

Abstract
Purpose
To investigate the association between PAX6 genotype and macular morphology in congenital aniridia.
Methods
The study included 37 participants (15 males) with congenital aniridia (aged 10–72 years) and 58 age-matched normal controls (18 males). DNA was isolated from saliva samples. PAX6 exons, intron/exon junctions, and known regulatory regions were amplified in PCR and sequenced. Multiplex ligation-dependent probe amplification (MLPA) was performed to detect larger deletions or duplications in PAX6 or known cis-regulatory regions. Spectral-domain optical coherence tomography images were acquired and segmented semiautomatically. Mean thicknesses were calculated for inner and outer retinal layers within the macula along nasal and temporal meridians.
Results
Mutations in PAX6 or regulatory regions were found in 97% of the participants with aniridia. Foveal hypoplasia was observed in all who had a mutation within the PAX6 gene. Aniridic eyes had thinner outer retinal layers than controls, but with large between-individual variation (mean ± SD, 156.3 ± 32.3 µm vs 210.8 ± 12.3 µm, P < 0.001). Parafoveal and perifoveal inner and outer retinal layers were thinner in aniridia. Participants with mutations in noncoding PAX6 regions had thicker foveal outer retinal layers than those with mutations in the PAX6 coding regions (P = 0.04) and showed signs of postnatal development and maturation. Mutations outside the PAX6 gene were associated with the mildest retinal phenotypes.
Conclusions
PAX6 mutations are associated with significant thinning of macular inner and outer retinal layers, consistent with misdirected retinal development resulting in abnormal foveal formation and reduced number of neurons in the macula, with mutations in PAX6 coding regions giving the worst outcome.
Keywords: aniridia, PAX6, retinal structure, optical coherence tomography, phenotype
Congenital aniridia is a rare genetic disorder disrupting normal development of the eye and affects an estimated 1 in 64,000–72,000 people worldwide.1,2 Heterozygous mutations within the PAX6 gene (paired box gene 6; OMIM # 607108) or associated regulatory regions are the most common cause of aniridia.3–6 These mutations reduce the expression of the PAX6 gene and lead to a shortage of functional PAX6 protein, which, among other effects, disrupts eye development.7 This can lead to a spectrum of ocular anomalies, including incomplete development of the iris, fovea, and optic nerve; severely impaired vision; and nystagmus. The progressive nature of aniridia frequently leads to secondary ocular complications such as cataract, glaucoma, and aniridia-associated keratopathy (AAK). The clinical phenotype is highly variable among individuals with different genotypes, as well as between individuals with the same genotype.8–10
While absence of the iris is considered the hallmark of aniridia, foveal hypoplasia is one of the most common ocular findings, observed even in cases where the iris may appear intact.8,11 PAX6 plays an important role in retinal development,12 including in cell type specification/differentiation13 and migration of cones toward the foveal center.14 However, little is known about PAX6's specific role in foveal maturation, and the reported variability in visual acuity and foveal hypoplasia in eyes with aniridia11,15 may be attributed to different mutations found in or around the PAX6 gene. It is therefore reasonable to hypothesize that more severe PAX6 mutations, which have a larger effect on the PAX6 protein dosage, will result in a thinner retina in the perifovea and parafovea, poorer foveal cone specialization, and more severe alteration of macular morphology. The current study tested this hypothesis. The aim, therefore, was to assess the contribution of each retinal layer to macular morphology in PAX6-associated aniridia, investigate the relationship between foveal cone specialization and visual acuity, and determine any genotype-phenotype relationships.
In addition to employing qualitative grading of foveal hypoplasia using optical coherence tomography (OCT),16 measures of inner and outer retinal layer thicknesses within the foveal, parafoveal, and perifoveal regions were assessed. Such assessment is warranted because of large between-individual variation, both between and within each OCT grade of foveal hypoplasia.10,11,17 This variation indicates that qualitative grading of foveal hypoplasia alone may be insufficient to fully characterize foveal formation in aniridia. Knowledge about individual variations in foveal formation and macular development in aniridia is particularly important for understanding each individual's potential of visual function and for predicting treatment outcomes.
Methods
Thirty-seven persons with congenital aniridia (24 familial, 13 sporadic; 15 males; aged 10–72 [mean ± SD, 35.8 ± 18.6] years) and 58 age-matched normal controls (18 males; aged 10–74 [35.7 ± 19.0] years, P = 0.99) participated in the study (Table 1). Those with aniridia were recruited through the Norwegian Association of Aniridia or via family members, whereas the normal controls were recruited through the National Centre for Optics, Vision and Eye Care, University of South-Eastern Norway. The study followed the principles in the Declaration of Helsinki and was approved by the Regional Committee for Medical and Health Research Ethics (Southern Norway Regional Health Authority). The purpose, procedures, and possible consequences of the study were explained to each participant and/or his or her guardians before data collection and written informed consent was obtained.
Table 1.
Distribution of Participants Within Each Age Group
| Normal Controls | Aniridia | |||||
|---|---|---|---|---|---|---|
| Age Group | n | Mean Age, y | Age Range, y | n | Mean Age, y | Age Range, y |
| <20 | 14 | 14.71 | 10–19 | 8 | 13.88 | 9–19 |
| 20–29 | 12 | 22.08 | 20–27 | 9 | 23.78 | 20–29 |
| 30–39 | 8 | 33.62 | 31–37 | 4 | 32.75 | 31–36 |
| 40–49 | 8 | 44.38 | 41–48 | 6 | 43.50 | 40–49 |
| 50–59 | 7 | 52.86 | 50–58 | 5 | 54.20 | 50–59 |
| >60 | 9 | 67.44 | 64–74 | 5 | 67.00 | 64–72 |
The participants underwent a comprehensive eye examination of the anterior and posterior segment as described previously.17 Best-corrected visual acuity was measured with a high-contrast logMAR acuity chart (TestChart 2000; Thomson Software Solutions, London, UK) at 6 m. If a reliable measurement could not be obtained at the longer distance, the test distance was reduced to 3 or 1 m and the logMAR value corrected accordingly. Refractive errors were classified based on spherical equivalent refraction (SER = sphere + ½ cylinder). Ocular axial length (AL) was measured with an optical interferometer (IOL Master 700; Carl Zeiss Meditec AG, Jena, Germany). Grading of AAK has been reported previously.17,18
Genetic Analysis
DNA, isolated from saliva samples (Oragene-DNA, DNA Self-Collection Kit; DNA Genotek, Inc., Ottawa, ON, Canada) from 35 of 37 participants with aniridia, was used in the PCR to amplify and sequence the exons (1–13) and intron/exon junctions of the PAX6 gene using previously described primers and conditions.9 Fluorescent DNA sequencing was performed on both DNA strands. PAX6 transcript reference sequence NM_00280.4 from the National Center for Biotechnology Information was used for nucleotide numbering. Variations were described according to conventional notations.19 Nucleotide numbering starts with 1 at the A of the ATG translation initiation codon.
For the participants for whom no abnormalities were found by intragenic PAX6 sequencing, the remote, ultra-conserved PAX6 enhancer SIMO, located 150 kb downstream from PAX6, was sequenced.4,20 Multiplex ligation-dependent probe amplification (MLPA) was performed using the SALSA MLPA reagent kit P219-B3 (MRC Holland, Amsterdam, the Netherlands) to detect larger deletions or duplications in the PAX6 genomic region, including several known PAX6 regulatory regions. The MLPA analysis included three control DNA samples. The deletions detected by the MLPA analysis were confirmed by TaqMan Copy Number Assays (Applied Biosystems, Foster City, CA) using quantitative real-time PCR to determine gene copy number. Identified PAX6 mutations were analyzed using online bioinformatics tools (Mutation Taster21) to predict their disease-causing potential. Mutations were also checked against entries of the Leiden Open Variation Database, ClinVar, Exome Aggregation Consortium, Human Gene Mutation Database, and a PubMed search.
PITX2 and FOXC1 genes were amplified and sequenced for participants who were negative for mutations in the PAX6 genomic region. The primer sequences and PCR conditions have been described previously.22
Optical Coherence Tomography
Heidelberg Spectralis OCT2 (Heidelberg Engineering GmbH, Heidelberg, Germany) was used to acquire spectral-domain optical coherence tomography (SD-OCT) scans of the macular region as described previously (512–1536 A-scans/B-scans, 49 B-scans over 20° × 20° or 30° × 10°; 30° × 5° for normal controls).17,23 The built-in eye-tracking system was used to compensate for eye-motion artifacts and 5–20 horizontal B-scans were averaged during imaging. The participants’ head posture and gaze direction were adjusted to minimize the amplitude of nystagmus during OCT imaging. If a reliable volume scan still was unattainable because of nystagmus, a horizontal scan line was moved manually across the macula to look for signs of foveal specialization.24,25 Several repetitive horizontal line scans (30° field of view) were acquired at the expected foveal location and above and below the central scan using the optic nerve head as a reference point26 to increase the probability of scanning through the center of the fovea. The image quality was verified and considered adequate when the signal was >15 dB.
Foveal hypoplasia was graded by two separate graders (authors HRP and RCB) according to the grading scheme suggested by Thomas et al.16: presence of inner layers at the foveal center (grade 1), absence of a foveal pit (grade 2), absence of outer segment lengthening (grade 3), and absence of outer nuclear layer widening (grade 4). Foveal hypoplasia grade was typically the same in both eyes of each individual, and thus only the dominant eye was included in analysis. The retinal layers were segmented using a semiautomatic active contour method,27 which sought to follow local image intensity gradients while a thin-plate constraint ensured that segmented contours were smooth and, therefore, robust to local noise. The operator could dynamically modify the contour via an interactive interface to correct for any segmentation errors that may have arisen due to image artifacts or noise (e.g., shadows cast from blood vessels). This method has been successfully applied to OCT image segmentation previously.10,28,29 The horizontal OCT B-scan through the foveal center, defined as the section with maximum photoreceptor layer thickness, was used for analysis (Fig. 1). In participants without the presence of outer segment (OS) lengthening, the expected foveal location was identified via maximum thickness of the outer nuclear layer (ONL). In those with grade 4 foveal hypoplasia, the expected foveal center was located based on doming of the inner retinal layers (if present) and/or the horizontal and vertical distance between fovea and optic disc as described previously.26 Because it is difficult to differentiate the ONL and Henle's fiber layer without capturing directional OCT,30 these were defined as one layer. The reflective bands corresponding to the photoreceptor OS–retinal pigment epithelium (RPE) interdigitation zone (IZ) and/or the external limiting membrane were not visible or clearly demarcated in 7 and 10 eyes, respectively. Hence, we used a combined measurement of ONL, photoreceptor inner and outer segments, and RPE to provide a more robust thickness measurement of the outer retinal layers. Measurements of the three innermost layers (i.e., the retinal nerve fiber layer [RNFL], ganglion cell layer [GCL], and inner plexiform layer [IPL]) were also combined. Thicknesses of all the component layers of the outer and inner retina were also calculated for those where the boundaries between the layers were visible. Mean thicknesses were calculated within five lateral regions: foveal center (central 50 µm), parafoveal region (0.5–1.5 mm retinal eccentricity), and perifoveal region (1.5–3.0 mm retinal eccentricity) along the nasal and temporal meridians. The lateral scale was corrected for between-individual AL differences by multiplying the lateral scale obtained from the instrument with the ratio between each individual's AL and the OCT AL setting (24 mm for a medium-long eye).
Figure 1.

Horizontal SD-OCT scan through the foveal center of a male with PAX6-related aniridia including an illustration of the segmented retinal layers and definition of the retinal layers. The foveal, parafoveal, and perifoveal regions along the nasal and temporal meridians are marked with red, yellow, and turquoise, respectively.
Data Analysis
Statistical analyses were performed using R statistical software,31 version 3.5.1. Normal distribution of the variables was verified using histograms, QQ-plots, and the Shapiro-Wilk test. Between-group differences were analyzed using two-tailed Student's or Welch's independent sample t-tests for equal and unequal variances, respectively. The Wilcoxon rank-sum test was applied for nonnormal data.
We performed a linear mixed-effects analysis, using the nmle R package,32 to examine the differences in retinal layer thicknesses between the participants with aniridia and normal controls. A random effect was entered to treat retinal layer as a within-subject variable (random intercept per subject). Weights were added to account for the difference in variance between the groups. Small deviations from homoscedasticity and normality of the residuals, when analyzing the foveal center, were accounted for by applying a square root transformation to the dependent variable. Likelihood ratio tests were performed to compare models. Differences were considered significant when P ≤ 0.05.
Holm-Bonferroni corrected pairwise t-tests were used to assess differences in outer retinal thickness between the different locations of the PAX6 mutations. Correlations were assessed using Pearson correlation coefficient (rp). Multiple linear regression was performed to assess the relationship between logMAR visual acuity, retinal layer thicknesses, and AAK grade. Significance level was set at 0.05. Weighted Cohen's κ was calculated to assess interrater agreement in grading of foveal hypoplasia.
Results
Table 2 shows an overview of the clinical phenotypes in aniridia. Iris anomaly severity varied from subtle structural anomalies to complete absence of the iris (more or less symmetrical in both eyes) as observed by slit-lamp examination. Best-corrected visual acuity ranged from logMAR 0.00–1.76 to hand movements at 0.5 m. The mean AL was similar in aniridic eyes (23.27; range, 19.48–28.30 mm) and control eyes (23.64; range, 20.63–26.14 mm, P = 0.29), although with a larger range in aniridia. Refractive errors in aniridic eyes ranged from SER –20.50 to +10.00 D. The normal controls were healthy with no systemic or ocular diseases and had visual acuity ≤0.10 logMAR.
Table 2.
Overview of Phenotypes in Aniridia
| ID | Test Eye* | VA (logMAR) | AL (mm) | Iris Hypoplasia | AAK Grade | Nystagmus | FH Grade | ON Hypoplasia | Glaucoma | Lens Status† |
|---|---|---|---|---|---|---|---|---|---|---|
| 5110 | OD | 1.00 (HM) | 21.29 (20.79) | Complete | 3 | Yes | N/A | Yes | Yes | P. phakic |
| 5118 | OD | 0.74 (CF) | 23.35 (24.80) | Complete | 3 | Yes | N/A | No | Yes | P. phakic |
| 5121 | OS | CF (CF) | 26.08 (N/A) | Partial | 3 | Yes | N/A | No | No | N5/C1/P1 |
| 5126 | OD | 0.80 (0.88) | 22.02 (21.68) | Complete | 1 | Yes | 4 | No | Yes | N1/C4/P4 |
| 5137 | OD | 0.70 (0.74) | 23.47 (23.20) | Partial | 1 (2) | Yes | 3 | No | No | N1/C3/P2 |
| 5138 | OD | 0.90 | 21.84 (21.64) | Complete | 1 | Yes | 4 | No | No | N1/C3/P1 |
| 5147 | OD | 0.50 (0.60) | 22.64 (23.09) | Complete | 2 | Yes | 3 | No | No | N1/C3/P1 |
| 5149 | OS | 0.90 (1.10) | 20.26 (20.21) | Partial | 2 (3) | Yes | 4 | N/A | No | N2/C5/P3 |
| 5151 | OS | CF (HM) | 23.35 (23.39) | Complete | 3 | Yes | N/A | N/A | Yes | N1/C2/NA |
| 5134 | OD | 0.18 | 21.66 (21.80) | Partial | 1 | No | 1 | No | No | P. phakic |
| 5154 | OD | 0.72 (0.80) | 22.27 (22.59) | Complete | 1 (2) | Yes | 2 | No | No | N1/C4/P2 |
| 5119 | OD | 1.00 (1.30) | 21.22 (N/A) | Complete | 1 (3) | Yes | 4 | No | Yes | N1/C3/P1 |
| 5124 | OD | 0.40 (0.60) | 24.01 (24.54) | Partial | 0 | No | 0 | No | No | N1/C1/P1 |
| 5113 | OD | 0.80 (1.00) | 21.14 (21.40) | Complete | 2 | Yes | 4 | No | No | P. phakic |
| 5129 | OS | 1.30 (CF) | 21.50 (23.37) | Complete | 3 | Yes | N/A | No | Yes | P. phakic |
| 5114 | OD | 0.86 (1.78) | 23.97 (24.04) | Complete | 1 (3) | Yes | 3 | Yes | Yes | Aphakic |
| 5116 | OD | 0.40 | 25.66 (25.41) | Partial | 2 | No | 2 | No | No | P. phakic |
| 5120 | OS | 0.22 (0.32) | 23.80 (23.93) | Partial | 1 | No | 2 | No | No | N2/C1/P1 |
| 5123 | OS | 0.50 (1.30‡) | 22.72 (24.63) | Almost complete | 2 (1) | No | 2 | No | No | N1/C3/P1 |
| 5135 | OD | 0.70 (0.80) | 24.05 (23.25) | Complete | 1 | Yes | 3 | Yes | Yes | N2/C4/P3 |
| 5148 | OS | 0.60 (0.64) | 21.04 (21.06) | Almost complete | 2 | No | 4 (3) | No | Yes | P. phakic |
| 5199 | OD | 0.20 (0.30) | 25.55 (25.36) | Near normal | 1 | No | 2 | No | No | N0/C2/P0 |
| 5125 | OD | 0.74 (0.76) | 24.13 (23.98) | Complete | 1 | Yes | 3 | No | Yes | N1/C2/P2 |
| 5127 | OD | 1.20 (1.10) | 25.37 (26.51) | Almost complete | 2 | Yes | 4 | No (Yes) | Yes | P. phakic |
| 5146 | OS | CF (LP) | 23.30 (N/A) | Complete | 2 (3) | Yes | N/A | N/A | Yes | P. phakic |
| 5131 | OD | 1.30 (CF) | 24.42 (24.37) | Partial | 1 | Yes | 4 | Yes | Yes | Aphakic |
| 5140 | OD | 0.70 (0.80) | 20.97 (20.88) | Complete | 2 (3) | Yes | 4 | No | No | N1/C4/P2 |
| 5141 | OD | 1.00 (0.90) | 28.30 (28.08) | Partial | 2 | Yes | 4 | No | No | N1/C3/P1 |
| 5144 | OD | 0.74 (1.00) | 23.55 (23.88) | Complete | 2 (3) | Yes | 3 | No | No (Yes) | P. phakic |
| 5145 | OD | 1.76 (1.84) | 23.45 (22.65) | Complete | 3 | Yes | N/A | N/A | No | P. phakic |
| 5117 | OD | 1.00 (1.10) | 23.34 (23.35) | Complete | 2 (1) | Yes | 4 | No | Yes | P. phakic |
| 5128 | OD | 0.90 (1.10) | 19.48 (19.44) | Complete | 2 (3) | Yes | N/A | N/A | No | N1/C4/P4 |
| 5152 | OD | 1.30 | 23.21 (N/A) | Complete | 3 | Yes | N/A | N/A | Yes | N1/C3/P1 |
| 5130 | OS | 1.30 (LP) | 26.59 (N/A) | Complete | 3 | Yes | N/A | Yes | Yes | Aphakic |
| 5132 | OS | 0.56 (0.30) | 25.74 (28.50) | Complete | 0 | No | 1 (2) | No | Yes | P. phakic |
| 5139 | OD | 0.00 (0.46§) | 22.95 (22.35) | Complete | 0 | No | 0 | No | No | N1/C1/P1 |
| 5155 | OS | N/A | N/A (N/A) | Complete | 0 | No | N/A | N/A | No | N/A |
Participants are sorted in the same way as in Table 3, that is, according to whether the genotype is sporadic (above the horizontal space) or familial. CF, counting fingers at 0.5 m; HM, hand motion; LP, light perception; N/A, not available; ON, optic nerve; P. phakic, pseudophakic; VA, visual acuity.
The test eye is the eye included in retinal layer thickness analyses. Data from the other eye are noted in parentheses if different from that of the test eye.
LOCS II grading.53
Subluxated lens OD, not corrected for during VA measurement.
Amblyopic OS.
Aniridia Genotypes
The 11 families (24 participants) showed mutations that followed an autosomal dominant pattern. Table 3 summarizes the details of the identified PAX6 mutations, including nucleotide change, amino acid change, type of mutation, and predicted functional outcome. Genetic data were not available from participants 5119 and 5124. Mutations that affect PAX6 were found in 34 of 35 participants with aniridia who provided a saliva sample (20 unique variants), employing PAX6 sequencing (n = 25) and MPLA analysis (n = 9). Three variants have not been reported previously. Figure 2 shows a schematic presentation of the deletions and mutations identified in PAX6 and adjacent downstream regulatory regions.
Table 3.
Summary of Genotypes
| ID | Inheritance | Family No. | Location | Nucleotide Change | Amino Acid Change | Type of Mutation* | Nonsense-Mediated Decay (NMD) Predicted |
|---|---|---|---|---|---|---|---|
| 5110 | Sporadic | Ex 5_6 | Deletion PAX6 Ex 5_6 | Deletion | Yes | ||
| 5118 | Sporadic | Ex 7 | c.485G>A | p.(Trp162*) | Nonsense | Yes | |
| 5121 | Sporadic | Ex 4 | Deletion PAX6 Ex 4 | Deletion | Yes | ||
| 5126 | Sporadic | Ex 8 | c.607C>T | p.(Arg203*) | Nonsense | Yes | |
| 5137 | Sporadic | Ex 5 | c.120C>A | p.(Cys40*) | Nonsense | Yes | |
| 5138 | Sporadic | Ex 8 | c.546delA§ | p.(Gly184Glufs*23) | Frameshift | Yes | |
| 5147 | Sporadic | — | Not found | — | — | — | |
| 5149 | Sporadic | Ex 13 | c.1268A>T | p.(*423Leuext15) | CTE | No | |
| 5151 | Sporadic | Ex 9 | c.718C>T | p.(Arg240*) | Nonsense | Yes | |
| 5134 | Sporadic | Ex 9_13; ELP4-Ex9 | Deletion PAX6, Ex 9_13 ELP4, Ex 9 | Deletion | No mRNA expected | ||
| 5154 | Sporadic | Up_Ex1_13; ELP4, Ex 9; DCDC1, Ex 1+4 | Deletion PAX6 Upstream_ DCDC1, Ex 4 | Deletion | No mRNA expected | ||
| 5113 | Familial | 1 | Ex 8 | c.538delC|| | p.(Gln180Argfs*27) | Frameshift | Yes |
| 5129 | 1 | Ex 8 | c.538delC|| | p.(Gln180Argfs*27) | Frameshift | Yes | |
| 5114|| | Familial | 2 | Int 2 | c.-128-2delA (IVS2-2delA) | Splice | Most likely | |
| 5116|| | 2 | Int 2 | c.-128-2delA (IVS2-2delA) | Splice | Most likely | ||
| 5120|| | 2 | Int 2 | c.-128-2delA (IVS2-2delA) | Splice | Most likely | ||
| 5123|| | 2 | Int 2 | c.-128-2delA (IVS2-2delA) | Splice | Most likely | ||
| 5135|| | 2 | Int 2 | c.-128-2delA (IVS2-2delA) | Splice | Most likely | ||
| 5148|| | 2 | Int 2 | c.-128-2delA (IVS2-2delA) | Splice | Most likely | ||
| 5199|| | 2 | Int 2 | c.-128-2delA (IVS2-2delA) | Splice | Most likely | ||
| 5125¶ | Familial | 3 | Int 3 | c.-52+1G>A (IVS3+1G>A) | Splice | Yes | |
| 5127 | Familial | 4 | Ex 6 | c.151G>T | p.(Gly51*) | Nonsense | Yes |
| 5146 | 4 | Ex 6 | c.151G>T | p.(Gly51*) | Nonsense | Yes | |
| 5131 | Familial | 5 | Ex 5 | c.112delC | p.(Arg38Glyfs*30) | Frameshift | Yes |
| 5140 | Familial | 6 | Ex 13 | c.1269A>T | p.(*423Tyrext15) | CTE | No |
| 5141 | Familial | 7 | Int 4 | c.11-2A>G (IVS4-2A>G) | Splice | Yes | |
| 5144 | Familial | 8 | Ex 9 | c.718C>T | p.(Arg240*) | Nonsense | Yes |
| 5145 | 8 | Ex 9 | c.718C>T | p.(Arg240*) | Nonsense | Yes | |
| 5117 | Familial | 9 | Ex 3_9 | Deletion PAX6, Ex 3_9 | Deletion | Yes | |
| 5128 | 9 | Ex 3_9 | Deletion PAX6, Ex 3_9 | Deletion | Yes | ||
| 5152 | 9 | Ex 3_9 | Deletion PAX6, Ex 3_9 | Deletion | Yes | ||
| 5130 | Familial | 10 | Ex 4_13 | Deletion PAX6, Ex 4_13 | Deletion | Yes | |
| 5132 | Familial | 11 | ELP4, Ex 9; DCDC1, Ex 1+4 | Deletion ELP4, Ex 9_DCDC1, Ex 4 | Deletion | Unknown | |
| 5139 | 11 | ELP4, Ex 9; DCDC1, Ex 1+4 | Deletion ELP4, Ex 9_DCDC1, Ex 4 | Deletion | Unknown | ||
| 5155 | 11 | ELP4, Ex 9; DCDC1, Ex 1+4 | Deletion ELP4, Ex 9_DCDC1, Ex 4 | Deletion | Unknown |
All genetic variants are predicted to be pathogenetic according to the ACMG classification scheme.54.
A 1013-bp deletion that removes the last 25 bp of exon 5 through the first 61 bp of exon 6.
There is a 17-pb deletion (GGCCCCCAGCCAGAGCC), followed by an A>T substitution that disrupts the Kozak sequence. This deletion has not been reported previously.
These single-nucleotide deletions have, to our knowledge, not been not reported previously. Slightly different nucleotide changes (c.551delG and c. 538C>T, respectively) are previously reported to give the same amino acid changes and are described as a cause of aniridia.
Participants previously reported.10
Participant harbors an additional variant in PAX6 Ex10: c.831G>A, does not alter amino acid.
Figure 2.

Schematic presentation of the deletions and mutations identified in PAX6 and adjacent downstream regulatory regions. (A) PAX6 is located at chromosome 11p13. The ruler at the top gives the nucleotide numbers for the region shown. By convention, the nucleotides that comprise each chromosome in the reference sequence are numbered consecutively starting at the tip of the petite (p) arm. The region shown is for the GRCh38/hg38 assembly of the human genome. The colored horizontal bars represent the size of the deletions identified in the participants with aniridia and are colored according retinal phenotype. Green: FH grades 0–1; yellow: FH grades 1–2. (B) Zoomed-in view of PAX6 gene structure. The gene has 14 exons and a 3′ UTR depicted as colored boxes (including exon 5a). The white boxes indicate introns. The protein coding region begins in exon 4 at position c.1. The protein domains encoded by the exons are color-coded (blue: paired domain; gray: linker region; orange: homeodomain; dark gray: proline-serine-threonine domain). Mutations involving only the PAX6 gene found in this study are indicated as red bars above the gene showing deletions larger than one nucleotide. The arrows below the gene show the locations of point mutations, including single-nucleotide deletions. The labeling convention for the arrows shows the coding region nucleotide number for the affected position, which are positive if after the translation start site or negative if in the 5′ untranslated exons. The nucleotide number is followed by the identity of the nucleotide found in the reference sequence followed by “>” and then by the nucleotide found in the mutant. Deletions are indicated by “del” followed by the identity of the deleted nucleotide. The arrows are color-coded as follows: turquoise: FH grades 2–4; black: FH grades 3–4.
PAX6 sequencing identified 15 different mutations, with five variants located in the paired domain, four in the linker region, one in the homeodomain, and two in the proline-serine-threonine rich region. Untranslated regions of the PAX6 gene (5′ untranslated region [5′ UTR]: exons/introns 1–3) were involved in three different mutation variants.
Thirteen participants had mutations that introduce a premature termination codon (PTC), with or without a frameshift. All of these occurred >50 base pairs (bp) upstream of the last exon/exon junction, and thus the mRNA are expected to be targeted for degradation in the nonsense-mediated decay pathway and result in haploinsufficiency.33,34 Two participants had mutations predicted to result in a PAX6 protein with a C-terminal extension (CTE) and nine had mutations predicted to cause splice errors. Partial deletions of PAX6 were detected in two participants with sporadic aniridia. One was a 17-bp deletion in exon 4 (GGCCCCCAGCCAGAGCC), followed by an A>T substitution that disrupts the Kozak sequence.35 This mutation has not been reported previously. Without the Kozak sequence, the absence of protein is expected. This participant had severe corneal and lens opacities that hindered retinal imaging. The second person has a deletion that removes the last 25 bp of exon 5 through the first 61 bp of exon 6, including exon 5A (previously described by Grønskov et al.36). Seven from one family (no. 2), who have a splice site mutation (IV2-2delA) in the 5′ UTR, have been described in detail previously.10
In nine participants, MPLA analysis identified five different multiple exon deletions, either within, upstream, or downstream of the PAX6 genomic region. One of these (ELP4-DCDC1) did not include any of the PAX6 exons but was located downstream of the PAX6 gene and contains 3′ regulatory elements for PAX6.37 Four of the large deletions included the PAX6 coding exons, but only two of them included the retina-specific enhancer located in DNaseI hypersensitive sites (HS2–3) within the downstream regulatory region (DRR) between the ELP4 and PAX6 genes.38 Without the enhancer, no mRNA is expected from the affected alleles.
No PAX6, FOXC1, or PITX2 mutations could be detected for one sporadic case who had a classical aniridia phenotype with iris hypoplasia, moderate AAK, and grade 3 foveal hypoplasia. No point mutations in the SIMO element were found for any of the participants.
Retinal Layer Thicknesses
SD-OCT imaging was obtained of 26 persons with aniridia and 58 controls. Severe ocular media opacities (AAK >2) limited the view of the posterior pole and/or severe nystagmus prohibited a reliable measurement or scan through the expected foveal center in 10 participants with aniridia. One participant (5155) was not available for OCT imaging.
Figure 3 shows the variability in foveal morphology, including variability of inner and outer retinal structures, within and across foveal hypoplasia grades among those with aniridia. There was a statistically significant agreement in grading of foveal hypoplasia between the two graders (κ = 0.883, P < 0.001). Foveal hypoplasia was observed in 24 of 26 (92.3%) participants. The central fovea was thicker in aniridia (mean ± SD, 311.6 ± 30.2 µm; range, 232.5–357.8 µm) compared with the controls (229.4 ± 15.9 µm; range, 198.2–275.6 µm, P < 0.001). In contrast, the mean nasal and temporal perifoveal and parafoveal retinal thicknesses were significantly thinner in aniridia compared with the controls (all regions P < 0.001; Fig. 4A). This was a consequence of thinner parafoveal and perifoveal inner (RNFL + GCL + IPL; Fig. 4B) and outer retinal layers in aniridia (summarized in Table 4), whereas parafoveal and perifoveal Inner nuclear layer (INL) and outer plexiform layer (OPL) were similar in both groups.
Figure 3.

Variability in foveal morphology in aniridia. SD-OCT horizontal line scans through the expected foveal location are shown for the participants with aniridia and one normal control. The images are ordered from thickest (upper left) to thinnest (lower right) outer retinal layers within groups of foveal hypoplasia grade: grade 0 (no foveal hypoplasia), grade 1, grade 2, grade 3, and grade 4. Scale bar: 200 µm.
Figure 4.
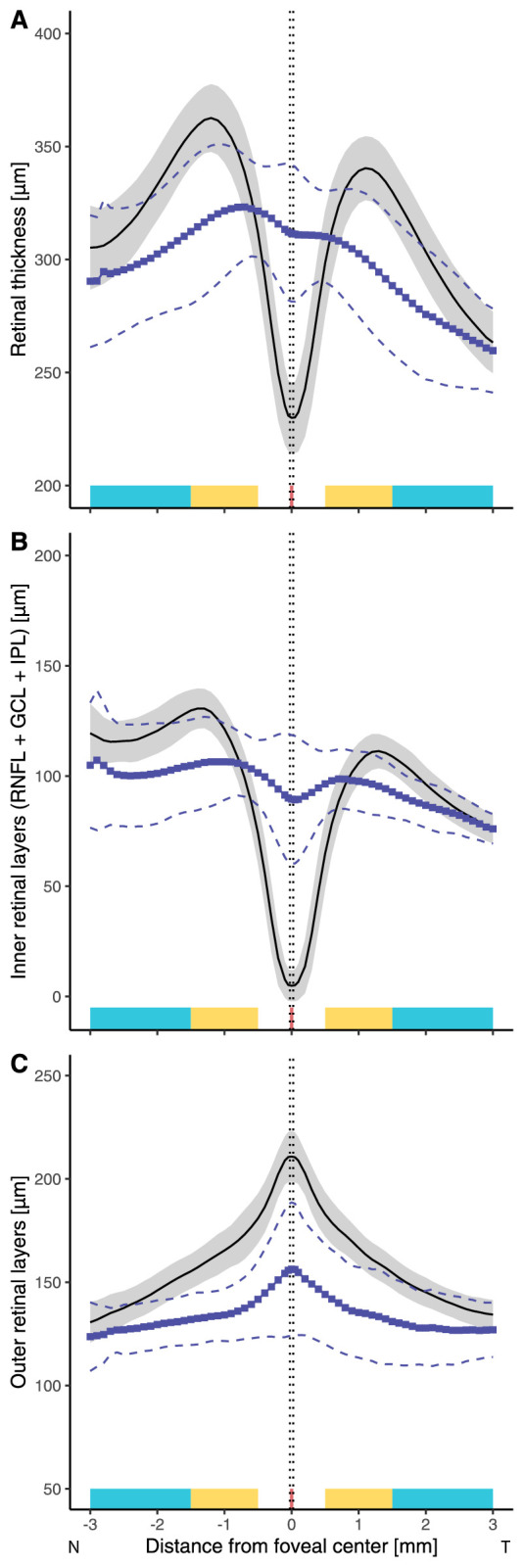
Graphs showing the variation in thickness of the (A) total retina, (B) inner retinal layers (Inner limiting membrane (ILM)-IPL), and (C) outer retinal layers (ONL + IS + OS + RPE) along the horizontal meridian in aniridia compared with normal control participants. Black solid lines and the shaded area represent the normal mean ± SD and lines with blue squares and dashed lines represent the mean ± SD for the participants with aniridia. The foveal, parafoveal, and perifoveal regions along the nasal and temporal meridians are marked with red, yellow, and turquoise, respectively.
Table 4.
Differences in Parafoveal (0.5–1.5 mm From the Foveal Center) and Perifoveal (1.5–3.0 mm From the Foveal Center) Retinal Layer Thicknesses Between the Participants With Aniridia (n = 26) and Normal Controls
| 95% Confidence Interval | 95% Confidence Interval | |||||||
|---|---|---|---|---|---|---|---|---|
| Retinal Layer | Mean Difference (µm) | Upper | Lower | P Value | Mean Difference (µm) | Upper | Lower | P Value |
| Nasal Parafovea | Temporal Parafovea | |||||||
| Inner retinal layers (RNFL + GCL + IPL) | −9.77 | −4.69 | −14.85 | <0.001 | −3.29 | 2.33 | −8.91 | 0.25 |
| Inner nuclear layer | 5.45 | 10.62 | 0.28 | 0.039 | 2.40 | 8.12 | −3.33 | 0.41 |
| Outer plexiform layer | 4.63 | 9.80 | −0.54 | 0.079 | 1.39 | 7.12 | −4.33 | 0.63 |
| Outer retinal layers (ONL + IS + OS + RPE) | −31.09 | −26.09 | −36.09 | <0.001 | −30.96 | −25.43 | −36.48 | <0.001 |
| Outer nuclear layer | −21.55 | −18.52 | −24.58 | <0.001 | −26.33 | −22.02 | −30.65 | <0.001 |
| Nasal Perifovea |
Temporal Perifovea | |||||||
| Inner retinal layers (RNFL + GCL + IPL) | −16.91 | −11.23 | −22.59 | <0.001 | −6.65 | −2.46 | −10.84 | 0.002 |
| Inner nuclear layer | 2.69 | 8.49 | −3.10 | 0.36 | −0.28 | 3.98 | −4.55 | 0.90 |
| Outer plexiform layer | 4.82 | 10.62 | −0.97 | 0.10 | 1.45 | 5.72 | −2.81 | 0.50 |
| Outer retinal layers (ONL + IS + OS + RPE) | −13.92 | −8.33 | −19.50 | <0.001 | −14.50 | −10.31 | −18.69 | <0.001 |
| Outer nuclear layer | −11.28 | −8.42 | −14.13 | <0.001 | −15.45 | −12.08 | −18.82 | <0.001 |
Outer nuclear thickness could only be calculated for a subset of the aniridia patients (n = 19). IS, inner segment.
The outer retinal layers were significantly thinner in aniridia, across the whole horizontal meridian, compared with the controls, particularly in the foveal center (mean ± SD, 156.3 ± 32.3 µm vs 210.8 ± 12.3 µm, P < 0.001; Fig. 4C). The mean (SD) central foveal thicknesses of two of the component layers of the outer retina were thinner in participants with aniridia than the controls: ONL (73.0 ± 21.7 vs 104.6 ± 12.9 µm, P < 0.001) and OS (29.0 ± 7.2 vs 44.0 ± 3.2 µm, P < 0.001). Inner segment thickness was similar in the two groups (31.3 ± 3.7 vs 33.7 ± 2.4 µm, P = 0.5) while the RPE tended to be slightly thicker in aniridia (32.4 ± 7.0 vs 28.5 ± 3.7 µm, P = 0.3). Outer retinal layer thinning in the foveal center was associated with thinner nasal and temporal parafoveal and perifoveal retinal thickness (nasal parafovea: rp = 0.74, P < 0.001; temporal parafovea: rp = 0.75, P < 0.001; nasal perifovea: rp = 0.51, P = 0.009; temporal perifovea: rp = 0.61, P = 0.001) and Inner limiting membrane (ILM)-IPL thickness (nasal parafovea: rp = 0.56, P = 0.004; temporal parafovea: rp = 0.45, P = 0.02; nasal perifovea: rp = 0.52, P = 0.009; temporal perifovea: rp = 0.59, P = 0.002).
We found a strong negative correlation between foveal outer retinal layer thickness and high-contrast logMAR visual acuity in aniridia (rp = –0.80, P > 0.001). Multiple linear regression also showed that foveal outer retinal layer thickness was the strongest predictor of high-contrast logMAR visual acuity when AAK was grade 2 or less (r2 = 0.66, P < 0.001; Fig. 5). No correlations were observed between AL and foveal retinal thickness, except when AL <21.5 mm, which was only observed with grade 4 foveal hypoplasia and thin outer retinal layers. Analysis of variance followed by post hoc Bonferroni-corrected pairwise t-tests revealed no significant differences in outer retinal thickness between a normal fovea and foveal hypoplasia grade 1 (P = 0.3), between grades 1 and 2 (P = 0.15), or between grades 2 and 3 (P = 0.2). Foveal phenotypes did not correlate with iris phenotype.
Figure 5.
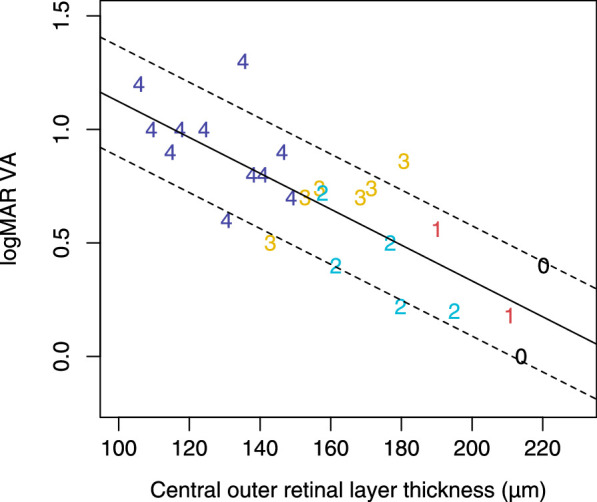
Relationship between foveal outer retinal layer thickness and high-contrast logMAR visual acuity in aniridia. The number of the datapoints for each participant corresponds to their grade of foveal hypoplasia.
Genotype-Phenotype Correlations
All participants with a PAX6 mutation had foveal hypoplasia. There was a large phenotypic variability among individuals with the same mutation, as well as between individuals with different mutations.
Participants with splice site mutations in the 5′ UTR of PAX6 (families 2 and 3) had, on average, thicker foveal outer retinal layers than those with mutations in the coding regions of the PAX6 gene (difference [range]: 27.7 [0.01–55.32] µm, P = 0.040; Fig. 6). Those with mutations in PAX6 coding regions that introduced a PTC and predicted haploinsufficiency due to Nonsense-mediated decay (NMD) had severe foveal hypoplasia (grade 3 or 4). In contrast, the participants who have large PAX6 deletions, including the retina-specific enhancer, had milder foveal hypoplasia (grades 1 and 2; Table 5). The family (no. 11) with a deletion in the 3′ regulatory region (ELP4-DCDC1), but an intact PAX6 transcriptional region, had complete iris hypoplasia but normal foveal shape or only mild foveal hypoplasia with outer retinal layer thickness within the normal range (Fig. 6). The two individuals with CTE mutations had complete foveal hypoplasia, thinning of outer retinal layers, and short AL <21 mm.
Figure 6.
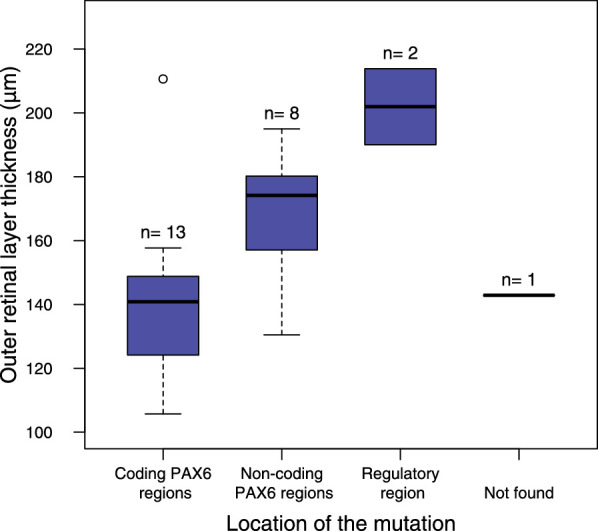
The boxplot shows the differences in foveal outer retinal layer thickness between participants who have mutations within PAX6 coding regions, PAX6 noncoding regions, and 3′ regulatory regions. One participant had no identified mutation.
Table 5.
Summary of the Types of Mutations and Retinal Phenotypes for the 26 Participants Who Were Imaged With OCT
| Mutation | ||||||
|---|---|---|---|---|---|---|
| ID | FH Grade | Central Retinal Thickness (µm) | Central Outer Retinal Thickness (µm) | Sex | Type | Remarks |
| 5124 | 0 | 239.27 | 220.22 | M | Not available | |
| 5139 | 0 | 232.48 | 213.88 | F | Deletion ELP4, DCD1 | Deletes retina enhancer |
| 5134 | 1 | 296.73 | 210.63 | F | Large deletion PAX6, ELP4 | Deletes retina enhancer |
| 5132 | 1 | 292.45 | 190.04 | F | Deletion ELP4, DCD1 | Deletes retina enhancer |
| 5199 | 2 | 338.12 | 194.98 | M | Splicing error PAX6 5′ UTR | |
| 5120 | 2 | 347.91 | 179.77 | M | Splicing error PAX6 5′ UTR | |
| 5123 | 2 | 323.30 | 176.84 | M | Splicing error PAX6 5′ UTR | |
| 5116 | 2 | 357.77 | 161.44 | M | Splicing error PAX6 5′ UTR | |
| 5154 | 2 | 300.10 | 157.69 | F | Large deletion PAX6, ELP4, DCD1 | Deletes retina enhancer |
| 5114 | 3 | 312.42 | 180.67 | F | Splicing error PAX6 5′ UTR | |
| 5125 | 3 | 343.52 | 171.49 | M | Splicing error PAX6 5′ UTR | |
| 5144 | 3 | 333.82 | 156.79 | F | PTC-PAX6 coding region | |
| 5135 | 3 | 342.37 | 152.71 | F | Splicing error PAX6 5′ UTR | |
| 5137 | 3 | 318.16 | 143.99 | F | PTC-PAX6 coding region | |
| 5147 | 3 | 307.81 | 142.82 | F | No mutation found | |
| 5140 | 4 | 316.55 | 148.83 | F | CTE-PAX6 coding region | |
| 5138 | 4 | 336.59 | 146.20 | F | PTC-PAX6 coding region | |
| 5113 | 4 | 325.71 | 140.84 | F | PTC-PAX6 coding region | |
| 5126 | 4 | 310.08 | 137.64 | M | PTC-PAX6 coding region | |
| 5131 | 4 | 340.81 | 135.18 | F | PTC-PAX6 coding region | |
| 5148 | 4 | 302.02 | 130.50 | F | Splicing error PAX6 5′ UTR | |
| 5141 | 4 | 284.63 | 124.15 | M | PTC-PAX6 coding region | |
| 5119 | 4 | 293.74 | 117.36 | F | Not available | |
| 5149 | 4 | 316.80 | 114.61 | F | CTE-PAX6 coding region | |
| 5117 | 4 | 282.13 | 109.26 | F | Large deletion PAX6 | Intact retina enhancer |
| 5127 | 4 | 305.73 | 105.72 | F | PTC-PAX6 coding region | |
The participants are ordered according to foveal hypoplasia grade corresponding to the OCT images in Figure 3.
Discussion
Mutations in the PAX6 gene or in one or several of its regulatory regions were found in 97% of the participants with aniridia, with 3 of 20 different pathogenic variants not reported previously. Comparison of the genotype with the detailed retinal phenotype in aniridia showed that all PAX6 mutations were associated with varying degrees of arrested foveal development, reduced outer retinal thickness, and altered macular morphology. There was an association between the location of the gene mutation and macular phenotype. The better-developed retinas were associated with mutations before the translational start codon (PAX6 5′ UTR), deletions including the 3′ regulatory region (ELP4-DCDC1) only, and in large PAX6 deletions when the downstream regulatory region retina enhancer was deleted. This underscores the role of PAX6 in foveal and macular development and indicates that residual PAX6 function may contribute to variation in phenotype depending on mutation location.
The mutations associated with the thickest foveal outer retinal layers and the mildest retinal phenotypes were mutations outside the PAX6 gene (3′ regulatory regions ELP4-DCDC1) followed by splice site mutations in the 5′ UTR of PAX6. Mutations at these locations were generally associated with a better-developed fovea than multiple PAX6 exon deletions and mutations within the PAX6 protein coding regions, which are predicted to cause haploinsufficiency due to nonsense-mediated decay of the mRNA. Deletions in 3′ regulatory regions may affect PAX6 expression through disruption of enhancer activity,20 whereas variants in the untranslated regions may affect PAX6 function by altering normal splicing or disrupting open reading frames.6 However, it is not known exactly how splice site mutations for the noncoding exons affect pre-mRNA splicing, mRNA expression level, or protein translation. The two variants in 5′ UTR in the present study (c.-128-2delA and c.52+1G>A) have been demonstrated to lead to skipping of exon 3 (in vitro) and exons 3 to 6 (reverse transcription PCR), respectively, 6,39 suggesting that the most likely outcome is haploinsufficiency.
It is clear that the contribution of each retinal layer to retinal thickness within the macula (central 6 mm) is considerably different in aniridia compared with the normal controls. During embryonic eye development, PAX6 induces the differentiation of progenitor cells into retinal neurons.40–42 The detailed analysis of the OCT images presented here shows that in eyes with aniridia, not only was the retina thicker and outer retinal layers thinner in the foveal center, but parafoveal and perifoveal inner and outer retinal layers were also significantly thinner than in the normal controls. This confirms previous findings in animal studies that loss off PAX6 expression leads to a hypocellular macula.40 It also corroborates the findings from other human studies,11,17 including a study using adaptive optics scanning light ophthalmoscopy, that revealed decreased macular cone photoreceptor density within one family with aniridia.10 Thinner foveal outer retinal layers suggest that cone specialization and migration have not occurred to a full extent, resulting in immature foveal cones and decreased cone density. Absence of the IZ band (the photoreceptor–RPE apical processes interdigitation zone), observed in some of those with aniridia, may indicate immature foveal cone outer segments resulting in shorter cones with less indentation into the RPE. This could result in a hyperreflective signal from the IZ that is more anterior than in a retina with longer and more mature cones (hence the tendency for the RPE to be thicker in aniridia).
The increased central foveal thickness in PAX6-associated aniridia appears to be caused by lack of centrifugal migration of the inner retinal layers away from the fovea. This is possibly related to the absence of a foveal avascular zone, which is known to prevent the formation of a foveal pit.25 PAX6 is important for the development of retinal ganglion cells43 and for RPE specification and pigmentation.44 The RPE and ganglion cells normally secrete pigment epithelium derive factor, which prevents retinal blood vessels from invading the foveal region during early development.25,45 EphA6, which plays a role in regulating astrocyte migration across the retina, is also highly expressed by ganglion cells.25 Thus, it is plausible that PAX6 mutations affect the expression of antiproliferative and antiangiogenic factors in the developing foveal region, which again lead to abnormal foveal vascularization.
Multiple exon deletions have previously been reported to result in a more severe corneal phenotype.46 The same was observed here as four of seven participants with large PAX6 deletions had corneal opacities that precluded retinal imaging. The three who were imaged (5134, 5154, 5117) had retinal phenotypes ranging from mild to severe (foveal hypoplasia [FH] grades 1–4). Interestingly, the one participant (5117) with a severe phenotype had a deletion that included the region covered by the MLPA probes from PAX6 exons 3–9, but left the DRR retina-specific enhancer intact. The mRNA from this deletion is likely to be unstable and be targeted to NMD. Thus, it appears that mutations that are predicted to target mRNA to destruction in NMD have a more severe phenotype than mutations that deleted the retina enhancer and thus do not produce any mRNA in the retina. The NMD pathway does not work with 100% efficiency, and there is variability in its efficiency even across family members with the same mutations.47 Thus, one explanation for the difference in severity for participants lacking the retina enhancer versus those with a mutation that would be transcribed, but the mRNA targeted to destruction, is that a small amount of the mRNA escapes NMD. The escaped mRNA is expected to be translated into a mutant protein that has a dominant negative effect. Variation in the efficiency of NMD across individuals may also contribute to the observed variability in phenotypes among family members with the same PAX6 mutation.
CTE and PTC mutations were associated with moderate to severe retinal phenotypes. Myopia has been reported to be a common feature of CTE mutations,8,48 but the two in our cohort were both hyperopic with a short AL (<21 mm). Aniridia has often been associated with small eyes, but our results show that there is large variability in ocular AL among those who have PAX6-associated aniridia. Only three participants were diagnosed with microphthalmia (AL <21 mm)49 accompanied by a severe retinal phenotype (FH grade 4, see 5140 and 5149 in Figure 3; ocular media opacities precluded OCT imaging in 5128). Normal eye growth is an important factor for emmetropization and is also thought to play a role in foveal maturation, including cone packing and elongation.50 In normally developing eyes, a large part of foveal maturation and cone packing occurs after birth and up to the age of at least 16 years.51 At birth, foveal photoreceptors are shorter than parafoveal and perifoveal photoreceptors, but they elongate as the eye grows and the fovea continues to mature.51 The increased outer retinal layer thickness in the foveal center relative to the perifovea observed in some persons with PAX6-associated aniridia therefore reflects photoreceptor development and foveal maturation after birth. This was evident in those who had a mutation in either 3′ regulatory regions (ELP4-DCDC1) or splice site mutations in the 5′ UTR of PAX6 but also in some participants with large PAX6 deletions including, the DRR (5134 and 5154). Longitudinal studies are required to understand the association between ocular axial growth and foveal maturation in aniridia, as well as the importance of refractive error correction and visual stimulation from an early age.
The effects of PAX6 mutations are dose dependent, and thus mutations on both alleles will cause a very severe phenotype that usually are lethal before birth.7 In the present study, participant 5125 exhibited classical aniridia and had two mutations, a splice site mutation (c.-52+1G>A) in intron 3 and a mutation in exon 10 (c.831 G>A). These two mutations are therefore likely to be present in one allele where the former one (c.-52+1G>A) first terminates the PAX6 protein. Alternatively, the mutation in exon 10 (c.831G>A) is a silent mutation because it does not alter amino acid as reported previously,39 although Mutation Taster21 predicted that it would cause a splicing defect and be disease causing.
No pathologic sequence changes were observed in one sporadic case with classical aniridia indistinguishable from PAX6-associated aniridia. Next-generation sequencing approaches such as whole-genome sequencing or whole-exome analysis might be able to uncover the mutation in this individual. Somatic PAX6 mosaicism has been described and might explain the presence of aniridia in persons without detectable PAX6 mutations.52 Mosaicism could also be a possible genetic factor to explain variable expressivity within families.52
A strength of this study was the detailed examination of foveal anatomy across a wide phenotypic and genotypic range of aniridia. Based on a reported prevalence of 1:72 000,1 the 37 participants in this study represent >50% of the persons who have aniridia in Norway and are therefore reasonably representative for persons with aniridia across the country. Despite limitations related to only being able to image with adequate quality in 26 participants with aniridia, the present analysis includes the full range of aniridia phenotypes.
Conclusions
The results presented here show significant variation in outer retinal layer thickness measurements within each grade of foveal hypoplasia. Importantly, outer retinal layer thickness was the structural measure with the highest correlation with visual acuity in participants without severe central AAK. Notable examples are participants 5154 and 5199, both with FH grade 2, but participant 5154 has 35-µm thinner outer retinal layers than participant 5199 with correspondingly poorer visual acuity (0.72 vs 0.20 logMAR, respectively). In clinical terms, this implies that qualitative grading (1–4) of foveal hypoplasia from OCT images is too coarse and may misclassify visual outcome for persons with aniridia.
PAX6 mutations were found to be associated with abnormal foveal formation and reduced number of neurons in the macula, with mutations in PAX6 coding regions giving the worst outcome. The observed variation underscores the importance of careful retinal phenotypic characterization, even when the PAX6 mutation is known, for predicting visual outcome after surgery, optical visual rehabilitation, and future gene therapy. If photoreceptor development extends after birth in some eyes with PAX6-associated aniridia, as the increased foveal outer retinal layer thicknesses observed here indicate, it suggests that early visual stimulation and optimization of visual function are as important as they are in normally developing eyes. This may be a factor in improving foveal maturation outcome in adult life.
Acknowledgments
The authors thank the Norwegian Association of Aniridia (Aniridi Norge) for the continued cooperation and support.
Supported by the University of South-Eastern Norway and Aniridia Norway. Work done at the University of Washington was supported by NEI Grants P30EY001730 and R01EY028118 and by unrestricted funds from Research to Prevent Blindness. The genetic analysis portion of this work was conducted by the University of Washington and was supported by Research to Prevent Blindness and National Institutes of Health/National Eye Institute grant NIH P30EY001730. HRP holds a PhD position funded by the Norwegian Ministry of Education and Research.
Disclosure: H.R. Pedersen, None; R.C. Baraas, None; E.C.S. Landsend, None; Ø.A. Utheim, None; T.P. Utheim, None; S.J. Gilson, None; M. Neitz, None
References
- 1. Edén U, Iggman D, Riise R, Tornqvist K. Epidemiology of aniridia in Sweden and Norway. Acta Ophthalmologica. 2008; 86: 727–729. [DOI] [PubMed] [Google Scholar]
- 2. Shaw MW, Falls HF, Neel JV. Congenital aniridia. Am J Hum Genet. 1960; 12: 389–415. [PMC free article] [PubMed] [Google Scholar]
- 3. Ton CC, Hirvonen H, Miwa H, et al.. Positional cloning and characterization of a paired box- and homeobox-containing gene from the aniridia region. Cell. 1991; 67: 1059–1074. [DOI] [PubMed] [Google Scholar]
- 4. Lauderdale JD, Wilensky JS, Oliver ER, Walton DS, Glaser T. 3' Deletions cause aniridia by preventing PAX6 gene expression. Proc Natl Acad Sci U S A. 2000; 97: 13755–13759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kleinjan DA, Seawright A, Schedl A, Quinlan RA, Danes S, van Heyningen V. Aniridia-associated translocations, DNase hypersensitivity, sequence comparison and transgenic analysis redefine the functional domain of PAX6. Hum Mol Genet. 2001; 10: 2049–2059. [DOI] [PubMed] [Google Scholar]
- 6. Plaisancie J, Tarilonte M, Ramos P, et al.. Implication of non-coding PAX6 mutations in aniridia. Hum Genet. 2018; 137: 831–846. [DOI] [PubMed] [Google Scholar]
- 7. Landsend ES, Utheim OA, Pedersen HR, Lagali N, Baraas RC, Utheim TP. The genetics of congenital aniridia-a guide for the ophthalmologist. Surv Ophthalmol. 2018; 63: 105–113. [DOI] [PubMed] [Google Scholar]
- 8. Hingorani M, Williamson KA, Moore AT, van Heyningen V. Detailed ophthalmologic evaluation of 43 individuals with PAX6 mutations. Invest Ophthalmol Vis Sci. 2009; 50: 2581–2590. [DOI] [PubMed] [Google Scholar]
- 9. Yokoi T, Nishina S, Fukami M, et al.. Genotype-phenotype correlation of PAX6 gene mutations in aniridia. Hum Genome Var. 2016; 3: 15052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pedersen HR, Neitz M, Gilson SJ, et al.. The cone photoreceptor mosaic in aniridia: within-family phenotype-genotype discordance. Ophthalmol Retina. 2019; 3: 523–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sannan NS, Gregory-Evans CY, Lyons CJ, et al.. Correlation of novel PAX6 gene abnormalities in aniridia and clinical presentation. Can J Ophthalmol. 2017; 52: 570–577. [DOI] [PubMed] [Google Scholar]
- 12. Gillespie FD. Aniridia cerebellar ataxia, and oligophrenia in siblings. Arch Ophthalmol. 1965; 73: 338–341. [DOI] [PubMed] [Google Scholar]
- 13. Remez LA, Onishi A, Menuchin-Lasowski Y, et al.. Pax6 is essential for the generation of late-born retinal neurons and for inhibition of photoreceptor-fate during late stages of retinogenesis. Dev Biol. 2017; 432: 140–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shaham O, Menuchin Y, Farhy C, Ashery-Padan R. Pax6: a multi-level regulator of ocular development. Prog Retin Eye Res. 2012; 31: 351–376. [DOI] [PubMed] [Google Scholar]
- 15. Casas-Llera P, Siverio A, Esquivel G, Bautista C, Alio JL. Spectral-domain optical coherence tomography foveal morphology as a prognostic factor for vision performance in congenital aniridia. Eur J Ophthalmol. 2020;30:58-65. [DOI] [PubMed] [Google Scholar]
- 16. Thomas MG, Kumar A, Mohammad S, et al.. Structural grading of foveal hypoplasia using spectral-domain optical coherence tomography a predictor of visual acuity? Ophthalmology. 2011; 118: 1653–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pedersen HR, Hagen LA, Landsend ECS, et al.. Color vision in aniridia. Invest Ophthalmol Vis Sci. 2018; 59: 2142–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Landsend ECS, Pedersen HR, Utheim OA, et al.. Meibomian gland dysfunction and keratopathy are associated with dry eye disease in aniridia. Br J Ophthalmol. 2019; 103: 119–124. [DOI] [PubMed] [Google Scholar]
- 19. den Dunnen JT, Dalgleish R, Maglott DR, et al.. HGVS recommendations for the description of sequence variants: 2016 update. Hum Mutat. 2016; 37: 564–569. [DOI] [PubMed] [Google Scholar]
- 20. Bhatia S, Bengani H, Fish M, et al.. Disruption of autoregulatory feedback by a mutation in a remote, ultraconserved PAX6 enhancer causes aniridia. Am J Hum Genet. 2013; 93: 1126–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014; 11: 361–362. [DOI] [PubMed] [Google Scholar]
- 22. Ansari M, Rainger J, Hanson IM, et al.. Genetic analysis of 'PAX6-negative' individuals with aniridia or Gillespie syndrome. PLoS One. 2016; 11: e0153757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Landsend ECS, Pedersen HR, Utheim OA, et al.. Characteristics and utility of fundus autofluorescence in congenital aniridia using scanning laser ophthalmoscopy. Invest Ophthalmol Vis Sci. 2019; 60: 4120–4128. [DOI] [PubMed] [Google Scholar]
- 24. Lee H, Purohit R, Patel A, et al.. In vivo foveal development using optical coherence tomography. Invest Ophthalmol Vis Sci. 2015; 56: 4537–4545. [DOI] [PubMed] [Google Scholar]
- 25. Provis JM, Dubis AM, Maddess T, Carroll J. Adaptation of the central retina for high acuity vision: cones, the fovea and the avascular zone. Prog Retin Eye Res. 2013; 35: 63–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rohrschneider K. Determination of the location of the fovea on the fundus. Invest Ophthalmol Vis Sci. 2004; 45: 3257–3258. [DOI] [PubMed] [Google Scholar]
- 27. Kass M, Witkin A, Terzopoulos D. Snakes: active contour models. International Journal of Computer Vision. 1988; 1: 321–331. [Google Scholar]
- 28. Mishra A, Wong A, Bizheva K, Clausi DA. Intra-retinal layer segmentation in optical coherence tomography images. Opt Express. 2009; 17: 23719–23728. [DOI] [PubMed] [Google Scholar]
- 29. González-López A, de Moura J, Novo J, Ortega M, Penedo MG. Robust segmentation of retinal layers in optical coherence tomography images based on a multistage active contour model. Heliyon. 2019; 5: e01271–e01271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lujan BJ, Roorda A, Croskrey JA, et al.. Directional optical coherence tomography provides accurate nuclear layer and Henle fiber layer measurements. Retina. 2015; 35: 1511–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. R Core Team. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2016. [Google Scholar]
- 32. Pinheiro JC, Bates DJ, DebRoy S, Sakar D; Core Team R. nmle: Linear and Nonlinear Mixed Effects Models R package version 3.1-141 2019, https://CRAN.R-project.org/package=nlme. [Google Scholar]
- 33. Frischmeyer PA, Dietz HC.. Nonsense-mediated mRNA decay in health and disease. Hum Mol Genet. 1999; 8: 1893–1900. [DOI] [PubMed] [Google Scholar]
- 34. Tzoulaki I, White IM, Hanson IM. PAX6 mutations: genotype-phenotype correlations. BMC Genet. 2005; 6: 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kozak M. The scanning model for translation: an update. J Cell Biol. 1989; 108: 229–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Grønskov K, Olsen JH, Sand A, et al.. Population-based risk estimates of Wilms tumor in sporadic aniridia: a comprehensive mutation screening procedure of PAX6 identifies 80% of mutations in aniridia. Hum Genet. 2001; 109: 11–18. [DOI] [PubMed] [Google Scholar]
- 37. Kikuta H, Laplante M, Navratilova P, et al.. Genomic regulatory blocks encompass multiple neighboring genes and maintain conserved synteny in vertebrates. Genome Res. 2007; 17: 545–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kleinjan DA, van Heyningen V. Long-range control of gene expression: emerging mechanisms and disruption in disease. Am J Hum Genet. 2005; 76: 8–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Grønskov K, Rosenberg T, Sand A, Brondum-Nielsen K. Mutational analysis of PAX6: 16 novel mutations including 5 missense mutations with a mild aniridia phenotype. Eur J Hum Genet. 1999; 7: 274–286. [DOI] [PubMed] [Google Scholar]
- 40. Klimova L, Kozmik Z.. Stage-dependent requirement of neuroretinal Pax6 for lens and retina development. Development. 2014; 141: 1292–1302. [DOI] [PubMed] [Google Scholar]
- 41. Oron-Karni V, Farhy C, Elgart M, et al.. Dual requirement for Pax6 in retinal progenitor cells. Development. 2008; 135: 4037–4047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Marquardt T, Ashery-Padan R, Andrejewski N, Scardigli R, Guillemot F, Gruss P. Pax6 is required for the multipotent state of retinal progenitor cells. Cell. 2001; 105: 43–55. [DOI] [PubMed] [Google Scholar]
- 43. Hsieh YW, Yang XJ.. Dynamic Pax6 expression during the neurogenic cell cycle influences proliferation and cell fate choices of retinal progenitors. Neural Dev. 2009; 4: 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Raviv S, Bharti K, Rencus-Lazar S, et al.. PAX6 regulates melanogenesis in the retinal pigmented epithelium through feed-forward regulatory interactions with MITF. PLoS Genet. 2014; 10: e1004360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kozulin P, Natoli R, Bumsted O'Brien KM, Madigan MC, Provis JM. The cellular expression of antiangiogenic factors in fetal primate macula. Invest Ophthalmol Vis Sci. 2010; 51: 4298–4306. [DOI] [PubMed] [Google Scholar]
- 46. Lagali N, Wowra B, Fries FN, et al.. PAX6 mutational status determines aniridia-associated keratopathy phenotype. Ophthalmology. 2020;127:273–275. [DOI] [PubMed] [Google Scholar]
- 47. Nguyen LS, Wilkinson MF, Gecz J. Nonsense-mediated mRNA decay: inter-individual variability and human disease. Neurosci Biobehav Rev. 2014; 46(pt 2): 175–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Souzeau E, Rudkin AK, Dubowsky A, et al.. PAX6 molecular analysis and genotype-phenotype correlations in families with aniridia from Australasia and Southeast Asia. Mol Vis. 2018; 24: 261–273. [PMC free article] [PubMed] [Google Scholar]
- 49. Harding P, Moosajee M.. The molecular basis of human anophthalmia and microphthalmia. J Dev Biol. 2019; 7:E16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Springer AD, Hendrickson AE.. Development of the primate area of high acuity, 3: temporal relationships between pit formation, retinal elongation and cone packing. Vis Neurosci. 2005; 22: 171–185. [DOI] [PubMed] [Google Scholar]
- 51. Vajzovic L, Hendrickson AE, O'Connell RV, et al.. Maturation of the human fovea: correlation of spectral-domain optical coherence tomography findings with histology. Am J Ophthalmol. 2012; 154: 779–789.e772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tarilonte M, Morin M, Ramos P, et al.. Parental mosaicism in PAX6 causes intra-familial variability: implications for genetic counseling of congenital aniridia and microphthalmia. Front Genet. 2018; 9: 479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chylack LT Jr, Wolfe JK, Singer DM, et al.. The Lens Opacities Classification System III. The Longitudinal Study of Cataract Study Group. Arch Ophthalmol. 1993; 111: 831–836. [DOI] [PubMed] [Google Scholar]
- 54. Richards S, Aziz N, Bale S, et al.. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015; 17: 405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]