

Abstract
Vector production scale-up is a major barrier in systemic adeno-associated virus (AAV) gene therapy. Many scalable manufacturing methods have been developed. However, the potency of the vectors generated by these methods has rarely been compared with vectors made by transient transfection (TT), the most commonly used method in preclinical studies. In this study, we blindly compared therapeutic efficacy of an AAV9 micro-dystrophin vector generated by the TT method and scalable herpes simplex virus (HSV) system in a Duchenne muscular dystrophy mouse model. AAV was injected intravenously at 5 × 1014 (high), 5 × 1013 (medium), or 5 × 1012 (low) viral genomes (vg)/kg. Comparable levels of micro-dystrophin expression were observed at each dose in a dose-dependent manner irrespective of the manufacturing method. Vector biodistribution was similar in mice injected with either the TT or the HSV method AAV. Evaluation of muscle degeneration/regeneration showed equivalent protection by vectors made by either method in a dose-dependent manner. Muscle function was similarly improved in a dose-dependent manner irrespective of the vector production method. No apparent toxicity was observed in any mouse. Collectively, our results suggest that the biological potency of the AAV micro-dystrophin vector made by the scalable HSV method is comparable to that made by the TT method.
Keywords: adeno-associated virus, herpes simplex virus, transient transfection, micro-dystrophin, Duchenne muscular dystrophy, mdx4cv, scale-up, manufacture, potency, toxicity
Graphical Abstract
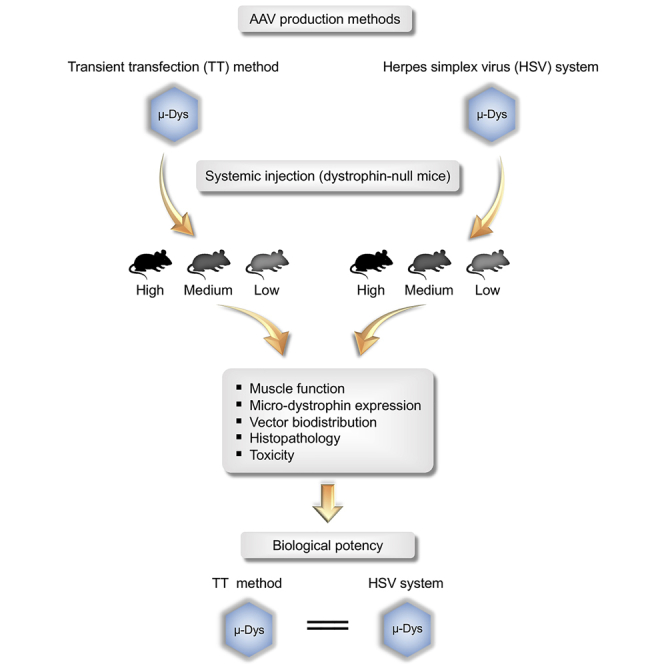
Hakim and colleagues compared AAV9 micro-dystrophin vectors produced by transient transfection and HSV-based methods in the murine DMD model by systemic delivery. They found that these vectors had comparable biological potency. Their findings paved the way to translate an HSV-based AAV production system for clinical application.
Introduction
Adeno-associated virus (AAV) is a non-enveloped single-stranded DNA virus.1,2 AAV-based gene therapy vectors have resulted in remarkable clinical successes in treating Leber congenital amaurosis and spinal muscular atrophy.3, 4, 5 Marketing approval of an AAV product by the US Food and Drug Administration (FDA) has already been achieved for these two conditions, and many more approvals are anticipated in the coming years. To date, AAV gene therapy has entered the clinical trial phase for many diseases. With the increased use of the AAV vector in human studies, there is an increased need for manufacturing high quantities of good manufacturing practice (GMP)-quality AAV vectors.
The reproductive life cycle of AAV requires helper viruses.6 Adenovirus and herpes simplex virus (HSV) are the two most commonly used helper viruses for AAV production. In terms of adenovirus, the helper function is accomplished by E1a, E1b, E2a, E4orf6, and virus-associated (VA) RNA.7 E1a and E1b are expressed in HEK293 cells.8 E2a, E4orf6, and VA RNA have been cloned into a helper plasmid.9,10 The development of the adenoviral helper plasmid has opened the door to a transfection-based, viral-free recombinant AAV production method called the transient transfection (TT) method.11,12 Briefly, three plasmids are co-transfected into HEK293 cells. These plasmids include the adenoviral helper plasmid, the AAV helper plasmid that encodes AAV replication proteins and capsids, and the vector plasmid that carries the transgene cassette. The adenoviral and AAV helper functions can also be provided from one plasmid to simplify the transfection protocol.13 In the vector plasmid, the therapeutic expression cassette is flanked by AAV inverted terminal repeats (ITRs). The ITR serves as the AAV replication origin and allows for selective packaging of the therapeutic expression cassette into AAV capsids. Following TT, mature AAV virions are harvested from culture medium and/or cell lysate and subsequently purified for in vivo gene therapy studies. AAV vectors generated using this method have been used extensively in preclinical studies to establish the proof of principle. The TT method has also been used to produce clinical-grade AAV vectors for human trials that require relatively low quantities of the vector.14
Systemic body-wide AAV delivery is essential for treating diseases such as Duchenne muscular dystrophy (DMD).15,16 In these cases, up to 1015–1016 viral genome (vg) particles of AAV vectors will likely be required for treating a single patient.17 It is unlikely that the cumbersome and labor-intensive TT method can generate enough AAV vectors to meet the needs of later-stage human studies and eventual commercialization. Hence, there is an urgent need to develop novel, large-scale AAV production methods that meet GMP regulation requirements.
Several systems are currently under development for large-scale AAV production. These include baculovirus-, HSV-, adenovirus-, and vaccinia virus-based systems, as well as producer cell lines.18, 19, 20, 21, 22 Of particular interest is the HSV-based system because this system is currently used to produce an AAV serotype-9 (AAV9) micro-dystrophin vector for systemic gene therapy in DMD patients.23
HSV is an enveloped, double-stranded DNA virus.24 A subset of HSV replication genes was found to provide helper functions for productive AAV replication.25,26 These include the UL5, UL8, UL9, UL29, UL30, UL42, and UL52 genes.27 Two HSV type 1-based AAV production systems have been developed, including the amplicon system and the recombinant HSV system.28,29 The latter was used to produce clinical-grade AAV for systemic micro-dystrophin gene therapy.23 Briefly, HEK293 cells were used for AAV production by co-infection with two replication-deficient recombinant HSV viruses, one carrying the AAV2 replication (rep) gene and the AAV9 capsid (cap) gene, and the other carrying the ITR-flanked micro-dystrophin expression cassette. After 2 days, the AAV9 micro-dystrophin vector was purified from infected cells.
While rodent studies suggest that AAV generated by the HSV system can effectively transduce tissues, it is unclear whether the biological potency of the AAV vectors are comparable when produced by the large-scale HSV-based system and the TT method. Lack of a side-by-side comparison greatly hinders translation and drug development. In this study, we blindly compared the biological activity of AAV9 micro-dystrophin vectors made by the HSV method and the TT method in the mdx4cv mouse, a commonly used DMD model. We found that AAV9 vectors made by the two methods yielded similar levels of micro-dystrophin expression and muscle protection at doses ranging from 5 × 1012 to 5 × 1014 vg/kg. Our results suggest that the HSV-based large-scale AAV production system has effectively retained the biological potency of the vector.
Results
Dystrophin Expression Was Not Influenced by the Manufacturing Method
To compare the biological activity of AAV vectors made by the TT method and the HSV method in a clinically relevant context, we performed a study in dystrophin-deficient mdx4cv mice using an AAV9 five-repeat micro-dystrophin vector that is in use in an ongoing clinical trial (ClinicalTrials.gov: NCT03368742).17,30,31 AAV stocks were manufactured and purified according to our recently published protocols at the University of Florida Powell Gene Therapy Center Vector Core.32,33 AAV vectors made by two methods showed similar purity on silver staining (Figure S1). The titers of the TT and HSV method AAV stocks were 5.68 × 1013 and 5.69 × 1013 vg/mL, respectively.
AAV9 micro-dystrophin vectors were injected blindly (without knowing whether the vectors were produced with the TT or HSV method) via the tail vein to young adult mdx4cv mice at clinically relevant doses. These include the high dose (n = 3 per production method, 1 × 1013 vg/mouse, 5.24–5.50 × 1014 vg/kg), medium dose (n = 6 per production method, 1 × 1012 vg/mouse, 5.22–5.30 × 1013 vg/kg), and low dose (n = 3 per production method, 1 × 1011 vg/mouse, 4.91–5.18 x1012 vg/kg) (Table 1). At the end of the study (3.34–3.77 months after injection), the diaphragm, extensor digitorum longus (EDL), heart, liver, quadriceps, and tibialis anterior (TA) were harvested (Table 1).
Table 1.
Summary of Experimental Mice
| Strain | Treatment Group | AAV Production Method | Sample Size (n) | Age at Injection (m) | Body Weight at Injection (g) | Dose (vg/Mouse) | Dose (vg/kg) | Age at Termination (m) | Study Duration (m) |
|---|---|---|---|---|---|---|---|---|---|
| BL6 | untreated | N/A | 3 | N/A | N/A | N/A | N/A | 5.00 ± 0.00 | N/A |
| mdx4cv | N/A | 7 | N/A | N/A | N/A | N/A | 5.43 ± 0.46 | N/A | |
| mdx4cv | high | TT | 3 | 1.83 ± 0.00 | 19.17 ± 0.83 | 1.00E+13 | 5.24E+14 | 5.17 ± 0.00 | 3.34 |
| HSV | 3 | 1.67 ± 0.00 | 18.20 ± 0.26 | 5.50E+14 | 5.19 ± 0.01 | 3.52 | |||
| medium | TT | 6 | 1.71 ± 0.04 | 19.15 ± 0.20 | 1.00E+12 | 5.22E+13 | 5.07 ± 0.04 | 3.36 | |
| HSV | 6 | 1.66 ± 0.01 | 19.07 ± 0.82 | 5.30E+13 | 5.20 ± 0.00 | 3.54 | |||
| low | TT | 3 | 2.03 ± 0.07 | 19.37 ± 0.74 | 1.00E+11 | 5.18E+12 | 5.60 ± 0.00 | 3.57 | |
| HSV | 3 | 1.90 ± 0.00 | 20.47 ± 0.95 | 4.91E+12 | 5.67 ± 0.00 | 3.77 |
Data are presented as mean ± standard error of the mean. N/A, not applicable; TT, AAV made with the transient transfection method; HSV, AAV made with the herpes simplex virus system.
Micro-dystrophin expression was first evaluated by immunofluorescence staining using a human dystrophin-specific antibody. Similar expression profiles were seen in mice injected with the TT method vectors and the HSV method vectors (Figure 1; Figure S2). At the high dose, we observed nearly saturated micro-dystrophin expression in all muscles we harvested. At the medium dose, we began to see a mosaic expression pattern. At the low dose, very few micro-dystrophin-positive fibers were detected (Figure 1; Figure S2). Quantification of micro-dystrophin-positive myofibers yielded similar results (Figure 2). Specifically, the high- and low-dose groups yielded the highest and lowest percentage of micro-dystrophin-positive myofibers, respectively, in all examined tissues. Except for the low-dose group in the TA muscle, in all other quantifications, no significant differences were detected between the TT method vectors and the HSV method vectors (Figure 2).
Figure 1.

Evaluation of Micro-dystrophin Expression by Immunostaining Using a Human Dystrophin-Specific Antibody
(A and B) Representative photomicrographs of dystrophin immunostaining in the (A) quadriceps and (B) heart. TT, AAV made with the transient transfection method; HSV, AAV made with the herpes simplex virus system. Cytosolic staining in the mdx4cv image and low dose images in panel A is due to cross-reaction of the secdary antibody with immunoglobulin infiltrated in damaged muscle cells
Figure 2.

Quantification of Micro-dystrophin Positive Myofibers
Percentage of myofibers expressing micro-dystrophin in the quadriceps, tibialis anterior (TA), diaphragm, extensor digitorum longus (EDL), and heart from mdx4cv mice injected with the AAV9 micro-dystrophin vector. TT, AAV made with the transient transfection method; HSV, AAV made with the herpes simplex virus system. ∗Significantly different from other dose groups. Data are presented as mean ± standard error of the mean. The difference was considered significant when p < 0.05.
To corroborate immunostaining results, we performed western blots on the quadriceps and heart (Figure 3). A strong micro-dystrophin band was detected in mice that received high-dose AAV vectors. The band intensity was reduced in mice treated with medium-dose vectors. Micro-dystrophin was not detected in low-dose group of mice (Figure 3A). On quantification, we observed the expected dose-response, with the high-, medium-, and low-dose groups yielding the highest, intermediate, and lowest micro-dystrophin levels, respectively (Figure 3B). With the exception of the medium-dose group in the quadriceps, no significant differences were detected between the TT method vectors and the HSV method vectors in the heart and quadriceps in other doses (Figure 3B).
Figure 3.

Evaluation of Micro-dystrophin Protein Expression by Western Blot
(A) Representative dystrophin western blot images showing abundant micro-dystrophin at the expected size in the quadriceps and heart of the treated mdx4cv mice. Vinculin was used as the load control. Fl-Dys, full-length dystrophin; μDys, micro-dystrophin. (B) LI-COR densitometry quantification of the intensity of the dystrophin band. ∗Significantly different from other dose groups. TT, AAV made with the transient transfection method; HSV, AAV made with the herpes simplex virus system. Please note that a nonspecific band was detected in heart western blot due to cross-reaction of the dystrophin antibody with an unknown protein in the mouse heart lysate. Data are presented as mean ± standard error of the mean. The difference was considered significant when p < 0.05.
AAV Vector Genome Biodistribution Was Not Altered by the Manufacturing Method
Next, we quantified AAV vector biodistribution in the diaphragm, heart, liver, and quadriceps (Figure 4). To accurately determine the AAV genome copy number in tissues, we designed two different TaqMan PCR assays targeting two unique junctional regions (R1-R16 and R17-R23) in the micro-dystrophin gene (Figure S3). The two TaqMan assays yielded consistent results in both heart and quadriceps (Figure S3). In subsequent analyses, data from the two PCR assays were pooled.
Figure 4.

TaqMan PCR Quantification of AAV Vector Genome Biodistribution
The AAV genome copy number in the heart, quadriceps, diaphragm, and liver from AAV-injected mdx4cv mice. TT, AAV made with the transient transfection method; HSV, AAV made with the herpes simplex virus system. ∗Significantly different from other dose groups. Data are presented as mean ± standard error of the mean. The difference was considered significant when p < 0.05.
In all dose groups, the highest vector copy number was found in the liver. It reached ∼1,300, ∼300, and ∼80 vg copies/diploid cell in the high-, medium-, and low-dose groups, respectively (Figure 4). The heart and quadriceps had similar amounts of the AAV genome. The diaphragm had the lowest AAV copy number. In the high-dose group, the diaphragm contained ∼50 vg copies/diploid cell, while the heart and quadriceps contained ∼80–100 vg copies/diploid cell (Figure 4). In all of the tissues studied, the vector genome copy number in the high-dose group was significantly higher than that of the medium- and low-dose groups. The medium-dose group appeared to have more vector genome copies than that of the low-dose group, but the difference did not reach statistical significance (Figure 4). There was no statistical difference in the AAV copy number between vectors made with the TT and HSV methods in the high-dose group. Interestingly, in the medium- and low-dose groups (except for the medium dose in the liver), the TT method vectors yielded a higher AAV copy number, which reached statistical significance for the medium and low dose in the quadriceps and the low dose in the liver (Figure 4).
The Manufacturing Method Did Not Affect Histological Rescue of the Muscle Disease
To study muscle pathology, we performed hematoxylin and eosin (H&E) staining and quantified myofiber centronucleation. Heart H&E staining was unremarkable (Figure S4). On skeletal muscle H&E staining, C57BL/6J (BL6) mice showed normal histological appearance, while untreated mdx4cv mice showed characteristic dystrophic pathology such as inflammation, muscle cell necrosis, centronucleation, and myofiber size variarion (Figure 5A). Qualitative evaluation of the H&E-stained images showed a clear trend of dose-dependent improvement of the histology in AAV-injected mdx4cv mice (Figure 5A). Inflammation and necrosis were absent and greatly reduced in mice treated with the high and medium dose, respectively. There was no apparent improvement in mice treated with the low dose. Nevertheless, similar histology was seen irrespective of the AAV manufacturing method (Figure 5A).
Figure 5.

Characterization of the Skeletal Muscle Histopathology
(A) Representative hematoxylin and eosin staining photomicrographs of the quadriceps from untreated BL6, untreated mdx4cv, and AAV-injected mdx4cv mice. TT, AAV made with the transient transfection method; HSV, AAV made with the herpes simplex virus system. (B) Quantification of myofibers with centrally localized myonuclei in the extensor digitorum longus (EDL), diaphragm, quadriceps, and tibialis anterior (TA) from mdx4cv mice injected with the TT method vectors and the HSV method vectors. ∗Significantly different from other dose groups. Data are presented as mean ± standard error of the mean. The difference was considered significant when p < 0.05.
The percentage of myofibers with centrally localized nuclei reflects muscle degeneration/regeneration. In the EDL, ∼55%, 63%, and 80% of the myofibers contained internal nuclei in the high-, medium-, and low-dose groups, respectively (Figure 5B). In the TA, ∼60%, 69%, and 75% of the myofibers contained internal nuclei in the high-, medium-, and low-dose groups, respectively. In the diaphragm, centronucleation was similar in low- and medium-dose AAV-treated mice (∼38%). However, a moderate reduction was seen in the diaphragm of high-dose AAV-treated mice (∼30%). In the quadriceps, the centronucleation rate was ∼72% in all treated mice. Overall, the dose response was seen in the EDL muscle (significantly different between the high and low dose) and TA muscle (significantly different between the high dose versus the other two doses) but not apparent in the quadriceps and diaphragm (Figure 5B). The levels of centrally nucleated myofibers were similar between mice treated with the TT method vector and the HSV method vector, with the exception of the low-dose group in the quadriceps (Figure 5B).
AAV Vectors Made with the TT Method and the HSV Method Were Equally Effective in Improving Muscle Function
To examine physiological outcomes, we measured the forces of the EDL muscle (Figure 6). The anatomical properties of the EDL muscle are shown in Table S1. As demonstrated by the elevated muscle weight and cross-sectional area, the untreated mdx4cv EDL muscle showed expected hypertrophy.34 EDL hypertrophy was ameliorated in a dose-dependent manner in mdx4cv mice that received AAV micro-dystrophin therapy. Vectors made with the TT method and HSV method showed similar treatment effects at each dose (Table S1).
Figure 6.

Quantitative Evaluation of Muscle Contractility in the Extensor Digitorum Longus
(A) Specific twitch (sPt) and tetanic (sPo) forces. *Significantly different from other groups. #Significantly different from that of BL6. (B) Eccentric contraction profiles. *BL6 significantly different from mdx4cv; #significantly different from mdx4cv; †significantly different from BL6. TT, AAV made with the transient transfection method; HSV, AAV made with the herpes simplex virus system. Data are presented as mean ± standard error of the mean. The difference was considered significant when p < 0.05.
Absolute twitch and tetanic forces of the untreated mdx4cv EDL muscle were comparable to those of the wild-type control BL6 EDL muscle (Table S1). However, specific twitch and tetanic forces of the untreated mdx4cv EDL muscle were significantly reduced (by ∼50%) compared to those of the BL6 EDL muscle (Figure 6A). The specific twitch force in all AAV-treated mdx4cv mice reached that of BL6 mice (Figure 6A). The TT method vector and the HSV method vector yielded comparable dose-dependent rescue of the specific tetanic force (Figure 6A). At the the high dose, both normalized the specific tetanic force. At the medium and low doses, both yielded specific tetanic forces significantly higher than those of untreated mdx4cv mice but lower than those of BL6 mice (Figure 6A).
Force reduction following consecutive cycles of eccentric contraction is a highly sensitive index to study dystrophic muscle function.30,34,35 The BL6 muscle was able to maintain ∼74% of the force following 10 cycles of eccentric contraction stress, while in the untreated mdx4cv muscle this value dropped to ∼22% (Figure 6B). After 10 cycles of eccentric contraction, the forces of the high-, medium-, and low-dose group mice were maintained at ∼70%, ∼50%, and ∼40%, respectively, of the starting level force (Figure 6B). At the high dose, the force response curves were identical between the TT method vector and the HSV method vector. Minor differences were seen at the medium and low doses, but did not reach statistical significance.
Neither the TT Method Vector nor the HSV Method Vector Induced Apparent Toxicity following Systemic Delivery
Irrespective of the vector dosage and preparation method, the intravenous AAV injection was well tolerated and no mortality occurred in any group following AAV injection. The mice were clinically observed daily throughout the entire study duration, and no apparent abnormality was noticed. Weekly bodyweight was recorded to evaluate body growth. Mice that received the TT method vector and the HSV method vector showed a similar growth rate (Figure 7A).
Figure 7.

Toxicity Assessment
(A) Weekly body weight of AAV-injected mdx4cv mice. TT, AAV made with the transient transfection method; HSV, AAV made with the herpes simplex virus system. (B) Blood chemistry. BUN, blood urea nitrogen; GLDH, glutamate dehydrogenase. ∗Significantly different from other dose groups. (C) Representative photomicrographs of hematoxylin and eosin staining of the liver from untreated BL6, untreated mdx4cv, and AAV injected mdx4cv mice. TT, AAV made with the transient transfection method; HSV, AAV made with the herpes simplex virus system. Data are presented as mean ± standard error of the mean. The difference was considered significant when p < 0.05.
Liver and kidney functions were examined using the plasma collected at the end of the study (Figure 7B). No significant difference was detected in the levels of blood urea nitrogen, creatinine, albumin, total protein, globulin, and glutamate dehydrogenase (GLDH) between TT and HSV vector-treated mice (Figure 7B). In the context of DMD, the commonly used aspartate aminotransferase and alanine aminotransferase cannot accurately reflect liver damage due to ongoing muscle disease,36,37 GLDH is recommended as the only reliable parameter to evaluate liver function in DMD studies,38,39 Interestingly, the GLDH level in the HSV method vector-treated mice appeared higher than that of the TT method vector-treated mice in all three dosages, although none reached statistical significance. To further investigate this, we examined liver histology. H&E staining revealed no difference in the histological appearance of the liver in all experimental mice (Figure 7C). No signs of hepatitis, cirrhosis, or necrosis were seen in mice treated with the HSV method vector.
Discussion
In this study, we blindly compared the biological potency of the AAV9 micro-dystrophin vector made by the TT method and the HSV method in mdx4cv mice. We observed similar levels of micro-dystrophin expression and disease rescue in a dose-dependent manner at doses ranging from 5 × 1012 to 5 × 1014 vg/kg. Our results suggest that the biological activity of a therapeutic AAV vector is not compromised when it is manufactured using the scalable HSV production system.
DMD is an X-linked lethal muscle disease caused by dystrophin deficiency.40 AAV micro-dystrophin therapy holds promise to significantly improve the life quality of DMD patients.16,23 However, because muscle accounts for ∼40% of the body mass and is distributed throughout the body, a gigantic quantity of AAV particles has to be injected into the circulation in order to treat all affected muscles, including the heart, in a single patient. In the canine DMD model, as high as 6 × 1014 vg/kg AAV vectors have been used.41 Several clinical trials are ongoing to evaluate the safety and potential therapeutic benefits of AAV micro-dystrophin gene therapy in DMD patients.23,42,43 In these human studies, the AAV dose ranges from 5 × 1013 to 3 × 1014 vg/kg.23,42 It is estimated that a clinically meaningful improvement may likely require mosaic dystrophin expression in 50% or more myofibers.23,44,45 This level of expression was detected only at the dose 2 × 1014 vg/kg or higher in ongoing clinical trials.42 Given the average body weight of a 5-year-old DMD patient be ∼20 kg,46,47 we will need to inject ≥4 × 1015 vg AAV particles/patient to affected boys that are 5 years or older. This dose greatly exceeds the doses used for treating many other diseases such as hemophilia and retinal diseases.
Ongoing systemic AAV micro-dystrophin trials are built on compelling efficacy data obtained from the rodent and canine DMD models.16,23 The AAV vector used in most of these studies was produced using the TT method. While the TT method can be readily adapted to make a variety of AAV vectors (such as different capsids and/or transgene cassettes) to meet the study needs in a research laboratory, the inherent scaling limitation of the TT method represents a significant challenge for large-scale production of GMP-grade vectors48, 49, 50, 51. According to an estimate, it requires more than 100 cell stacks to produce 1 × 1015 vg of clinical vectors in a typical GMP manufacturing campaign with the TT method.50 Multiple production batches have to be combined to treat even a single DMD patient.
Unlike the TT method, the HSV method has the capacity to produce >1 × 1016 vg AAV particles in a single production campaign using a 100-L bioreactor.50 The scalability of the HSV method offers a clear advantage in producing large quantities of the micro-dystrophin vector for future commercialization if this vector is proven safe and clinically effective. Given that the HSV method was not used to generate the micro-dystrophin vector for preclinical animal studies, it is imperative to prove the vectors made the HSV and TT methods are functionally equivalent.
To stringently test vectors made by two different methods, we have taken several precautions in our study. First, the same facility was used to produce the AAV vector to avoid inter-facility differences. Second, all vectors were purified using the same protocol at the University of Florida Powell Gene Therapy Center to minimize purification protocol-associated bias. Third, AAV purity was confirmed by silver staining. Fourth, the AAV titer was independently verified at the University of Missouri to ensure the accuracy of dosing. Fifth, a human dystrophin-specific antibody was used to distinguish the expression of human micro-dystrophin from that of endogenous revertant mouse dystrophin. Sixth and most importantly, we carefully followed the pre-established blinding protocol throughout the study to avoid unintended interferences of investigators. Specifically, the vector manufacturing and animal studies are carried out in two independent institutions; the vector production method was blinded to investigators who performed animal studies, the experimental mice were randomly assigned, the investigators involved in AAV delivery did not participate in physiological assays, and all tissue samples were coded during the analysis.
To thoroughly evaluate the biological potency of the vector, we performed the study in three different doses. Based on the literature and our own experience,15 we intentionally chose 5 × 1014 vg/kg (1 × 1013 vg/mouse) as the high dose, 5 × 1013 vg/kg (1 × 1012 vg/mouse) as the medium dose, and 5 × 1012 vg/kg (1 × 1011 vg/mouse) as the low dose (Table 1). The high, medium, and low dose should yield saturated, mosaic, and nominal transduction, respectively. On dystrophin immunostaining, we indeed observed the expected dose-dependent expression pattern (Figures 1 and 2; Figure S2). Western blot confirmed the dose dependence of the micro-dystrophin level in the whole muscle lysate (Figure 3). Interestingly, the total micro-dystrophin level in the lysate only reached ∼30% of normal (the full-length dystrophin level in wild-type mice), even in the high-dose group (Figure 3B). In nearly all quantitative analyses (Figures 2 and 3B), we did not detect significant differences between vectors made by the TT method and the HSV method. The only exception is the quadriceps western blot data in the medium-dose group (Figure 3B). Here, significantly more micro-dystrophin was detected in mice injected with the TT method vector. We currently do not have an explanation for this unexpected finding.
Micro-dystrophin expression is the end product of AAV transduction. To more directly compare the transduction efficiency of the TT method vector and the HSV method vector, we quantified the vector genome copy number (Figure 4). Consistent with dystrophin quantification data, we did not see a significant difference between the two AAV manufacturing methods in the high-dose groups. Intriguingly, in the medium- and low-dose experimental groups, mice that received the TT method vector consistently showed a higher AAV copy number in tissues we examined (except for the medium dose in the liver). In some cases, the difference was statistically significant (the medium and low dose in the quadriceps and low dose in the liver) (Figure 4). Future studies are needed to understand this observation.
The ultimate goal of our study is to treat muscle disease and improve muscle function. To this end, we compared the disease rescue between the TT method vector and the HSV method vector (Figures 5 and 6; Table S1). With H&E staining, muscle inflammation and necrosis were markedly attenuated in mice treated with the high- and medium-dose vectors, but not the low-dose vectors (Figure 5A). To quantify histological changes, we counted the percentage of myofibers with centrally localized nuclei (Figure 5B). In the EDL and TA muscle, we saw a clear trend of the dose response. However, the dose response was not observed in the quadriceps. The variation seen in different muscles was not unexpected. We previously studied AAV9 micro-dystrophin therapy in the D2-mdx mouse, a different mouse model for DMD.30 In that study, we also found that micro-dystrophin significantly reduced centronucleation in the EDL and TA muscle, but not the quadriceps.30 Although in regard to centronucleation, the response to micro-dystrophin therapy varied among different muscles, we did not see a significant difference between the TT method vector and the HSV method vector in the same muscle, except for the low-dose group in the quadriceps (Figure 5B).
In muscle function assays, the dose responses were apparent in terms of the muscle weight, specific tetanic force, and eccentric contraction profile (Figure 6; Table S1). The best rescue was seen in high-dose AAV-treated mice, and the least rescue was seen in low-dose AAV-treated mice. Eccentric contraction challenge has been considered a more sensitive assay to detect functional deficiency in dystrophic muscle.52 Indeed, the specific tetanic force and eccentric contraction profile were normalized in mice treated with high-dose AAV (Figure 6). Interestingly, mice treated with low-dose AAV showed significant improvement in the specific forces and eccentric contraction profiles although nominal micro-dystrophin was detected in these mice (Figures 2, 3, and 6). This observation corroborates a finding that has been noticed in many studies before.34,53, 54, 55 Specifically, even residual level dystrophin expression may still offer therapeutic benefits. Despite the observed dose responses, consistent with what we saw in other assays (micro-dystrophin expression, AAV genome copy number, and muscle histology data), the vectors made with the TT method and HSV method yielded similar levels of physiological protection.
Safety is paramount in any gene therapy study. Although this is not the primary focus of the current study, some preliminary analyses were explored. In particular, we did not see clinically dramatic differences between mice injected with the TT method vector and the HSV method vector. Similar growth curves were observed in AAV-treated mice irrespective the method of AAV manufacturing (Figure 7A). In addition, we did not see a significant difference in blood parameters for the liver or kidney function (Figure 7B). Histology examination of the liver also did not show apparent abnormalities in AAV-injected mice (Figure 7C).
The outcome of a gene therapy study is decided by many factors such as the nature of the therapeutic gene, the level, timing, and location of expression, the quantity of the vector, and the route of vector administration. While the method of vector manufacturing will no doubt influence the outcome, very few studies have investigated this critical factor. In this study, we compared the biological activity of AAV9 micro-dystrophin vectors made by the TT and HSV methods in a DMD mouse model. We showed that these vectors had similar biological potency. Our results suggest that the scalable HSV method represents a viable option to overcome the AAV production scale-up barrier in systemic DMD gene therapy. Additional characterization of the product-related impurities (such as empty capsids and encapsidation of non-vector DNA) will further demonstrate the suitability of the HSV method.56,57
Materials and Methods
Experimental Mice
All animal experiments were approved by the Animal Care and Use Committee of the University of Missouri and were in accordance with NIH guidelines. All animal experiments were conducted at the University of Missouri. Dystrophin-deficient mdx4cv mice (stock no. 002378) and normal control BL6 mice (stock no. 000664) were originally purchased from The Jackson Laboratory (Bar Harbor, ME, USA). Experimental mice were generated in-house in a specific pathogen-free barrier facility at the University of Missouri using founders from The Jackson Laboratory. The genotype of the mice was confirmed using our published protocol.58 Only female mice were used in the study. All mice were maintained in a specific-pathogen free animal care facility on a 12-h light (25 lux)/12-h dark cycle with access to PicoLab rodent diet 20 (#5053; LabDiet, St. Louis, MO, USA) and autoclaved municipal tap water ad libitum. The room temperature and relative humidity were maintained at 68°F ± 2°F and 50% ± 20%, respectively. All animals were observed daily for their general condition and well-being. All mice had a unique identification number (ear tag) that was randomly assigned at the time of weaning.
Cell Lines
All cell culture, plasmid and recombinant HSV stock generation, and AAV production and purification were conducted at the University of Florida Powell Gene Therapy Center Vector Core.32 HEK293 cells (catalog no. CRL-1537, American Type Culture Collection, Manassas, VA, USA) were maintained in Dulbecco’s modified Eagle’s medium (HyClone, GE Healthcare, Logan, UT, USA) supplemented with 5% fetal bovine serum (Corning Life Sciences, Corning, NY, USA) and 1% antibiotic/antimycotic (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) in 10-stack Corning CellSTACK culture chambers (Corning Life Sciences, Tewksbury, MA, USA).
Plasmids
The AAV9 helper plasmid, pDG-UF9-KanR, was described before.32 It contains the AAV2 rep gene and the AAV9 cap gene under their endogenous promoters. It also contains the adenoviral E2a gene, E4 gene, and VA RNA.13 The micro-dystrophin plasmid, pTR-CK8-hDys5, was described before.31 It contains a codon-optimized human micro-dystrophin gene under transcriptional regulation of the muscle-specific CK8 promoter and a 49-bp synthetic pA signal. The micro-dystrophin expression cassette is flanked by AAV2 ITRs. The micro-dystrophin contains the dystrophin N-terminal domain and cysteine-rich domain, hinges 1 and 4, and spectrin-like repeats 1, 16, 17, 23, and 24.30,31 Plasmids used in the study were manufactured by Aldevron (Fargo, ND, USA).
Recombinant HSV Stock
Recombinant herpes viruses rHSV-UF AAV2/9 and rHSV-CK8-hDys5 were generated by homologous recombination on the backbone of d27-1, a replication-defective HSV vector according to a previously published method.29,32 The rHSV-UF AAV2/9 virus carries the AAV2 rep gene and the AAV9 cap gene. The rHSV-CK8-hDys5 virus carries the same micro-dystrophin expression cassette and flanking ITRs as in described in pTR-CK8-hDys5.
AAV Production by the TT Method
The standard calcium phosphate transfection protocol was used.32,59 Briefly, plasmid pTR-CK8-hDys5 and pDG-UF9-KanR were transfected at a 1:1 molar ratio in 70% confluent HEK293 cells and incubated for 72 h. On the day of harvest, cells were detached using 1× Dulbecco’s phosphate-buffer saline containing 5 mM ethylenediaminetetraacetic acid, rinsed with 1× Dulbecco’s phosphate-buffer saline, and pelleted at 526 × g at 4°C for 15 min. The pellet was collected and stored in −80°C for subsequent purification.
AAV Production by the HSV Method
HEK293 cells were infected when cells reached 80%–100% confluency. Cells were counted on the day of infection. Recombinant HSV stocks were mixed with fresh Dulbecco’s modified Eagle’s medium and applied to HEK293 cells at the multiplicity of infection of 2 for rHSV-CK8-hDys5 and 12 for rHSV-UF AAV2/9. At 48–52 h after infection, cells were dislodged from the flask by gentle shaking. Cells were pelleted at 935 × g at 4°C for 20 min. The cell pellet was resuspended in phosphate-buffer saline and re-pelleted using the same condition. The pellet was collected and stored in −80°C for subsequent purification.
AAV Purification
The pellet was thawed at 37°C and processed for virus purification as previously described.32,33 Briefly, the cell pellet was resuspended in sodium citrate buffer (final concentration 30.63 mM, prepared in water for injection) containing 1 mM MgCl2 and 30 U/mL Benzonase (EMD Chemicals, Billerica, MA, USA) for 1 h at 37°C with regular swirling. Citric acid (prepared in water for injection) was added to acidify slurry and initiate protein flocculate formation. The protein flocculate was clarified by centrifugation at 3,729 × g at 22°C for 10 min. The virus-containing supernatant was loaded onto a sanitized HiPrep SP fast flow (FF) column (20-mL bed volume, GE Healthcare, Pittsburg, PA, USA) that had been pre-equilibrated with buffer A (8 mM sodium citrate, 16 mM citric acid) and buffer B (42.8 mM sodium citrate, 7.2 mM citric acid, 0.5 M sodium chloride). AAV was eluted as a discrete peak (SP peak, 40% buffer B) in a final formulation of 22 mM sodium citrate, 13.1 mM citric acid, and 200 mM sodium chloride (∼pH 4.8). The sample was then sterile filtered with a 0.22-μm Millex-GV syringe filter (EMD Millipore). Finally, AAV was concentrated by tangential flow filtration using a 100K nominal molecular weight cutoff (NMWC) MidGee hollow-fiber cartridge (GE Healthcare), filter sterilized, and shipped to the University of Missouri on dry ice for animal studies. Different lot numbers (lot nos. 120215 and 121815) were assigned to AAV vectors made by the TT method and AAV made by the HSV method at the University of Florida. Prior to animal studies, AAV vectors were dialyzed in HEPES buffer (150 mM NaCl, 20 mM HEPES [pH 7.4]) as described before.12 The corresponding AAV preparation method of the AAV lot was blinded to investigators at the University of Missouri until all data were collected and analyzed.
AAV Titer Determination
The AAV titer was determined by quantitative PCR (qPCR) using the iTaq Fast SYBR Green supermix with ROX (Bio-Rad, Hercules, CA) in an ABI 7900 HT qPCR machine (Applied Biosystems, Foster City, CA, USA). A set of primers were designed to amplify a fragment in the CK8 promoter. The forward primer is 5′-AGCCCCTCCTGGCTAGTCAC-3′, and the reverse primer is 5′-CCTGAGTGTCTGTCTGTGCTGTG-3′. The qPCR reaction was carried out under the following conditions: 20 s at 95°C, followed by 40 cycles: 15 s at 95°C and 60 s at 60°C. A dissociation curve step was applied at the end of the reaction to confirm the primer efficiency using the following conditions: 15 s at 95°C, 15 s at 60°C, and 15 s at 95°C. The threshold cycle (Ct) value of each reaction was converted to the vector genome copy number by measuring against plasmid standard series representing 1 × 106 to 1 × 1011 vg/μL at a log10 increments.
AAV Purity Evaluation
AAV purity was examined by silver staining. Briefly, purified AAV vectors were denatured at 95°C for 5 min and then chilled on ice for 2 min. Denatured AAV vectors were electrophoresed on a 4% stacking/8% separating sodium dodecyl sulfate (SDS)-polyacrylamide gel at 60 V for ∼10 min until the dye line entered the separating gel. Electrophorese was continued at 140 V for approximately 1 more h until the dye line reached the bottom of the separating gel. The electrophoresed gel was stained with the Pierce silver stain kit (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions.
AAV Administration
AAV vectors were injected systemically via the tail vein to a mouse during a period of 60 s accordingly to our published protocol.30 Delivery was confirmed by the absence of resistance during injection and a lack of swelling of the tail after injection. Three AAV dose groups were included in the study including the high (1 × 1013 vg/mouse), medium (1 × 1012 vg/mouse), and low (1 × 1011 vg/mouse) doses (Table 1). A total of eight group mice were assigned to the study. Three groups were mdx4cv mice treated with the lot no. 120215 vector at the high (three mice), medium (six mice), and low (three mice) doses. Three groups were mdx4cv mice treated with the lot no. 121815 vector at the high (three mice), medium (six mice), and low (three mice) doses. One group was un-injected mdx4cv mice (seven mice) and one more group was BL6 mice (three mice) that received the excipient only (Table 1).
Tissue Collection
Mice were euthanized at the end of the study according to the protocols approved by the University of Missouri Animal Care and Use Committee. The TA, EDL, quadriceps, diaphragm, heart, and liver were carefully dissected out and snap-frozen in liquid nitrogen for protein and DNA analysis or in liquid nitrogen-cooled isopentane in the optimal cutting temperature compound (Sakura Finetek, Torrance, CA, USA) for morphological analysis.
Morphological Analysis
10-μm cryosections were used for staining. General muscle histopathology was revealed with H&E staining. Dystrophin expression was evaluated by immunofluorescence staining using Dys-3 (1:20, Vector Laboratories, Peterborough, UK), a species-specific dystrophin monoclonal antibody that recognizes the hinge 1 region of human dystrophin but does not cross-react with mouse dystrophin. Slides were viewed at the identical exposure setting using a Nikon E800 fluorescence microscope. Images were taken with a QImaging Retiga 1300 camera. Centrally nucleated myofibers were determined from digitalized H&E-stained images using the Fiji imaging software (https://fiji.sc).60 The percentage of dystrophin-positive cells was quantified from digitalized dystrophin immunostaining images using the Fiji imaging software (https://fiji.sc).
Western Blot
Heart and quadriceps muscle were homogenized in a homogenization buffer containing 10% SDS, 5 mM ethylenediaminetetraacetic acid, 62.5 mM Tris-HCl (pH 6.8), and 2% protease inhibitor (Roche, Indianapolis, IN, USA) using a tissue homogenizer (Bullet Blender Storm 24, Next Advance, Troy, NY, USA) at the speed set 12 in the machine for 8 min at 4°C. The homogenate was centrifuged at 14,000 rpm for 3 min in an Eppendorf centrifuge (model 5417C; Brinkmann Instruments, Westbury, NY, USA). The total protein concentration in the supernatant was measured using the DC (detergent-compatible) assay kit (Bio-Rad, Hercules, CA, USA). 100–150 μg of protein was denatured at 95°C for 5 min, chilled on ice for 2 min, and then separated on a 3% stacking/6% separating SDS-polyacrylamide gel at 100 V. Proteins were transferred to a 0.45-μm polyvinylidene fluoride (PVDF) membrane at 50 V for 10 h at 4°C in Towbin’s buffer containing 10% methanol. The blot was blocked for 1 h at room temperature with 5% non-fat dry milk in Tris-buffered saline (TBS) with Tween 20 (TBST) solution (containing 1× TBS and 0.1% Tween 20) for 1 h at room temperature. Subsequently, the PVDF membrane was incubated with Mannex44A, a mouse monoclonal antibody raised against the repeat 17 of human dystrophin (1:100, a gift from Dr. Glenn Morris, The Robert Jones and Agnes Hunt Orthopaedic Hospital, UK) in 5% milk/TBST overnight at 4°C.61 The membrane was washed in TBST three times for 10 min each and then incubated with horseradish peroxidase-conjugated goat anti-mouse immunoglobulin G (IgG) secondary antibody (1:2,000 dilution in TBST, Santa Cruz, Dallas, TX, USA) for 1 h at room temperature. After another round of TBST wash (three times, 10 min each), signals were detected using the Clarity Western enhanced chemiluminescence (ECL) substrate (Bio-Rad, Hercules, CA, USA) and visualized using the Li-COR Odyssey imaging system. The membrane was then washed in TBST three times for 10 min each at room temperature. The membrane was then incubated with a rabbit polyclonal antibody against vinculin (1:2,000, ab155120, Abcam, Cambridge, MA, USA) in 5% milk/TBST overnight at 4°C. The membrane was washed in TBST three times for 10 min each and then incubated with horseradish peroxidase-conjugated goat anti-rabbit IgG secondary antibody (1:1,000 dilution in TBST, MilliporeSigma, Burlington, MA, USA) for 1 h at room temperature. After another round of TBST wash (three times, 10 min each), signals were detected using the Clarity Western ECL substrate (Bio-Rad, Hercules, CA, USA) and visualized using the LI-COR Odyssey imaging system. Densitometry quantification of the band intensity was performed using the Li-COR Image Studio version 5.0.21 software (https://www.licor.com). The relative intensity of the full-length dystrophin and micro-dystrophin protein bands were normalized to the corresponding vinculin band (loading control) in the same blot. The relative intensity of the micro-dystrophin band was normalized to that of untreated BL6 control.
Tissue AAV Vector Genome Copy Number Quantification
Genomic DNA was extracted from liquid nitrogen-frozen tissue samples. DNA concentration was quantified with a Qubit double-stranded DNA (dsDNA) high-sensitivity (HS) assay kit (Thermo Fisher Scientific, Waltham, MA, USA). Quantitative TaqMan PCR assays were performed using the TaqMan Universal PCR master mix (Thermo Fisher Scientific, Waltham, MA, USA) in an ABI 7900HT qPCR machine (Applied Biosystems, Foster City, CA, USA) to detect either the R1-R16 or R17-R23 junction in the vector genome (Figure S4). For the R1-R16 junction PCR reaction, the forward primer is 5′-TGGCCAGCATGGAAAAGCA-3′, the reverse primer is 5′-GGTGATCTCGGTCAGGTAGGT-3′, and the probe is 5′-CAACCTGCACAGCTACG-3′. For the R17-23 junction PCR reaction, the forward primer is 5′-TGCAAGCAGCTGTCCGA-3′, the reverse primer is 5′-AGATGCAGCCGCTTCCA-3′, and the probe is 5′-CTGGTCGCTCTGTTCTT-3′. The qPCR reaction was carried out under the following conditions: 10 min at 95°C, followed by 40 cycles: 15 s at 95°C and 1 min at 60°C. The Ct value of each reaction was converted to the vector genome copy number by measuring against the copy number standard curve of known amount of the pTR-CK8-hDys5 plasmid. The data were reported as the vector genome copy number per diploid genome.
Skeletal Muscle Function Assay
The function of the EDL muscle (twitch force, tetanic force, and eccentric contraction profile) was evaluated ex vivo according to our published protocols.62,63 Experimental mice were anesthetized via intraperitoneal injection of a cocktail containing 25 mg/mL ketamine, 2.5 mg/mL xylazine, and 0.5 mg/mL acepromazine at 2.5 μL/g body weight. The EDL muscle was gently dissected and mounted to a muscle test system (Aurora Scientific, Aurora, ON, Canada). Muscle force was evaluated with a 305B dual-mode servomotor transducer (Aurora Scientific, Aurora, ON, Canada). The optimal muscle length (Lo) was determined based on the isometric twitch force. Briefly, twitch stimulation was applied while the muscle was strained at different lengths. The length that yielded the highest force was defined as Lo, and the muscle length was measured with an electronic digital caliper (McMaster, Chicago, IL, USA). After the identification of Lo, the absolute twitch force (Pt) was measured at 1 Hz and the optimal maximal isometric tetanic force was measured at 150 Hz. After a 2-min rest, the percentage of the force drop through 10 repetitive cycles of eccentric contraction was determined. In each cycle the muscle was stimulated for 500 ms using the frequency that produced the absolute tetanic force (Po). After a 300-ms stimulation, the muscle was stretched by 10% Lo at 0.5 Lo/s for 200 ms. The muscle was rested for 1 min between each cycle. At the end of the experiment, the EDL muscle was gently removed and the muscle weight was measured. Data acquisition and analysis were performed with the Dynamic Muscle Control and Analysis software (Aurora Scientific). The specific muscle force was calculated by dividing the absolute muscle force with the muscle cross-sectional area (CSA). Muscle CSA was calculated according to the following equation: CSA = (muscle mass, in g)/[(muscle density, in g/cm3) × (length ratio) × (optimal muscle length, in cm)]. 1.06 g/cm3 is used as the muscle density.64 The length ratio refers to the ratio of the optimal fiber length to the optimal muscle length. The length ratio for the EDL muscle is 0.44,65,66
Clinical Chemistry Analysis
Plasma was collected at the end of the study and stored at −80°C. The clinical chemistry analysis was performed at the University of Missouri Veterinary Medical Diagnostic Laboratory (University of Missouri, Columbia, MO, USA) using a Beckman Coulter AU480 analyzer (Beckman Coulter, Brea, CA, USA) and in accordance with the standard protocols. Parameters were determined, including blood urea nitrogen, creatinine, albumin, total protein, globulin, and GLDH.
Statistical Analysis
Data are presented as mean ± standard error of mean (SEM). One-way ANOVA or two-way ANOVA with Tukey’s multiple comparison test were performed for statistical analysis for more than two-group comparisons. An unpaired t test was used for two-group comparisons. All statistical analyses were performed using GraphPad Prism software version 7.0 (GraphPad, La Jolla, CA, USA. The difference was considered significant when p < 0.05.
Author Contributions
Conceived and designed experiments: C.H.H., J.S.S., B.J.B., and D.D. AAV production: N.C., L.A.-S., and B.J.B. Performed the experiments: C.H.H., N.C., L.P.W., H.T.Y., Y.Y., K.Z., K.K., and X.P. Analyzed the data: C.H.H., L.P.W., H.T.Y., J.S.S., N.N.Y., and D.D. Provided critical reagents: J.S.C. Wrote the paper: C.H.H. and D.D. All authors edited the paper and approved the submission.
Conflicts of Interest
J.S.S. is an employee of Solid Biosciences. D.D. and J.S.C. are members of the scientific advisory board for Solid Biosciences and equity holders of Solid Biosciences. D.D., J.S.C., and Y.Y. are inventors on patents that were licensed to Solid Biosciences. The Duan Laboratory and Chamberlain Laboratory have received research support from Solid Biosciences. The Duan Laboratory has received research support unrelated to this project from Edgewise Therapeutics. The remaining authors declare no competing interests.
Acknowledgments
This work was supported in part by Solid Biosciences and by grants from the National Institutes of Health (NS-90634 to D.D. and AR-40864 to J.S.C.); the Intramural/Extramural Research Program of the National Center for Advancing Translational Sciences (to C.H.H. and N.N.Y.); the Department of Defense (MD130014 to D.D.); Jesse’s Journey—The Foundation for Gene and Cell Therapy (to D.D.); and by the Jackson Freel DMD Research Fund (to D.D.). We thank Jacqueline Louderman, Samantha Metzger, Alex Hinken, and Matthew Burke for technical assistance.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtm.2020.07.004.
Supplemental Information
References
- 1.Flotte T.R., Berns K.I. Adeno-associated virus: a ubiquitous commensal of mammals. Hum. Gene Ther. 2005;16:401–407. doi: 10.1089/hum.2005.16.401. [DOI] [PubMed] [Google Scholar]
- 2.Carter B.J. Adeno-associated virus and the development of adeno-associated virus vectors: a historical perspective. Mol. Ther. 2004;10:981–989. doi: 10.1016/j.ymthe.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 3.Russell S., Bennett J., Wellman J.A., Chung D.C., Yu Z.F., Tillman A., Wittes J., Pappas J., Elci O., McCague S. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet. 2017;390:849–860. doi: 10.1016/S0140-6736(17)31868-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mendell J.R., Al-Zaidy S., Shell R., Arnold W.D., Rodino-Klapac L.R., Prior T.W., Lowes L., Alfano L., Berry K., Church K. Single-dose gene-replacement therapy for spinal muscular atrophy. N. Engl. J. Med. 2017;377:1713–1722. doi: 10.1056/NEJMoa1706198. [DOI] [PubMed] [Google Scholar]
- 5.Wang D., Tai P.W.L., Gao G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 2019;18:358–378. doi: 10.1038/s41573-019-0012-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Geoffroy M.C., Salvetti A. Helper functions required for wild type and recombinant adeno-associated virus growth. Curr. Gene Ther. 2005;5:265–271. doi: 10.2174/1566523054064977. [DOI] [PubMed] [Google Scholar]
- 7.Janik J.E., Huston M.M., Rose J.A. Locations of adenovirus genes required for the replication of adenovirus-associated virus. Proc. Natl. Acad. Sci. USA. 1981;78:1925–1929. doi: 10.1073/pnas.78.3.1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Graham F.L., Smiley J., Russell W.C., Nairn R. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J. Gen. Virol. 1977;36:59–74. doi: 10.1099/0022-1317-36-1-59. [DOI] [PubMed] [Google Scholar]
- 9.Xiao X., Li J., Samulski R.J. Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J. Virol. 1998;72:2224–2232. doi: 10.1128/jvi.72.3.2224-2232.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matsushita T., Elliger S., Elliger C., Podsakoff G., Villarreal L., Kurtzman G.J., Iwaki Y., Colosi P. Adeno-associated virus vectors can be efficiently produced without helper virus. Gene Ther. 1998;5:938–945. doi: 10.1038/sj.gt.3300680. [DOI] [PubMed] [Google Scholar]
- 11.Su Q., Sena-Esteves M., Gao G. Production of recombinant adeno-associated viruses (rAAVs) by transient transfection. Cold Spring Harb. Protoc. 2020;2020:095596. doi: 10.1101/pdb.prot095596. [DOI] [PubMed] [Google Scholar]
- 12.Shin J.-H., Yue Y., Duan D. Recombinant adeno-associated viral vector production and purification. Methods Mol. Biol. 2012;798:267–284. doi: 10.1007/978-1-61779-343-1_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grimm D., Kern A., Rittner K., Kleinschmidt J.A. Novel tools for production and purification of recombinant adenoassociated virus vectors. Hum. Gene Ther. 1998;9:2745–2760. doi: 10.1089/hum.1998.9.18-2745. [DOI] [PubMed] [Google Scholar]
- 14.Wright J.F. Transient transfection methods for clinical adeno-associated viral vector production. Hum. Gene Ther. 2009;20:698–706. doi: 10.1089/hum.2009.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Duan D. Systemic delivery of adeno-associated viral vectors. Curr. Opin. Virol. 2016;21:16–25. doi: 10.1016/j.coviro.2016.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chamberlain J.R., Chamberlain J.S. Progress toward gene therapy for Duchenne muscular dystrophy. Mol. Ther. 2017;25:1125–1131. doi: 10.1016/j.ymthe.2017.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Duan D. Micro-dystrophin gene therapy goes systemic in Duchenne muscular dystrophy patients. Hum. Gene Ther. 2018;29:733–736. doi: 10.1089/hum.2018.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Virag T., Cecchini S., Kotin R.M. Producing recombinant adeno-associated virus in foster cells: overcoming production limitations using a baculovirus-insect cell expression strategy. Hum. Gene Ther. 2009;20:807–817. doi: 10.1089/hum.2009.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Clément N., Knop D.R., Byrne B.J. Large-scale adeno-associated viral vector production using a herpesvirus-based system enables manufacturing for clinical studies. Hum. Gene Ther. 2009;20:796–806. doi: 10.1089/hum.2009.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang H., Xie J., Xie Q., Wilson J.M., Gao G. Adenovirus-adeno-associated virus hybrid for large-scale recombinant adeno-associated virus production. Hum. Gene Ther. 2009;20:922–929. doi: 10.1089/hum.2009.125. [DOI] [PubMed] [Google Scholar]
- 21.Wang Q., Wu Z., Zhang J., Firrman J., Wei H., Zhuang Z., Liu L., Miao L., Hu Y., Li D. A robust system for production of superabundant VP1 recombinant AAV vectors. Mol. Ther. Methods Clin. Dev. 2017;7:146–156. doi: 10.1016/j.omtm.2017.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chadeuf G., Salvetti A. Stable producer cell lines for adeno-associated virus (AAV) assembly. Cold Spring Harb. Protoc. 2010;2010:5496. doi: 10.1101/pdb.prot5496. [DOI] [PubMed] [Google Scholar]
- 23.Duan D. Systemic AAV micro-dystrophin gene therapy for Duchenne muscular dystrophy. Mol. Ther. 2018;26:2337–2356. doi: 10.1016/j.ymthe.2018.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McGeoch D.J., Cook S., Dolan A., Jamieson F.E., Telford E.A. Molecular phylogeny and evolutionary timescale for the family of mammalian herpesviruses. J. Mol. Biol. 1995;247:443–458. doi: 10.1006/jmbi.1995.0152. [DOI] [PubMed] [Google Scholar]
- 25.Mishra L., Rose J.A. Adeno-associated virus DNA replication is induced by genes that are essential for HSV-1 DNA synthesis. Virology. 1990;179:632–639. doi: 10.1016/0042-6822(90)90130-j. [DOI] [PubMed] [Google Scholar]
- 26.Weindler F.W., Heilbronn R. A subset of herpes simplex virus replication genes provides helper functions for productive adeno-associated virus replication. J. Virol. 1991;65:2476–2483. doi: 10.1128/jvi.65.5.2476-2483.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ward P., Falkenberg M., Elias P., Weitzman M., Linden R.M. Rep-dependent initiation of adeno-associated virus type 2 DNA replication by a herpes simplex virus type 1 replication complex in a reconstituted system. J. Virol. 2001;75:10250–10258. doi: 10.1128/JVI.75.21.10250-10258.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Conway J.E., Zolotukhin S., Muzyczka N., Hayward G.S., Byrne B.J. Recombinant adeno-associated virus type 2 replication and packaging is entirely supported by a herpes simplex virus type 1 amplicon expressing Rep and Cap. J. Virol. 1997;71:8780–8789. doi: 10.1128/jvi.71.11.8780-8789.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Conway J.E., Rhys C.M., Zolotukhin I., Zolotukhin S., Muzyczka N., Hayward G.S., Byrne B.J. High-titer recombinant adeno-associated virus production utilizing a recombinant herpes simplex virus type I vector expressing AAV-2 Rep and Cap. Gene Ther. 1999;6:986–993. doi: 10.1038/sj.gt.3300937. [DOI] [PubMed] [Google Scholar]
- 30.Hakim C.H., Wasala N.B., Pan X., Kodippili K., Yue Y., Zhang K., Yao G., Haffner B., Duan S.X., Ramos J. A five-repeat micro-dystrophin gene ameliorated dystrophic phenotype in the severe DBA/2J-mdx model of Duchenne muscular dystrophy. Mol. Ther. Methods Clin. Dev. 2017;6:216–230. doi: 10.1016/j.omtm.2017.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ramos J.N., Hollinger K., Bengtsson N.E., Allen J.M., Hauschka S.D., Chamberlain J.S. Development of novel micro-dystrophins with enhanced functionality. Mol. Ther. 2019;27:623–635. doi: 10.1016/j.ymthe.2019.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Adamson-Small L., Potter M., Falk D.J., Cleaver B., Byrne B.J., Clément N. A scalable method for the production of high-titer and high-quality adeno-associated type 9 vectors using the HSV platform. Mol. Ther. Methods Clin. Dev. 2016;3:16031. doi: 10.1038/mtm.2016.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Potter M., Lins B., Mietzsch M., Heilbronn R., Van Vliet K., Chipman P., Agbandje-McKenna M., Cleaver B.D., Clément N., Byrne B.J., Zolotukhin S. A simplified purification protocol for recombinant adeno-associated virus vectors. Mol. Ther. Methods Clin. Dev. 2014;1:14034. doi: 10.1038/mtm.2014.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li D., Yue Y., Duan D. Preservation of muscle force in mdx3cv mice correlates with low-level expression of a near full-length dystrophin protein. Am. J. Pathol. 2008;172:1332–1341. doi: 10.2353/ajpath.2008.071042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu M., Yue Y., Harper S.Q., Grange R.W., Chamberlain J.S., Duan D. Adeno-associated virus-mediated microdystrophin expression protects young mdx muscle from contraction-induced injury. Mol. Ther. 2005;11:245–256. doi: 10.1016/j.ymthe.2004.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McMillan H.J., Gregas M., Darras B.T., Kang P.B. Serum transaminase levels in boys with Duchenne and Becker muscular dystrophy. Pediatrics. 2011;127:e132–e136. doi: 10.1542/peds.2010-0929. [DOI] [PubMed] [Google Scholar]
- 37.Wright M.A., Yang M.L., Parsons J.A., Westfall J.M., Yee A.S. Consider muscle disease in children with elevated transaminase. J. Am. Board Fam. Med. 2012;25:536–540. doi: 10.3122/jabfm.2012.04.110183. [DOI] [PubMed] [Google Scholar]
- 38.Flanigan K.M., Voit T., Rosales X.Q., Servais L., Kraus J.E., Wardell C., Morgan A., Dorricott S., Nakielny J., Quarcoo N. Pharmacokinetics and safety of single doses of drisapersen in non-ambulant subjects with Duchenne muscular dystrophy: results of a double-blind randomized clinical trial. Neuromuscul. Disord. 2014;24:16–24. doi: 10.1016/j.nmd.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Church R.J., Watkins P.B. The transformation in biomarker detection and management of drug-induced liver injury. Liver Int. 2017;37:1582–1590. doi: 10.1111/liv.13441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kunkel L.M. 2004 William Allan award address. cloning of the DMD gene. Am. J. Hum. Genet. 2005;76:205–214. doi: 10.1086/428143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yue Y., Pan X., Hakim C.H., Kodippili K., Zhang K., Shin J.-H., Yang H.T., McDonald T., Duan D. Safe and bodywide muscle transduction in young adult Duchenne muscular dystrophy dogs with adeno-associated virus. Hum. Mol. Genet. 2015;24:5880–5890. doi: 10.1093/hmg/ddv310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Verhaart I.E.C., Aartsma-Rus A. Therapeutic developments for Duchenne muscular dystrophy. Nat. Rev. Neurol. 2019;15:373–386. doi: 10.1038/s41582-019-0203-3. [DOI] [PubMed] [Google Scholar]
- 43.Crudele J.M., Chamberlain J.S. AAV-based gene therapies for the muscular dystrophies. Hum. Mol. Genet. 2019;28:R102–R107. doi: 10.1093/hmg/ddz128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wells D.J. What is the level of dystrophin expression required for effective therapy of Duchenne muscular dystrophy? J. Muscle Res. Cell Motil. 2019;40:141–150. doi: 10.1007/s10974-019-09535-9. [DOI] [PubMed] [Google Scholar]
- 45.Chamberlain J.S. Dystrophin levels required for correction of Duchenne muscular dystrophy. Basic Appl. Myol. 1997;7:251–255. [Google Scholar]
- 46.West N.A., Yang M.L., Weitzenkamp D.A., Andrews J., Meaney F.J., Oleszek J., Miller L.A., Matthews D., DiGuiseppi C. Patterns of growth in ambulatory males with Duchenne muscular dystrophy. J. Pediatr. 2013;163:1759–1763.e1. doi: 10.1016/j.jpeds.2013.08.004. [DOI] [PubMed] [Google Scholar]
- 47.Sarrazin E., von der Hagen M., Schara U., von Au K., Kaindl A.M. Growth and psychomotor development of patients with Duchenne muscular dystrophy. Eur. J. Paediatr. Neurol. 2014;18:38–44. doi: 10.1016/j.ejpn.2013.08.008. [DOI] [PubMed] [Google Scholar]
- 48.Kotin R.M. Large-scale recombinant adeno-associated virus production. Hum. Mol. Genet. 2011;20(R1):R2–R6. doi: 10.1093/hmg/ddr141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.van der Loo J.C., Wright J.F. Progress and challenges in viral vector manufacturing. Hum. Mol. Genet. 2016;25(R1):R42–R52. doi: 10.1093/hmg/ddv451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Clément N., Grieger J.C. Manufacturing of recombinant adeno-associated viral vectors for clinical trials. Mol. Ther. Methods Clin. Dev. 2016;3:16002. doi: 10.1038/mtm.2016.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Penaud-Budloo M., François A., Clément N., Ayuso E. Pharmacology of recombinant adeno-associated virus production. Mol. Ther. Methods Clin. Dev. 2018;8:166–180. doi: 10.1016/j.omtm.2018.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Petrof B.J., Shrager J.B., Stedman H.H., Kelly A.M., Sweeney H.L. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc. Natl. Acad. Sci. USA. 1993;90:3710–3714. doi: 10.1073/pnas.90.8.3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li D., Yue Y., Duan D. Marginal level dystrophin expression improves clinical outcome in a strain of dystrophin/utrophin double knockout mice. PLoS ONE. 2010;5:e15286. doi: 10.1371/journal.pone.0015286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.van Putten M., Hulsker M., Nadarajah V.D., van Heiningen S.H., van Huizen E., van Iterson M., Admiraal P., Messemaker T., den Dunnen J.T., ’t Hoen P.A., Aartsma-Rus A. The effects of low levels of dystrophin on mouse muscle function and pathology. PLoS ONE. 2012;7:e31937. doi: 10.1371/journal.pone.0031937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.van Putten M., Hulsker M., Young C., Nadarajah V.D., Heemskerk H., van der Weerd L., ’t Hoen P.A., van Ommen G.J., Aartsma-Rus A.M. Low dystrophin levels increase survival and improve muscle pathology and function in dystrophin/utrophin double-knockout mice. FASEB J. 2013;27:2484–2495. doi: 10.1096/fj.12-224170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kotin R.M., Wright J.F. Recombinant adeno-associated virus quality control for non-clinical and clinical vectors: how an unregulated commercial sector can compromise development of new gene therapies. Hum. Gene Ther. 2019;30:1447–1448. doi: 10.1089/hum.2019.29099.rmk. [DOI] [PubMed] [Google Scholar]
- 57.Schnödt M., Büning H. Improving the quality of adeno-associated viral vector preparations: the challenge of product-related impurities. Hum. Gene Ther. Methods. 2017;28:101–108. doi: 10.1089/hgtb.2016.188. [DOI] [PubMed] [Google Scholar]
- 58.Shin J.-H., Hakim C., Zhang K., Duan D. Genotyping mdx, mdx3cv, and mdx4cv mice by primer competition polymerase chain reaction. Muscle Nerve. 2011;43:283–286. doi: 10.1002/mus.21873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kingston R.E., Chen C.A., Okayama H. Calcium phosphate transfection. Curr. Protoc. Cell Biol. 2003;Chapter 20:Unit 20.3. doi: 10.1002/0471143030.cb2003s19. [DOI] [PubMed] [Google Scholar]
- 60.Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kodippili K., Vince L., Shin J.H., Yue Y., Morris G.E., McIntosh M.A., Duan D. Characterization of 65 epitope-specific dystrophin monoclonal antibodies in canine and murine models of duchenne muscular dystrophy by immunostaining and western blot. PLoS ONE. 2014;9:e88280. doi: 10.1371/journal.pone.0088280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hakim C.H., Wasala N.B., Duan D. Evaluation of muscle function of the extensor digitorum longus muscle ex vivo and tibialis anterior muscle in situ in mice. J. Vis. Exp. 2013;(72):50183. doi: 10.3791/50183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hakim C.H., Li D., Duan D. Monitoring murine skeletal muscle function for muscle gene therapy. Methods Mol. Biol. 2011;709:75–89. doi: 10.1007/978-1-61737-982-6_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mendez J., Keys A. Density and composition of mammalian muscle. Metabolism. 1960;9:184–188. [Google Scholar]
- 65.Burkholder T.J., Fingado B., Baron S., Lieber R.L. Relationship between muscle fiber types and sizes and muscle architectural properties in the mouse hindlimb. J. Morphol. 1994;221:177–190. doi: 10.1002/jmor.1052210207. [DOI] [PubMed] [Google Scholar]
- 66.Brooks S.V., Faulkner J.A. Contractile properties of skeletal muscles from young, adult and aged mice. J. Physiol. 1988;404:71–82. doi: 10.1113/jphysiol.1988.sp017279. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.