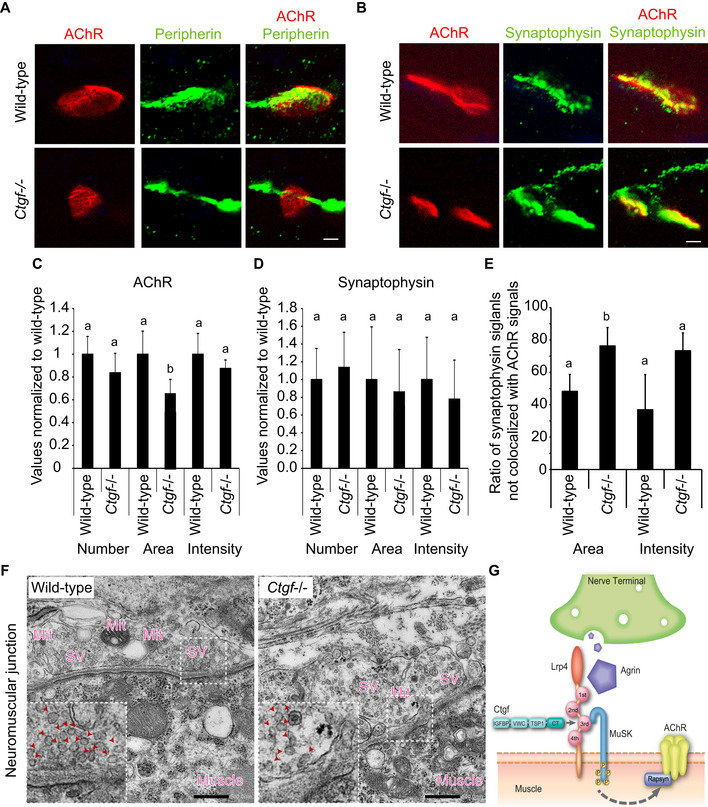
-
ARepresentative confocal images of the left diaphragm at E18.5 labeled with α‐bungarotoxin (red) and anti‐peripherin antibody (green) to visualize AChRs and the motor axon, respectively. The endplates of Ctgf −/− diaphragm were smaller (lower left panel) than those of the wild‐type diaphragm (upper left panel). In the Ctgf −/− diaphragm, 10.0 ± 3.2% (mean and SD, n = 105 in 5 embryos) of peripherin‐positive axons passed through the AChR‐positive areas. In contrast, in the wild‐type diaphragm, 2.0 ± 1.4% (mean and SD, n = 102 in 5 embryos) of peripherin‐positive axons passed through the AChR‐positive areas. Quantification was performed in a blinded manner. Scale bar = 5 μm.
-
BRepresentative confocal images of the left diaphragm at E18.5 labeled with α‐bungarotoxin (red) and anti‐synaptophysin antibody (green) to visualize AChRs and the nerve terminal, respectively. Synaptophysin signals mostly overlapped with α‐bungarotoxin signals in the wild‐type diaphragm, whereas synaptophysin signals extended into the axon in the Ctgf −/− diaphragm. Scale bar = 5 μm.
-
C–EBlinded morphometric analysis of AChR signals (C), synaptophysin signals (D), and the ratio of synaptophysin signals not colocalized with AChR signals (E) of confocal images shown in (A) and (B). AChR areas (C), but not synaptophysin‐positive areas (D), were decreased in Ctgf −/− mice at E18.5. (E) A large fraction of synaptophysin signals were not colocalized with AChR signals in Ctgf −/− diaphragm. Mean and SD (n = 40–50 NMJs in 6 left diaphragms) are indicated. P‐value < 0.05 by one‐way ANOVA for (C and E), but not for (D). P < 0.05 by post hoc Tukey test is indicated by a single letter representing each group.
-
FRepresentative electron micrographs of the NMJs in the diaphragm of wild‐type and Ctgf −/− mice at E18.5. In Ctgf −/− mice, synaptic vesicles are sparser and the number of mitochondria at the nerve terminals is reduced compared to those in wild‐type mice. See Table 1 for morphometric analysis. Regions indicated by rectangles are enlarged in the insets. Arrowheads point to synaptic vesicles (SV). Mit, mitochondria. Blinded morphometric measurements are shown in Table 1. Scale bar = 500 nm.
-
GSchematic showing binding of CTGF to the agrin/Lrp4/MuSK complex and induction of acetylcholine receptor (AChR) clustering at the neuromuscular junctions. The CT domain of CTGF binds to the 3rd β‐propeller domain of LRP4 to enhance binding of LRP4 to MuSK and facilitate agrin‐mediated MuSK phosphorylation and AChR clustering.
Figure EV5. Ctgf −/− diaphragm shows mild to moderate electrophysiological abnormalities and different expression patterns for CCN family genes.
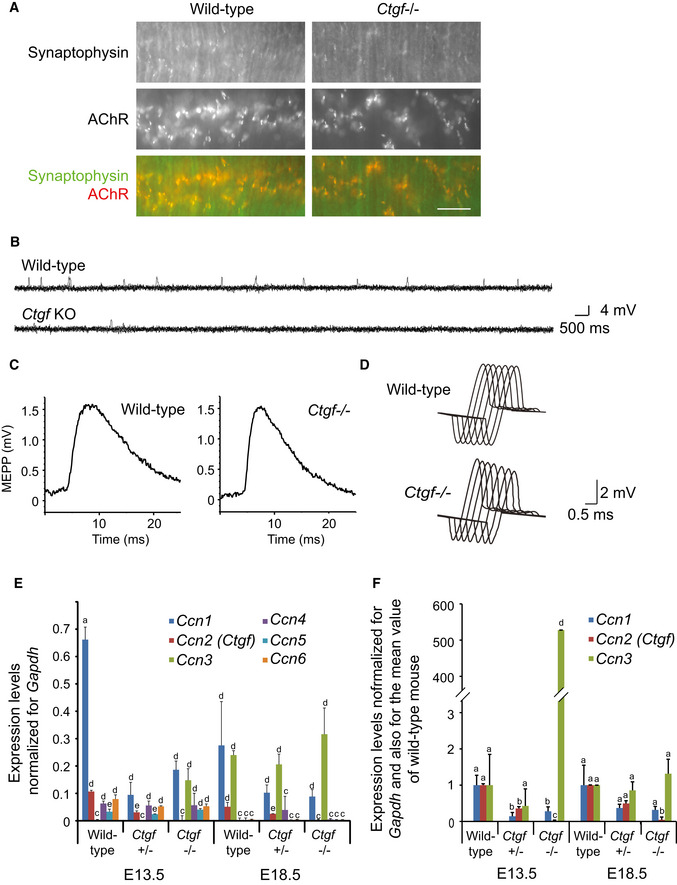
-
ARepresentative low magnification images of the whole‐mount left diaphragms at E18.5 stained for synaptophysin (green) and AChR (α‐bungarotoxin, red). Confocal images of the NMJ are indicated in Fig 5B. Scale bar = 20 μm.
-
BRepresentative raw MEPP traces for 7 s of wild‐type and Ctgf −/− mice at E18.5.
-
CRepresentative averaged MEPP recordings of wild‐type and Ctgf −/− mice at E18.5.
-
DRepresentative CMAP traces of wild‐type and Ctgf −/− mice at E18.5 in response to repetitive nerve stimulation at 2 Hz. Quantifications of MEPP and CMAP recordings are indicated in Table 2.
-
E, FQuantitative RT–PCR of the CCN family genes, Ccn1‐6, in the mouse diaphragm at E13.5 and E18.5. (F) Expressions of Ccn1‐3 are indicated by additional normalization for the mean value of wild‐type mice. Note that Ccn3 is highly expressed in E13.5 Ctgf −/− embryos. Mean and SD are indicated (n = 9 including 3 independent experiments for 3 diaphragm). P‐value < 0.05 by two‐way repeated measures ANOVA for both (E and F). P < 0.05 by post hoc Tukey test is indicated by a single letter representing each group.
Table 1.
Morphometric analysis of the NMJ ultrastructure of the diaphragm of wild‐type and Ctgf −/− mice at embryonic day (E) 18.5 shown in Fig 5F
| Wild type | Ctgf −/− | Ratio | P | |
|---|---|---|---|---|
| Nerve terminal area (μm2) | 3.25 ± 1.37 (n = 15) | 3.81 ± 3.87 (n = 16) | 1.17 | 0.223 |
| Area of mitochondria/area of nerve terminal (%) | 7.55 ± 3.23 (n = 15) | 2.93 ± 2.47 (n = 16) | 0.38 | 0.123 |
| Number of presynaptic mitochondria (/synapse) | 3.00 ± 1.51 (n = 15) | 0.63 ± 0.81 (n = 16) | 0.21 | 4.45E‐6a |
| Diameter of synaptic vesicles (nm) | 40.15 ± 12.23 (n = 936) | 44.36 ± 11.35 (n = 836) | 1.10 | 0.234 |
| Density of synaptic vesicles (/μm2) | 20.45 ± 10.43 (n = 15) | 16.83 ± 8.73 (n = 16) | 0.82 | 0.0423a |
| Width of the synaptic cleft (μm) | 146.23 ± 98.64 (n = 150) | 152.53 ± 129.25 (n = 160) | 1.04 | 0.154 |
| Number of active zones (/synapse) | 2.53 ± 1.14 (n = 15) | 1.01 ± 0.70 (n = 16) | 0.86 | 0.0261a |
| Number of postsynaptic folds (/synapse) | 1.20 ± 0.86 (n = 15) | 0.60 ± 0.41 (n = 15) | 0.50 | 0.0567 |
Blinded morphometric measurements were performed on electron microscopic images of the NMJs in the diaphragms at E18.5. Four wild‐type and four Ctgf −/− mice were analyzed. Mean and SD are indicated. Ratio indicates the mean value of Ctgf −/− mice divided by that of wild‐type mice. Statistical significance was calculated by two‐way repeated measures ANOVA.
P < 0.05 by post hoc Tukey test compared to wild type.
To evaluate NMJ signal transduction in the Ctgf −/− mice, we analyzed the miniature endplate potentials (MEPPs) of the diaphragm and the compound muscle action potentials (CMAPs) of the tibialis anterior (TA) muscle. The MEPP frequencies were significantly reduced in the Ctgf −/− diaphragms compared to those in the wild‐type diaphragms (Table 2 and Fig EV5B and C). Defective NMJ signal transmission was also indicated by abnormally decreased CMAP amplitudes of the TA muscle after repetitive stimulation of the sciatic nerve (Table 2 and Fig EV5D), which is a diagnostic hallmark of myasthenia gravis and congenital myasthenic syndromes (Ohno et al, 2012). The Ctgf −/− diaphragms thus showed mild morphological abnormalities at the light and electron microscopy levels, as well as mild to moderate electrophysiological abnormalities. Expression profiles of six Ccn family genes at E13.5 and E18.5 showed that other CCN family genes were expressed in the diaphragm during embryogenesis (Fig EV5E and F). One or more of these CCN proteins might have partially compensated for the absence of CTGF at the NMJ.
Table 2.
Microelectrode studies and repetitive nerve stimulation of Ctgf +/+, Ctgf +/−, and Ctgf −/− mice at embryonic day (E) 18.5
| Ctgf +/+ | Ctgf +/− | Ctgf −/− | P (Ctgf +/+ vs. −/−) | |
|---|---|---|---|---|
| MEPP amplitude (mV) | 3.24 ± 0.49 (n = 18) | 3.14 ± 0.47 (n = 26) | 3.89 ± 0.70 (n = 20) | 0.06 |
| MEPP frequency (s−1) | 0.69 ± 1.03 (n = 18) | 0.65 ± 1.02 (n = 26) | 0.09 ± 0.10 (n = 20) | 0.01a |
| The fifth CMAP area (%) | 98.75 ± 5.87 (n = 3) | 102.94 ± 15.45 (n = 5) | 55.66 ± 16.05 (n = 3) | 0.04a |
Miniature endplate potentials (MEPPs) were recorded from the left diaphragms of Ctgf +/+, Ctgf +/−, and Ctgf −/− mice at E18.5. Relative area of the fifth compound muscle action potential (CMAP) compared to the first CMAP at 2‐Hz stimulation of the sciatic nerve is indicated. Mean and SEM are indicated. Statistical significance was calculated by one‐way ANOVA.
P < 0.05 by post hoc Tukey test.
Discussion
We report that CTGF is a binding partner of LRP4 at the NMJ and enhances the roles of LRP4 on the formation of AChR clusters at the postsynaptic motor endplate. CTGF is also critical for the maturation and function of the presynaptic nerve terminal.
The CTGF protein, a 40‐kDa extracellular molecule, contains the IGFBP, VWC, TSP1, and CT domains (Takigawa, 2017). As stated in the introduction, each domain binds to specific molecules (Malik et al, 2015; Takigawa, 2017). The presence of a plethora of molecules that bind to CTGF supports the notion that CTGF orchestrates mutual coordination of extracellular matrix (ECM) proteins and binding of ECM proteins to their cell surface receptors (Yeger & Perbal, 2007).
We showed that CTGF binds to LRP4 (Fig 1A–D), enhances the binding of LRP4 to MuSK (Figs 1E and EV1A), and increases the amount of LRP4 on the sarcoplasmic membrane (Fig 2). Functional analysis revealed that CTGF enhances agrin‐induced MuSK phosphorylation and AChR clustering in myotubes (Fig 3A and B). Amyloid precursor protein (APP) and its family proteins, APLP1 and APLP2, bind to LRP4 and coordinate the development of NMJ in mice (Choi et al, 2013). Similarly, sarcoglycan α (SGα), a component of the dystrophin–glycoprotein complex, interacts with LRP4, stabilizes LRP4, and potentiates LRP4‐mediated AChR clustering in adult skeletal muscles (Zhao et al, 2018). LRP4 thus binds to APPs, SGα, and CTGF in addition to agrin and MuSK. Interestingly, two membrane‐bound proteins (APPs and SGα) and an ECM protein (CTGF) have similar effects on AChR clustering. These proteins (APPs, SGα, CTGF, and agrin) may form a large complex along with transmembrane proteins (LRP4 and MuSK) on the sarcoplasmic membrane to enable efficient clustering of AChRs at the NMJ. Spatially restricted expression of LRP4 protein at the NMJ may be achieved by these binding partners.
We showed that neuregulin‐1 and CTGF itself increase Ctgf expression by 1.6‐fold (Fig 4A and B). Although the increased expression of Ctgf is mild, CTGF protein is concentrated at the NMJ in embryonic (Fig 4C) and adult (Sun et al, 2008) skeletal muscles. We previously reported that the ectopically expressed ECM proteins, collagen Q (Ito et al, 2012) and biglycan (Ito et al, 2017), are anchored to the NMJ via their proprietary binding partners (MuSK and the dystrophin–glycoprotein complex, respectively) at the NMJ (Ito & Ohno, 2018). Because CTGF binds to integrins including integrin α3 (Ross et al, 2017) and HSPGs including perlecan (Arikawa‐Hirasawa et al, 2002; Nishida et al, 2003), both of which are accumulated at the NMJ, CTGF may anchor to the NMJ via binding to these ECM proteins.
CCN proteins have been extensively studied in the context of osteochondrogenesis, angiogenesis, and carcinogenesis. Indeed, Ctgf‐deficient mice die immediately after birth due to respiratory failure caused by severe chondrodysplasia (Ivkovic et al, 2003). However, the physiological roles of CCN proteins expressed in skeletal muscle remain to be elucidated. We demonstrated here that Ctgf is required for AChR clustering in C2C12 myotubes (Fig 3C and D). In vivo, the lack of CTGF has no gross effect on muscle differentiation (Fig EV3A and B) or branching of the phrenic nerve (Fig EV3C, D and F). However, at the NMJ, the lack of CTGF causes a mild defect in AChR clustering (Fig 5A–C) and moderate to severe defects in the spatial confinement of synaptic vesicles at the nerve terminal (Fig 5B, D and E) and in ultrastructural organization of the nerve terminal (Fig 5F and Table 1). Previous studies demonstrated that a lack of muscle LRP4 leads to defective AChR clustering in skeletal muscle, as well as sparse synaptic vesicles and fewer active zones in the presynaptic nerve terminal (Kim et al, 2008; Zhang et al, 2008; Wu et al, 2012; Yumoto et al, 2012). Although the lack of CTGF induces more prominent defects at the nerve terminal than at the motor endplate, both presynaptic and postsynaptic defects are milder compared to those in the lack of muscle LRP4. As stated above, the lack of APPs (Choi et al, 2013) and SGα (Zhao et al, 2018) induces presynapse‐dominant defects at the NMJ, as we observed in Ctgf −/− mice. APPs, SGα, and CTGF enhance the AChR clustering activity mediated by LRP4 at the motor endplate. Similarly, APPs, SGα, and CTGF enhance the organization of the nerve terminal mediated by LRP4, and the effects of these modulators are more prominent at the nerve terminal than those at the motor endplate. LRP4 at the postsynaptic skeletal muscle induces the clustering of synaptic vesicles and active zones (Wu et al, 2012; Yumoto et al, 2012). However, binding partner(s) for this LRP4 remain elusive. CTGF is one of candidate molecules that may enhance the functions of LRP4 in pre‐ and postsynaptic regions.
Materials and Methods
Expression vectors, luciferase reporter vectors, and lentiviral vectors
Constructs for co‐immunoprecipitation and cell‐free plate‐binding assays were generated as follows: pMuSKect‐Flag (Zhang et al, 2008), which was kindly provided by Dr. Lin Mei at Augusta University, included the ectodomain of mouse MuSK cDNA (codons 1–454) upstream of the 3xFlag epitope in a mammalian expression vector, p3xFlag‐CMV‐14 (Sigma‐Aldrich). phLRP4ect‐Flag, which we previously generated (Ohkawara et al, 2014), carried the extracellular domain (codons 1–1,722) of human LRP4 cDNA between the HindIII and XbaI sites upstream of the 3xFlag epitope in a mammalian expression vector p3xFlag‐CMV‐14. phLRP4ect‐Flag was used for constructing Flag‐tagged human LRP4 lacking one of the four β‐propeller domains via the site‐directed mutagenesis kit (BioLabs, E0554S). Human LRP4‐Δ1 lacked codons 456–717, LRP4‐Δ2 lacked codons 761‐022, LRP4‐Δ3 lacked codons 1,069–1,330, and LRP4‐Δ4 lacked codons 1,373–1,634. Human full‐length CTGF cDNA was PCR‐amplified and cloned into a pcDNA3.1(+) mammalian expression vector to generate phCTGF. CTGF lacking both TSP1 and CT domains (ΔTSP1_ΔCT construct retaining codons 1–190) and CTGF lacking the CT domain (ΔCT construct retaining codons 1–252) were similarly generated from phCTGF. cDNAs for the ectodomain of mouse MuSK carrying codons 1–454, partial rat Agrin carrying codons 1,141–1,937 (Ohkawara et al, 2014), mouse full‐length CTGF carrying codons 1–348, mouse CTGF‐ΔCT retaining codons 1–252, mouse CTGF‐CT carrying codons 252–348, and human C‐terminal‐deleted RSPO2 carrying codons 1–218 as a control were cloned into APtag‐5 (GenHunter) at HindIII and SnaBI sites to generate MuSKect/pAPtag‐5, Agrin/pAPtag‐5, CTGF/pAPtag‐5, CTGF‐ΔCT/pAPtag‐5, CTGF‐CT/pAPtag‐5, and Control/pAPtag‐5, respectively. The pAPtag‐5 clones carried the Igk‐originated signal peptide upstream of the insert and the myc‐tag/alkaline phosphatase downstream of the insert. cDNAs for mouse full‐length CTGF, mouse CTGF‐ΔCT, mouse CTGF‐CT, and human C‐terminal‐deleted RSPO2 were also cloned into pcDNA3.1/3× myc at EcoRI sites to generate myc‐CTGF/pcDNA3.1, myc‐CTGF‐ΔCT/pcDNA3.1, myc‐CTGF‐CT/pcDNA3.1, and myc‐Control/pcDNA3.1, respectively.
For constructing the lentiviral vector expressing shRNA against Ctgf, double‐stranded oligonucleotides (sense, 5′‐gatcccgcgagatcatgaaaaagaattcaagagattctttttcatgatcacgctttttggaaa‐3′ and antisense, 5′‐agcttttccaaaaagcgagatcatgaaaaagaatctcttgaattctttttcatgatctcgcgg‐3′ for shCtgf‐1; and sense, 5′‐gatccccgaactcattagactataattcaagagattatagtctaatgagttcgtttttggaaa‐3′ and antisense, 5′‐agcttttccaaaaacgaactcattagactataatctcttgaattatagtctaatgagttcggg‐3′ for shCtgf‐2) were cloned into a lentiviral vector pLenti CMV GFP ×2 DEST, which was kindly provided by Dr. Eric Campeau at University of Massachusetts Medical School (Campeau et al, 2009).
Lack of PCR artifacts was verified by sequencing the entire insert for all clones.
Cell cultures, transfections, and lentivirus production
HEK293 and C2C12 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS). Transfection was performed with Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocols.
Lentiviruses expressing shRNA were prepared as previously reported (Campeau et al, 2009). Briefly, HEK293 cells were plated in a 150‐mm dish on the day before transfection. HEK293 cells were introduced with pLP1, pLP2, and pLP/VSVG plasmids (ViraPower Packaging Mix, Invitrogen) as well as pLenti vector using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocols. To increase the number of viral particles, the media were collected at both 48 and 96 h after transfection and combined. All media containing the virus particles were filtered using the Millex‐HV 0.45‐μm PVDF filter (Millex). The viruses were concentrated by centrifugation using a Beckman SW28 rotor at 75,600 × g for 2 h at 4°C and resuspended in 4 ml of Hank's Buffered Saline solution (HBS, Invitrogen). After the second ultracentrifugation using a Beckman 55Ti rotor at 75,600 × g for 90 min, the viral pellet was resuspended in 100 μl of HBS. The lentivirus was added to the medium for mouse C2C12 myotubes. After 48 h, we confirmed that more than ~ 70% of the myotubes were positive for GFP signals driven by the CMV promoter in the pLenti vector.
Immunoprecipitation and Western blotting
For the immunoprecipitation assay, HEK293 cells were transfected with the indicated constructs. To detect Lrp4 phosphorylation for Fig 2B, C2C12 myoblasts were seeded on a culture plate coated with collagen I (BD Biosciences) and were differentiated into myotubes in 2% horse serum (Gibco) in DMEM medium (Gibco) for 5 days. Then, the myotubes were treated with recombinant rat agrin protein (R&D Systems, 550‐AG‐100) with or without human CTGF protein (ProSpec, cyt‐541) for 3 h. The cells were lysed with PLC buffer containing 50 mM HEPES pH 7.0, 150 mM NaCl, 10% glycerol, 1% TritonX‐100, 1.5 mM MgCl2, 1 mM EGTA, 100 mM NaF, 10 mM sodium pyrophosphate, 1 μg/μl aprotinin, 1 μg/μl leupeptin, 1 μg/μl pepstatin A, 1 mM PMSF, and 1 mM sodium orthovanadate with PhosSTOP (Sigma‐Aldrich). The cell lysates were subjected to immunoprecipitation using 1–2 μg of anti‐Flag M2 (Sigma‐Aldrich, F1804), anti‐CTGF (1:200, Santa Cruz Biotechnology, sc‐14939), anti‐LRP4 (R&D Systems, 741704), or anti‐MuSK (Santa Cruz Biotechnology, C‐19) antibody attached to protein G Sepharose beads (GE Healthcare).
Total or precipitated protein was dissolved in 1 × Laemmli buffer, separated by electrophoresis on a 10% or 7.5% SDS–polyacrylamide gel in Tris‐Glycine or Tris‐Tricine buffer, and transferred onto a polyvinylidene difluoride membrane (PVDF, Immobilon‐P, 0.45 or 0.2 μm, Millipore). The membrane was washed in Tris‐buffered saline containing 0.05% Tween 20 (TBS‐T) and blocked for 1 h at room temperature (RT) in TBS‐T with 3% bovine serum albumin (BSA). The membrane was incubated overnight at 4°C either with mouse monoclonal anti‐myc (Abcam, 9E10), anti‐MuSK (1:1,000, Santa Cruz Biotechnology, sc‐6009), anti‐LRP4 (1:2,000, Abcam, ab85679), anti‐CTGF (1:200, Santa Cruz Biotechnology, sc‐14939), anti‐Flag M2 (1:4,000, Sigma‐Aldrich, F1804), anti‐transferrin receptor (TfR, 1:1,000, Abcam, ab84036), anti‐phospho Y755 MuSK (1:1,000, Abcam, ab192583), anti‐phosphotyrosine (1:1,000, Millipore, 05‐321), or anti‐β‐actin (1:200, Santa Cruz Biotechnology, sc‐47778) antibody. The membranes were washed three times for 10 min with TBS‐T and incubated with secondary goat anti‐mouse IgG (1:6,000, GE Healthcare, NA9310), donkey anti‐rabbit IgG (1:6,000, GE Healthcare, NA9340V), or mouse anti‐goat IgG (1:6,000, Santa Cruz Biotechnology, sc‐2354) conjugated to horseradish peroxidase (HRP) for 1 h at RT. The blots were visualized using Amersham ECL Western blotting detection reagents and quantified with ImageJ software.
Purification of tagged proteins and cell‐free plate‐binding assay
LRP4ect‐Flag, MuSKect‐Flag, CTGF‐myc, CTGF‐ΔCT‐myc, CTGF‐mycAP, CTGF‐CT‐mycAP, CTGF‐ΔCT‐mycAP, agrin‐mycAP, Rspo2‐mycAP, or MuSKect‐mycAP protein in the conditioned medium (CM) of transfected HEK293 cells was concentrated ~ 100‐fold using an Amicon Ultra‐4 filter (Millipore). Then, we further purified the LRP4ect‐Flag and MuSKect‐Flag proteins with the Anti‐DYKDDDDK‐tag Antibody Beads (Wako). agrin‐mycAP, MuSKect‐mycAP, Rspo2‐mycAP, and CTGF‐mycAP were purified using the c‐myc‐Tagged Protein Mild Purification Kit ver. 2 (MBL). The purified proteins were detected using anti‐myc (Abcam, 9E10) and anti‐Flag (Sigma‐Aldrich, M2) antibodies, respectively. We also measured the concentration of each protein by SDS–PAGE followed by protein staining with SYBRO Ruby Protein Gel Stain (Molecular Probes) using BSA as a standard.
For the cell‐free plate‐binding assay, a Nunc MaxiSorp flat‐bottom plate (Thermo Fisher Scientific) was coated with 0.15 μg (1.5 picomole) of LRP4ect‐Flag or MuSKect‐Flag at 4°C overnight and incubated with a blocking buffer (1% BSA in PBS) at RT for 1 h. For the binding assay, 2 picomole of Rspo2‐mycAP, CTGF‐mycAP, CTGF‐CT‐mycAP, CTGF‐ΔCT‐mycAP, agrin‐mycAP, or MuSKect‐mycAP in 80 μl was added to each well containing the blocking buffer. After incubating for 2 h at RT, the wells were washed twice with PBS. Bound AP activity was measured using LabAssay ALP (Wako). For all experiments, Rspo2‐mycAP was used as a control.
Ctgf‐deficient (Ctgf −/−) mice
We previously reported the osteochondrogenic features of Ctgf knockout mice (Ivkovic et al, 2003). Mouse experiments were approved by the Animal Care and Use Committee of the Nagoya University and were performed in accordance with all relevant guidelines.
Biotinylation and purification of plasma membrane proteins of C2C12 myotubes and the mouse skeletal muscle
To biotinylate plasma membrane proteins of C2C12 myotubes, the cells were treated with 1 ng/ml rat recombinant agrin and/or 10 ng/ml CTGF proteins for 2 days, washed twice with PBS containing 0.1 mM CaCl2 and 1 mM MgCl2 (PBS/CM), and incubated with 0.5 mg/ml sulfo‐NHS‐SS‐biotin (Pierce) in PBS/CM at RT for 30 min. The cells were then washed once with PBS/CM and incubated with 10 mM monoethanolamine for quenching free biotin. The cells were washed several times with ice‐cold PBS and then lysed with PLC buffer containing 50 mM HEPES pH 7.0, 150 mM NaCl, 10% glycerol, 1% Triton X‐100, 1.5 mM MgCl2, 1 mM EGTA, 100 mM NaF, 10 mM sodium pyrophosphate, 1 μg/μl aprotinin, 1 μg/μl leupeptin, 1 μg/μl pepstatin A, 1 mM PMSF, and 1 mM sodium orthovanadate with PhosSTOP (Sigma‐Aldrich). The cell lysates were incubated with streptavidin Sepharose beads (GE healthcare) to purify the cell membrane proteins.
The lower limb muscles of mice at E18.5 were dissected and crushed using the Multi‐Beads Shocker (Yasui Kikai Corp.). The Minute Plasma Membrane Isolation Kit (Invent Biotechnologies) was used to isolate the total and plasma membrane protein fractions from the crushed muscles. All procedures were performed on ice according to the manufacturer's protocols.
Total or precipitated proteins were dissolved in 1× Laemmli buffer, separated on a 10% or 7.5% SDS‐polyacrylamide gel, and transferred onto a PVDF membrane (Immobilon‐P, Millipore). The membrane was washed in Tris‐buffered saline containing 0.05% Tween 20 (TBS‐T) and blocked for 1 h at RT in TBS‐T with 3% bovine serum albumin. The primary antibodies were against LRP4 (1:1,000, Abcam, ab85697), MuSK (1:500, Santa Cruz Biotechnology, sc‐6009), transferrin receptor (TfR) (1:1,000, Abcam, ab84036), and β‐actin (1:200, Santa Cruz Biotechnology, sc‐47778). The secondary antibodies were goat anti‐mouse IgG (1:6,000, GE Healthcare, LNA931V/AG) and goat anti‐rabbit IgG (1:6,000, CST, #7074S), both of which were conjugated to horseradish peroxidase.
Assays for detecting MuSK phosphorylation and AChR clusters in C2C12 myotubes
C2C12 myoblasts were seeded on a culture plate coated with collagen I (BD Biosciences) and were differentiated to myotubes in 2% horse serum (Gibco) in DMEM medium (Gibco) for 5 days. Then, the myotubes were infected with lentivirus for 12 h, followed by treatment with 2 mg/ml doxycycline (ICN Biomedicals) for 2 days to induce shRNA expression. For a MuSK phosphorylation assay, the myotubes were treated with recombinant rat agrin protein (R&D Systems, 550‐AG‐100), purified agrin‐mycAP, purified CTGF‐mycAP, and/or CTGF‐ΔCT‐mycAP for 3 h, and then lysed with the buffer for immunoprecipitation. For an AChR clustering assay, the myotubes were treated with recombinant rat agrin protein, purified agrin‐mycAP, purified CTGF‐mycAP, and/or CTGF‐ΔCT‐mycAP for 18 h. Thirty minutes before fixation in 4% paraformaldehyde, the cells were incubated with 10 μg/ml Alexa 594‐conjugated α‐bungarotoxin (Invitrogen) for 30 min to label AChRs. Fluorescence images were obtained using an Olympus XL71 fluorescence microscope and analyzed with MetaMorph software (Molecular Devices). The lengths of the AChR clusters and myotubes were defined as the longest axes of Alexa 594 signals and GFP signals, respectively, in C2C12 myotubes stained with the CellMask green plasma membrane stain (Invitrogen, C37608) in Fig 3B or in C2C12 myotubes infected with lentivirus expressing shRNA and GFP in Fig 3F. AChR clusters with an axis length of less than 4 μm were excluded from the analysis of AChR clusters.
Immunohistochemistry of the NMJ
Frozen sections (20 μm thickness) of the thigh muscles of mouse embryos were fixed with acetone for 10 min at −20°C, washed three times with PBS, and then covered with PBS containing 1% goat serum for 60 min. For staining, the sections were incubated with rabbit polyclonal anti‐CTGF antibody (1:100, Abcam, ab73761) overnight at 4°C in a humidified chamber. After removing the primary antibody by washing twice with PBS containing 0.05% Tween‐20 (PBS‐T), the sections were incubated with a polyclonal antibody against goat IgG conjugated with FITC, and α‐bungarotoxin conjugated with Alexa 594 for 1.5 h, and repeatedly washed in PBS‐T to remove the residual secondary antibody and α‐bungarotoxin. Finally, the sections were mounted in the VectaShield containing 1.5 μg/ml DAPI (Vector Laboratories) and visualized using the A1Rsi confocal microscope (Nikon).
Quantitative RT–PCR
In wild‐type and Ctgf −/− mouse embryos, Ccn1‐6 mRNAs in the diaphragm and the spinal cord, as well as Myod1, Myh1, Musk, and Lrp4 mRNAs in the diaphragm, were quantified by qRT‐PCR. C2C12 myotubes were treated with 10 ng/ml rat recombinant agrin, 10 ng/ml human CTGF, and/or 10 ng/ml human neuregulin‐1 (R&D Systems, 396‐HB) for 2 days, and Ctgf, Chrne, Musk, and Lrp4 mRNAs were quantified by qRT‐PCR. Gene expression levels were normalized to that of glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) mRNA. Total RNA was extracted using TRIzol reagent (Invitrogen) and was reverse‐transcribed using Oligo (dT)20 Primer (Toyobo) and ReverTra Ace reverse transcriptase (Toyobo). qRT‐PCR was performed in triplicate using SYBR Premix Ex Taq (Takara) on LightCycler 480 (Roche). Primer pairs are shown in Table EV2.
Diaphragm staining
The diaphragms of Ctgf −/− embryos at E18.5 were fixed in 2% paraformaldehyde in PBS for 4 h at 4°C and rinsed with PBS. After dissection of the connective tissue, the muscles were permeabilized with 0.5% Triton X‐100 in PBS for 10 min and then incubated with α‐bungarotoxin conjugated with biotin‐XX (1:600, Invitrogen, B1606), anti‐peripherin antibody (1:800, Millipore, AB1530), anti‐S100 beta antibody (1:100, Abcam, ab52642), and anti‐synaptophysin antibody (1:100, Invitrogen, 180130) overnight. After washing, the sections were incubated with streptavidin conjugated with Alexa 564 (1:500, Invitrogen) or anti‐mouse IgG conjugated with Alexa 488 (1:500, Invitrogen). Fluorescence images were obtained using an Olympus FSX100 fluorescence microscope for the whole‐mount diaphragms (n = 10–20 images for each left diaphragm, 5 diaphragms), and Zeiss LSM710 or Nikon A1Rsi confocal microscope for high magnification images of the NMJ (n = 40–50 images for each left diaphragm, 6 diaphragms). The numbers, intensities, and areas of AChR signals and synaptophysin signals were blindly quantified using images taken by Olympus FSX100. Ratios of synaptophysin signals not colocalized with AChR signals were quantified using images taken by Zeiss LSM710 or Nikon A1Rsi confocal microscope. All signal values were quantified by two blinded researchers using MetaMorph software (Molecular Devices).
Electron microscopy
Ultrastructure of the left diaphragm at E18.5 was analyzed as previously described (Nakashima et al, 2016). The diaphragm and the thorax of E18.5 embryos were isolated together and fixed in 4% paraformaldehyde for 3 h, while applying physiological tension to the diaphragm using the thorax. The middle portion of the diaphragm muscle fibers, where the NMJs are located, was isolated and minced into 0.2‐to‐0.3‐mm blocks. The excised blocks were fixed with 2% glutaraldehyde for 2 h, treated with 1% OsO4, dehydrated in ethanol, and embedded in Epon 812 (TAAB). As the terminal of the phrenic nerve could not be identified even in wild‐type embryos, every second block was stained for cholinesterase using the Ellman method to confirm that a series of the excised blocks included the NMJs. Ultrathin sections were made from blocks that were not stained for cholinesterase. Ultrathin sections (60–70 nm) were stained with uranyl acetate and lead citrate. We identified the NMJs by inspecting the entire ultrathin sections using a JEM‐1400 transmission electron microscope. The following morphometric parameters were measured according to a previous report (Brandon et al, 2003): nerve terminal area in μm2, synaptic vesicle density in μm2 at the nerve terminal area, area of mitochondria/area of nerve terminal (%), the number of active zones, the diameter of synaptic vesicles, and the width of the synaptic cleft. The active zone was defined as the site with a cluster of five or more synaptic vesicles gathered at the presynaptic membrane (Brandon et al, 2003). The postsynaptic fold was defined as an indentation in the postsynaptic membrane where the fold depth was more than 70 nm and the width of the fold aperture was less than half of the fold depth. The numbers of active zones and postsynaptic folds were counted for each nerve terminal. Images were quantified using the ImageJ software.
Electrophysiology
Miniature and evoked endplate potentials (MEPPs) were recorded essentially as described previously (Engel et al, 1993). Ctgf −/− mice and their wild‐type littermates were delivered by cesarean section of anesthetized pregnant mice. The diaphragm was dissected in Tyrode solution containing 135 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 1.3 mM Na2HPO4, 12.5 mM NaHCO3, and 11 mM D‐glucose at pH 7.2, and pinned to a Sylgard‐coated dish. The solution was continuously gassed with 95% O2 and 5% CO2 at 37°C. The endplate region was inserted with a glass micropipette (20–40 MΩ) filled with 2 M KCl. The signal was amplified by an AxonClamp 900A amplifier (Molecular Devices), digitized at 10 kHz by Digidata 1550B (Molecular Devices), and was analyzed with AxoGraph × 1.5.0 (AxoGraph Scientific). To record compound muscle action potentials (CMAPs), the delivered E18.5 embryos were kept at 37°C with manual respiratory assist. The stimulation needle electrode (Inter Medical, IMK2‐1001) was inserted near the sciatic nerve in the right thigh. The recording needle electrode was inserted into the calf muscle and was connected to a Neuropack S1 system (Nihon Kohden, MEB‐9404). The stimulation of the sciatic nerve was triggered with a series of 10 stimuli at 2 Hz, and CMAP amplitudes at the first and fifth stimuli were measured.
Cell surface‐binding assays
Full‐length human LRP4 cDNA (Open Biosystems) was cloned into the EcoRI sites of the pcDNA3.1 mammalian expression vector to make phLRP4. Full‐length human CTGF cDNA was PCR‐amplified and cloned into pcDNA3.1 vector to make phCTGF. For the cell surface‐binding assay, COS cells in 24‐well plates were transfected with phLRP4 (0.5 μg or 1.0 μg per a well) and phCTGF (0.5 μg or 1.0 μg per a well) using FuGENE 6 (Roche). The cells were incubated for 24 h with medium containing either purified MuSKect‐mycAP or agrin‐mycAP for 1.5 h at RT. The cells were washed with HABH buffer (0.5 mg/ml bovine serum albumin, 0.1% NaN3, and 20 mM HEPES pH 7.0 in Hank's balanced salt solution) and fixed with 60% acetone for 10 min on ice, followed by 4% paraformaldehyde in 20 mM HEPES pH 7.0 in Hank's balanced salt solution for 10 min on ice. The fixed cells were washed once with 20 mM HEPES pH 7.0 and 150 mM NaCl, incubated at 65°C for 30 min, washed with 0.1 M Tris–HCl pH 8.0, and then washed with water. Bound AP activity was measured using LabAssay ALP (Wako).
ATF‐2 luciferase assay in HEK293 cells
The mouse Musk cDNA in pExpress‐1 was purchased from Open Biosystems. Partial rat Agrin (codons 1,141–1,937; Ohkawara et al, 2014) was cloned into APtag‐5 (GenHunter) at the HindIII and SnaBI sites. ATF2‐Luc to quantify the JNK signaling activity (van der Sanden et al, 2004; Ohkawara et al, 2014) and phRL‐TK Renilla luciferase vector (Promega) were used to monitor agrin‐Lrp4‐MuSK signaling. HEK293 cells (1 × 104 cells) in a 96‐well plate were transfected with 5 ng ATF2‐Luc and 0.5 ng phRL‐TK, along with 1, 2, or 5 ng pExpress‐1‐Musk encoding full‐length MuSK, 5 ng phLRP4 encoding full‐length LRP4, and 5 ng Agrin/pAPtag‐5 encoding rat partial Agrin. The amounts of these constructs were optimized to lower levels to enable the detection of the effect of CTGF. The cells were cultured for 24 h in the presence of CTGF (250 pM, ProSpec, cyt‐541).
Statistical analysis
Statistical analyses including unpaired Student's t‐test, one‐way or two‐way ANOVA, and post hoc Tukey test were performed using SPSS ver. 23 (IBM Corp). P values less than 0.05 were considered to be statistically significant. After one‐way or two‐way ANOVA, statistically similar items were grouped together according to the post hoc Tukey test (P < 0.05), and each group was labeled by an identical single lowercase letter.
Author contributions
BO, AK, and KO conceived the study and interpreted the results. SKa and BO quantified the results as blinded researchers. BO and AK contributed to the experiments using Ctgf‐deficient mice. TH, SKu and MT created plasmids carrying wild‐type and deleted forms of human CTGF cDNA. KML generated the Ctgf −/− mice. MT, AM, NI, and KO supervised the project. NI and KO provided financial support. BO, AK, and KO prepared the paper with assistance from the other authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Table EV2
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Acknowledgements
We would like to thank Drs. Hideki Hiraiwa and Yusuke Kawamura at Nagoya University for their technical supports. pMuSKect‐Flag (Zhang et al, 2008) and pLenti CMV GFP ×2 DEST (Campeau et al, 2009) were kindly provided by Dr. Lin Mei at the Augusta University and Dr. Eric Campeau at the University of Massachusetts Medical School, respectively. This study was supported by Grants‐in‐Aids from the Japan Society for the Promotion of Science (JP15H05015, JP17K19757, JP19H03817, JP18K06058, JP17K07094, and JP18K06483); the Ministry of Health, Labour, and Welfare of Japan (H29‐Nanchi‐Ippan‐030); the Japan Agency for Medical Research and Development (JP19gm1010002, JP19ek0109230, JP19ek0109281, and JP19bm0804005); and the Intramural Research Grant for Neurological and Psychiatric Disorders of NCNP (29‐4).
EMBO Reports (2020) 21: e48462
References
- Arikawa‐Hirasawa E, Rossi SG, Rotundo RL, Yamada Y (2002) Absence of acetylcholinesterase at the neuromuscular junctions of perlecan‐null mice. Nat Neurosci 5: 119–123 [DOI] [PubMed] [Google Scholar]
- Borges LS, Yechikhov S, Lee YI, Rudell JB, Friese MB, Burden SJ, Ferns MJ (2008) Identification of a motif in the acetylcholine receptor beta subunit whose phosphorylation regulates rapsyn association and postsynaptic receptor localization. J Neurosci 28: 11468–11476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandon EP, Lin W, D'Amour KA, Pizzo DP, Dominguez B, Sugiura Y, Thode S, Ko CP, Thal LJ, Gage FH et al (2003) Aberrant patterning of neuromuscular synapses in choline acetyltransferase‐deficient mice. J Neurosci 23: 539–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burden SJ (2002) Building the vertebrate neuromuscular synapse. J Neurobiol 53: 501–511 [DOI] [PubMed] [Google Scholar]
- Burden SJ (2011) SnapShot: neuromuscular junction. Cell 144: 826 e821 [DOI] [PubMed] [Google Scholar]
- Campeau E, Ruhl VE, Rodier F, Smith CL, Rahmberg BL, Fuss JO, Campisi J, Yaswen P, Cooper PK, Kaufman PD (2009) A versatile viral system for expression and depletion of proteins in mammalian cells. PLoS ONE 4: e6529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camurdanoglu BZ, Hrovat C, Durnberger G, Madalinski M, Mechtler K, Herbst R (2016) MuSK kinase activity is modulated by A serine phosphorylation site in the kinase loop. Sci Rep 6: 33583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi HY, Liu Y, Tennert C, Sugiura Y, Karakatsani A, Kroger S, Johnson EB, Hammer RE, Lin W, Herz J (2013) APP interacts with LRP4 and agrin to coordinate the development of the neuromuscular junction in mice. Elife 2: e00220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel AG, Nagel A, Walls TJ, Harper CM, Waisburg HA (1993) Congenital myasthenic syndromes: I. Deficiency and short open‐time of the acetylcholine receptor. Muscle Nerve 16: 1284–1292 [DOI] [PubMed] [Google Scholar]
- Fischbach GD, Rosen KM (1997) ARIA: a neuromuscular junction neuregulin. Annu Rev Neurosci 20: 429–458 [DOI] [PubMed] [Google Scholar]
- Gautam M, Noakes PG, Mudd J, Nichol M, Chu GC, Sanes JR, Merlie JP (1995) Failure of postsynaptic specialization to develop at neuromuscular junctions of rapsyn‐deficient mice. Nature 377: 232–236 [DOI] [PubMed] [Google Scholar]
- Gonzalez D, Brandan E (2019) CTGF/CCN2 from skeletal muscle to nervous system: impact on neurodegenerative diseases. Mol Neurobiol 56: 5911–5916 [DOI] [PubMed] [Google Scholar]
- Hall ZW, Sanes JR (1993) Synaptic structure and development: the neuromuscular junction. Cell 72(Suppl): 99–121 [DOI] [PubMed] [Google Scholar]
- Holbourn KP, Acharya KR, Perbal B (2008) The CCN family of proteins: structure‐function relationships. Trends Biochem Sci 33: 461–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito M, Suzuki Y, Okada T, Fukudome T, Yoshimura T, Masuda A, Takeda S, Krejci E, Ohno K (2012) Protein‐anchoring strategy for delivering acetylcholinesterase to the neuromuscular junction. Mol Ther 20: 1384–1392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito M, Ehara Y, Li J, Inada K, Ohno K (2017) Protein‐anchoring therapy of biglycan for mdx mouse model of duchenne muscular dystrophy. Hum Gene Ther 28: 428–436 [DOI] [PubMed] [Google Scholar]
- Ito M, Ohno K (2018) Protein‐anchoring therapy to target extracellular matrix proteins to their physiological destinations. Matrix Biol 68–69: 628–636 [DOI] [PubMed] [Google Scholar]
- Ivkovic S, Yoon BS, Popoff SN, Safadi FF, Libuda DE, Stephenson RC, Daluiski A, Lyons KM (2003) Connective tissue growth factor coordinates chondrogenesis and angiogenesis during skeletal development. Development 130: 2779–2791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun JI, Lau LF (2011) Taking aim at the extracellular matrix: CCN proteins as emerging therapeutic targets. Nat Rev Drug Discov 10: 945–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim N, Stiegler AL, Cameron TO, Hallock PT, Gomez AM, Huang JH, Hubbard SR, Dustin ML, Burden SJ (2008) Lrp4 is a receptor for Agrin and forms a complex with MuSK. Cell 135: 334–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krupska I, Bruford EA, Chaqour B (2015) Eyeing the Cyr61/CTGF/NOV (CCN) group of genes in development and diseases: highlights of their structural likenesses and functional dissimilarities. Hum Genomics 9: 24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik AR, Liszewska E, Jaworski J (2015) Matricellular proteins of the Cyr61/CTGF/NOV (CCN) family and the nervous system. Front Cell Neurosci 9: 237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercurio S, Latinkic B, Itasaki N, Krumlauf R, Smith JC (2004) Connective‐tissue growth factor modulates WNT signalling and interacts with the WNT receptor complex. Development 131: 2137–2147 [DOI] [PubMed] [Google Scholar]
- Nakashima H, Ohkawara B, Ishigaki S, Fukudome T, Ito K, Tsushima M, Konishi H, Okuno T, Yoshimura T, Ito M et al (2016) R‐spondin 2 promotes acetylcholine receptor clustering at the neuromuscular junction via Lgr5. Sci Rep 6: 28512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo ST, Cole RN, Sunn N, Phillips WD, Noakes PG (2012) Neuregulin‐1 potentiates agrin‐induced acetylcholine receptor clustering through muscle‐specific kinase phosphorylation. J Cell Sci 125: 1531–1543 [DOI] [PubMed] [Google Scholar]
- Nishida T, Kubota S, Fukunaga T, Kondo S, Yosimichi G, Nakanishi T, Takano‐Yamamoto T, Takigawa M (2003) CTGF/Hcs24, hypertrophic chondrocyte‐specific gene product, interacts with perlecan in regulating the proliferation and differentiation of chondrocytes. J Cell Physiol 196: 265–275 [DOI] [PubMed] [Google Scholar]
- Ohkawara B, Cabrera‐Serrano M, Nakata T, Milone M, Asai N, Ito K, Ito M, Masuda A, Ito Y, Engel AG et al (2014) LRP4 third beta‐propeller domain mutations cause novel congenital myasthenia by compromising agrin‐mediated MuSK signaling in a position‐specific manner. Hum Mol Genet 23: 1856–1868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno K, Ito M, Engel AG (2012) Congenital myasthenic syndromes – molecular bases of congenital defects of proteins at the neuromuscular junction In: Neuromuscul Disord, Zaher A. (ed.), pp. 175–200. InTechOPen: London, UK [Google Scholar]
- Ross JA, Webster RG, Lechertier T, Reynolds LE, Turmaine M, Bencze M, Jamshidi Y, Cetin H, Muntoni F, Beeson D et al (2017) Multiple roles of integrin‐alpha3 at the neuromuscular junction. J Cell Sci 130: 1772–1784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Sanden MH, Meems H, Houweling M, Helms JB, Vaandrager AB (2004) Induction of CCAAT/enhancer‐binding protein (C/EBP)‐homologous protein/growth arrest and DNA damage‐inducible protein 153 expression during inhibition of phosphatidylcholine synthesis is mediated via activation of a C/EBP‐activating transcription factor‐responsive element. J Biol Chem 279: 52007–52015 [DOI] [PubMed] [Google Scholar]
- Sun G, Haginoya K, Wu Y, Chiba Y, Nakanishi T, Onuma A, Sato Y, Takigawa M, Iinuma K, Tsuchiya S (2008) Connective tissue growth factor is overexpressed in muscles of human muscular dystrophy. J Neurol Sci 267: 48–56 [DOI] [PubMed] [Google Scholar]
- Takigawa M (2017) The CCN proteins: an overview. Methods Mol Biol 1489: 1–8 [DOI] [PubMed] [Google Scholar]
- Wu H, Xiong WC, Mei L (2010) To build a synapse: signaling pathways in neuromuscular junction assembly. Development 137: 1017–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Lu Y, Shen C, Patel N, Gan L, Xiong WC, Mei L (2012) Distinct roles of muscle and motoneuron LRP4 in neuromuscular junction formation. Neuron 75: 94–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeger H, Perbal B (2007) The CCN family of genes: a perspective on CCN biology and therapeutic potential. J Cell Commun Signal 1: 159–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeger H, Perbal B (2016) CCN family of proteins: critical modulators of the tumor cell microenvironment. J Cell Commun Signal 10: 229–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yumoto N, Kim N, Burden SJ (2012) Lrp4 is a retrograde signal for presynaptic differentiation at neuromuscular synapses. Nature 489: 438–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Luo S, Wang Q, Suzuki T, Xiong WC, Mei L (2008) LRP4 serves as a coreceptor of agrin. Neuron 60: 285–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao K, Shen C, Li L, Wu H, Xing G, Dong Z, Jing H, Chen W, Zhang H, Tan Z et al (2018) Sarcoglycan alpha mitigates neuromuscular junction decline in aged mice by stabilizing LRP4. J Neurosci 38: 8860–8873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong Y, Zhang B, Gu S, Lee K, Zhou J, Yao G, Figueiredo D, Perry K, Mei L, Jin R (2012) Structural basis of agrin‐LRP4‐MuSK signaling. Genes Dev 26: 247–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Table EV2
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3