

Abstract
Macrophages, a kind of innate immune cells, derive from monocytes in circulation and play a crucial role in the innate and adaptive immunity. Under the stimulation of the signals from local microenvironment, macrophages generally tend to differentiate into two main functional phenotypes depending on their high plasticity and heterogeneity, namely, classically activated macrophage (M1) and alternatively activated macrophage (M2). This phenomenon is often called macrophage polarization. In pathological conditions, chronic persistent inflammation could induce an aberrant response of macrophage and cause a shift in their phenotypes. Moreover, this shift would result in the alteration of macrophage polarization in some vascular dermatoses; e.g., an increase in proinflammatory M1 emerges from Behcet's disease (BD), psoriasis, and systemic lupus erythematosus (SLE), whereas an enhancement in anti-inflammatory M2 appears in infantile hemangioma (IH). Individual polarized phenotypes and their complicated cytokine networks may crucially mediate in the pathological processes of some vascular diseases (vascular dermatosis in particular) by activation of T cell subsets (such as Th1, Th2, Th17, and Treg cells), deterioration of oxidative stress damage, and induction of angiogenesis, but the specific mechanism remains ambiguous. Therefore, in this review, we discuss the possible role of macrophage polarization in the pathological processes of vascular skin diseases. In addition, it is proposed that regulation of macrophage polarization may become a potential strategy for controlling these disorders.
1. Introduction
Macrophages are a group of innate immune cells coming from peripheral blood monocytes. Owing to their multifunctional activities, macrophages potently work in homeostasis maintenance, inflammation, angiogenesis, wound healing, etc. [1]. In general, macrophages differentiate into two functional phenotypes at the microenvironment signal stimulus, namely, classically activated macrophage (M1) and alternatively activated macrophage (M2); this is known as macrophage polarization [2, 3]. M1, an inflammatory phenotype, is dominated by Toll-like receptor (TLR) 4 ligand, lipopolysaccharide (LPS), or Th1 cytokines [e.g., IFN-γ, TNF-α, and granulocyte-macrophage colony-stimulating factor (GM-CSF)], being marked by CD40, CD80, and CD86 and possessing proinflammatory, tumoricidal, and antimicrobial activity [4, 5]; inversely, M2 is an anti-inflammatory phenotype polarized by Th2 cytokines [e.g., IL-4, IL-10, IL-13, and macrophage colony-stimulating factor (M-CSF)], specifically expressing the markers of CD163, CD206, CD209, and CD301 and being linked to wound healing, inflammation elimination, angiogenesis, and tumorigenesis [6, 7].
In fact, macrophage polarization often occurs in an inflammatory process and therefore many studies about inflammatory disorders or inflammation-related vascular diseases always focus on these two polarized macrophages [8–10]. Recently, it is found macrophage polarization heavily functions in some immune-mediated inflammatory vascular dermatoses, like Behcet's disease (BD), psoriasis, and systemic lupus erythematosus (SLE) [11–13]. There is an upregulation of M1 polarized macrophages in above disorders, which not only causes disproportions in Th1/Th2 and Th17/Treg cells but also leads to a worsening state of oxidative stress (OS), while M2 polarized macrophages markedly increase in angiogenic disorders, such as infantile hemangioma (IH) [14, 15]. Thus, macrophage polarization imbalance would be a major contributor to these dermatoses, and thereby, regulation of macrophage polarization may be a potential target for vascular skin disease treatment.
2. Macrophage Polarization
Macrophage polarization has profound impacts on various physiological and pathological conditions, such as angiogenesis, wound repair, inflammation, and tumorigenesis. Regardless of physiological or pathological process, a serial of signaling pathways and diverse mediators (e.g., cytokines, chemokines, transcriptional factors) are heavily implicated in macrophage polarization. Signals from the local microenvironment are modulated by various receptors on the macrophages to initiate multiple pathways of macrophage polarization.
2.1. Related Signal Pathways in Macrophage Polarization
On exposure to local microenvironment, macrophages convert into two phenotypes (M1 and M2) via activating related signaling pathways. In the process of M1 macrophage polarization, two well-known signals, namely, IFN-γ and LPS, are mainly involved [16–18]. After binding to their corresponding receptors (IFNGR and TLR4), IFN-γ and LPS recruit the adaptors of Janus kinase 1/2 (JAK1/2), TLR domain-containing adapter protein [interferon-β (TRIF) and myeloid differentiation factor 88 (MyD88)], further activating the downstream factors of interferon regulatory factor 3 (IRF3), IL-1 receptor-associated kinase 4 (IRAK-4), TNF receptor-associated factor 6 (TRAF-6), and inhibitor of nuclear factor kappa B kinase (IKK-β), ultimately resulting in the activation of signal transducer and activator of transcription 1 (STAT1) and nuclear factor kappa B (NF-κB) [19–21]. These factors contribute to M1 polarization that promotes the expression of inflammatory genes including TNF-α, B lymphocyte stimulator (BAFF), IL-1B, cyclooxygenase 2 (COX2), CXCL9, CXCL10, IL-6, and IL-12p40 [22–24]. On the other hand, IL-4, IL-10, and IL-13, respectively, combine with their corresponding receptors to activate JAK1/3, STAT3, and STAT6 [25–27]. Both activated STAT3 and STAT6 encourage M2 polarization and elicit the production of anti-inflammatory cytokines (shown in Figure 1).
Figure 1.
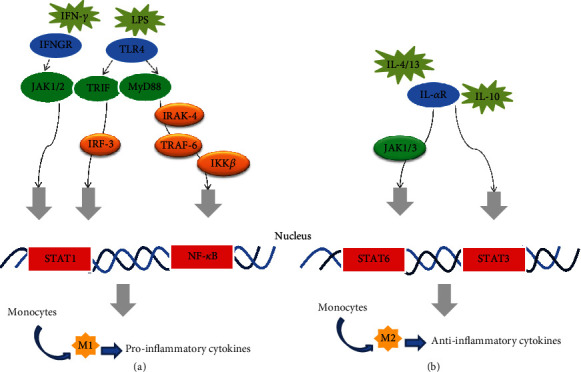
Several signaling pathways mediate in macrophage polarization. (a) M1 macrophage polarization and (b) M2 macrophage polarization are shown with some signal pathways or factors involved in their development. Although this graph displays two categories of macrophage, in fact a dynamic spectrum of polarization often occurs. Abbreviations: IFN-γ: interferon gamma; LPS: lipopolysaccharide; IFNGR: interferon gamma receptor; TLR4: Toll-like receptor-4; JAK1/2/3: Janus kinase1/2/3; TRIF: TLR domain-containing adapter protein inducing interferon-β; MyD88: myeloid differentiation factor 88; IL-4/10/13: interleukin 4/10/13; IL-αR: interleukin receptor; IRF-3: interferon regulatory factor 3; IRAK-4: interleukin-1 receptor-associated kinase 4; TRAF-6: tumor necrosis factor receptor-associated factor 6; IKK-β: inhibitor of nuclear factor kappa B kinase; STAT1/3/6: signal transducer and activator of transcription 1/3/6; NF-κB: nuclear factor kappa B.
But it is notable that there is a dynamic spectrum of polarization occurring in macrophages and the direction of macrophage polarization is modulated by some special signal pathways. More importantly, the phosphatidylinositol 3-kinase/protein kinase B (PI3K/Akt) pathway and its downstream targets recently emerge as the central regulators of activated phenotype in macrophages [28]. This pathway mediates multiple signals (chemokines, LPS, IL-10, and IL-4) from a variety of receptors mainly involving TLR4 and cytokine receptors (IL-αR). The PI3K is initially activated by the above signals via binding to their respective receptors, but the activation of different Akt isoforms (the downstream of PI3K, namely, Akt1 isoform and Akt2 isoform) greatly switches the direction of macrophage polarization. Some studies showed that Akt1 activation could inhibit M1 polarization; conversely, Akt2 activation prevented M2 polarization [29, 30]. Until now, however, the specific mechanisms still remain unclear [28].
2.2. Inflammatory Mediators from M1 or M2
Different polarized macrophages perform their unique function via secreting a variety of inflammatory mediators, i.e., cytokines, chemokines, transcriptional factors. These mediators in turn actively participate in macrophage polarization. Basing on macrophage phenotypes, the mediators from macrophages are classified into two categories, namely, M1-derived mediators and M2-derived mediators. M1 activation contributes to the production of proinflammatory cytokines (e.g., TNF-α, IFN-γ, IL-6, IL-10, IL-23), chemokines (MCP-1, CCL2-4, CXCL8-11, and GM-CSF), nitric oxide (NO), and reactive oxygen species (ROS). Moreover, antigen-presenting molecule MHC-II highly express on M1 and benefit M1 polarization [17, 18]. In the presence of M2 activation, however, the following mediators are markedly upregulated, involving anti-inflammatory cytokines [transforming growth factor-β (TGF-β), IL-4, and IL-13], chemokines (CCL17, 18, 22, 24, and M-CSF, and proangiogenic factors [vascular endothelial growth factor A (VEGF-A), VEGF-C, platelet-derived growth factor (PDGF),epidermal growth factor (EGF), and basic fibroblast growth factor 2 (FGF-2)] [31–33]. As a result, all these inflammatory mediators further mediate in physiological or pathological process and encourage a normal or morbid switch.
3. Macrophage Polarization in Physiological and Pathological Conditions
Macrophage polarization serves as a crucial role in physiology and disease status, such as embryonic development, individual growth, homeostasis maintenance, immunity defence, inflammation, and trauma. At the stage of embryonic development, fetal macrophages display an M2 phenotype and energetically promote angiogenesis, tissue growth, and organ formation, angiogenesis in particular [33]. With the maturation of the innate immune system, the M1/M2 population tends to balance and maintains body homeostasis [34]. In pathological conditions, however, when inflammation occurs, vascular tissue is the major response place where macrophages have a switch from M1 to M2 [35]. M1 actively participates in the initial process of vascular inflammation, which benefits to the establishment of a proinflammatory response. If M1 phase persistently exists, tissue damage would occur. Instead, the sequential presence of M2 macrophages facilitates damage termination and tissue repair in the later phase [35, 36]. However, in many vascular disorders especially in vascular dermatosis, the vascular microenvironment fills with various inflammatory mediators from infiltrated lymphocytes and resident parenchymal cells. The interaction of these cells with the mediators initiates a serial of proinflammatory signals to trigger the persistence M1 polarization. The predominance of M1 vs. M2 would result in extensive vascular damage, abnormal repair, and clinical deterioration. On the contrary, the persistence of M2 polarization leads to the occurrence and development of angiogenic diseases.
4. Possible Mechanisms of Macrophage Polarization in Vascular Complaints
Although the specific mechanism of macrophage polarization in vascular disorders keeps obscure, it is considered that T cell dysregulation, oxidative stress damage, and angiogenesis are probably involved in macrophage polarization-mediated vascular diseases, especially in some vascular skin complaints, e.g., BD, psoriasis, SLE, and IH (summarized in Figure 2).
Figure 2.
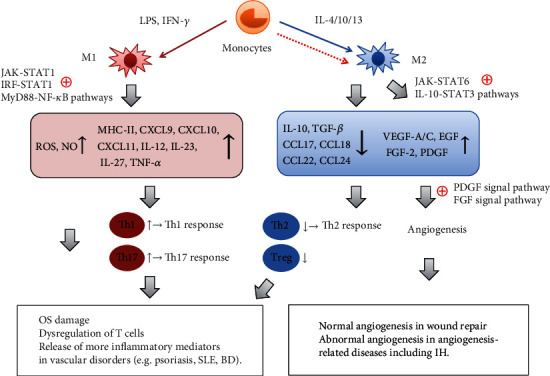
Possible mechanisms of different macrophage polarizations in vascular disorders. Upon the different stimuli, monocytes tend to differentiate into M1 or M2 macrophages via different signaling pathways. In most vascular inflammatory diseases, M1 activation is dominant, whereas M2 activation is relatively inhibited. Through activation of the pathways (JAK/STAT1, IRF/STAT1, and MyD88/NF-κB), activated M1 macrophages release various inflammatory mediators, such as MHC-II, chemokines (CXCL10 and CXCL11), and inflammatory cytokines (TNF-α, IL-12, IL-23, and IL-27), to encourage the activation of Th1/Th17 cells and trigger Th1/Th17 response. Apart from that, M1 macrophages produce substantial ROS and NO. As the key factors that regulate the differentiation and chemotaxis of Th2/Treg cells, however, M2-secreted chemokines (CCL17, CCL18, CCL22, and CCL24) and anti-inflammatory cytokines (TGF-β, IL-10) markedly decrease along with the inhibition of activated M2. As a result, these events may contribute to the appearance of T cell dysregulation, OS damage, and increased inflammatory mediators in the pathological process of vascular inflammatory disorders or dermatoses, e.g., psoriasis, SLE, and BD. On the other hand, activated M2 macrophages secrete abundant angiogenic factors (e.g., VEGF-A/C, FGF-2, EGF, and PDGF) via stimulating the JAK/STAT6 and IL-10/STAT3 pathways, which not only facilitate normal angiogenesis in wound repair but also promote abnormal angiogenesis in angiogenesis-related diseases including IH by activating the PDGF and FGF signal pathways. Notes: the red and blue solid arrows indicate normal activation, while the red dotted arrow indicates relative inactivation; ⊕indicates “activation,” ↑ indicates “upregulation,” and ↓ indicates “downregulation.” Abbreviations: LPS: lipopolysaccharide; IFN-γ: interferon gamma; IL-4/10/13: interleukin 4/10/13; M1: classically activated macrophage; M2: alternatively activated macrophage; ROS: reactive oxygen species; NO: nitric oxide; MHC-II: major histocompatibility complex-II; CXCL9/10/11: chemokine (c-x-c motif) ligand 9/10/11; IL-10/12/23/27: interleukin 10/12/23/27; TNF-α: tumor necrosis factor alpha; CCL17/18/22/24: chemokine (c-c motif) ligand 17/18/22/24; VEGF-A/C: vascular endothelial growth factor A/C; EGF: epidermal growth factor; FGF-2: fibroblast growth factor 2; PDGF: platelet-derived growth factor; Th: T helper; Treg: regulatory T cells; OS: oxidative stress; SLE: systemic lupus erythematosus; BD: Behcet's disease; IH: infantile hemangioma
4.1. M1 Polarization Causes T Cell Dysregulation
In vascular inflammation site, macrophages and T cells always coexist and interact with each other [37, 38]. The dynamic equilibrium of M1/M2 benefits to balance inflammatory T cells (Th1 and Th17 cells) and anti-inflammatory T cells (Th2 and Treg cells) in quantity, proportion, and function [39–41]. In certain vascular inflammatory diseases, e.g., psoriasis, BD, and atherosclerosis, however, this balance is broken [42–44]. Under the stimulation from chronic inflammatory signals, macrophages are provoked mainly as M1, whereas M2 activation is relatively inhibited [42, 45]. Activated M1 in turn impels T cell activation and differentiation [46]. CD4+ T cells, as macrophage-stimulated effector cells, usually function as immune modulators by differentiating into various functional subtypes (e.g., Th1/Th2/Th17 cells and Treg cells) [47]. Accordingly, M1-produced chemokines (CXCL9-11) attract Th1/Th17 cells to the vascular inflammatory site [48]; beyond that, some proinflammatory factors from M1 (IL-12, IL-23, IL-27, etc.) encourage Th1/Th17 differentiation and Th1/Th17-derived factor secretion (IL-17A, IFN-γ, IL-17F, IL-21, and IL-26) [49, 50]. Consequently, Th1 and Th17 cells promote the progress of vascular inflammation and accelerate vascular tissue damage through secreting above inflammatory cytokines [51–53]. Worse yet, Th1 cells could recruit more M1 to the vascular inflammatory site and prevent M2 activation, thereby forming a vicious circle [54–56]. Just owing to M2 inactivation, the differentiation of anti-inflammatory cells (Th2/Treg cells) is arrested, which may contribute to T cell dysregulation and aggravate vascular tissue damage [57]. Besides, the decrease of Treg cells that skew the macrophages toward M2 would encourage the state of M1 polarization and in turn exacerbate vascular injury [18, 58, 59].
4.2. M1 Polarization Aggravates OS Damage
ROS and NO, the crucial mediators of vascular inflammation, are mostly derived from macrophages and potently involved in various vascular inflammatory diseases such as BD, SLE, atherosclerosis, and cancer [60–63]. During the early period of vascular inflammation, ROS and NO from macrophages benefit to eliminate the foreign pathogens [64–66]. As the aggravation of inflammation, however, excessive ROS or NO from macrophages could induce OS and result in OS damage [67]. ROS, the major contributor to OS, are mainly generated by nicotinamide adenine dinucleotide phosphate (NADPH) oxidase in inflammatory responses [68]. Fuchs et al. (2019) indicated that the metabolite profile of M1 displayed an increase in superoxide whereas a decrease in antioxidant; M1 macrophages appeared to consume more NADPH than inactive macrophages or M2 macrophages; instead, increase of taurine (an essential amino acid in biological processes) in M2 macrophages could neutralize and scavenge excessive or harmful ROS [69]. Apart from that, some proinflammatory factors from the vascular microenvironment could drive macrophages to polarize towards M1 [8], further to accelerate the overproduction of ROS/NO and trigger OS [68]. Sustained M1 dominant in vascular diseases/vascular dermatoses (psoriasis, BD, and SLE) in turn induces continuous accumulation of ROS and NO [70–72]. These excessive prooxidants go beyond the antioxidant defense of cells, then to encourage OS initiation and OS state [79–81]; in the persistence of OS state, prooxidants directly hurt tissue biomolecules to form oxidized lipids and denatured proteins, further broke nucleic acids and consequently damage vascular endothelial cells/tissue [73]. Besides, high-level ROS actively work in a feedback-loop switching macrophage to M1 through complex mechanisms involving that ROS-induced TNF-α production promotes M1 macrophage activation, and ROS-stimulated MAPK/NF-κB signals mediate proinflammatory genes to reprogram macrophages towards M1, thereby contributing to OS exacerbation and vascular endothelial cells/tissue injury [74–76].
4.3. M2 Polarization Promotes Angiogenesis
As one key member of the angiogenic-promoting cells, M2 dominates in wound healing, oncogenic angiogenesis, and blindness-related aberrant angiogenesis [77–79]. M2 macrophages not only facilitate the endothelial cell progenitors to differentiate into endothelial cells [80, 81] but also interfere with all the stages of angiogenesis via release of various proangiogenic growth factors, e.g.,VEGF-A, VEGF-C, PDGF, EGF, and FGF-2 [82–84]. They act as “bridge cells” or “cellular chaperones” that guide the fusion of endothelial tip cells (vascular anastomosis) and facilitate vascular sprouting [85]; they also produce heparinases and plasmin to degrade the extracellular matrix, bind the growth factor to its receptor on endothelial cells, and promote the transmission of growth factor signals [14, 86]. In an experiment on mice, it found that M2-produced IL-10 was a crucial factor that positively derived abnormal angiogenesis, whereas angiogenesis would decrease once IL-10 absence [87]. Moreover, the study in vitro showed that angiogenic factor expression on M2 surpassed those expressions on M1 [88]; M2 instead of M1 could induce angiogenesis in vivo and in vitro, mainly involving the PDGF and FGF signal pathways. Besides, macrophage polarization is closely associated with angiogenesis in tumor growth. Circulating monocytes are recruited into the tumor stroma and then differentiate into tumor-associated macrophages (TAMs). These TAMs release a serial of growth factors (e.g., VEGFA, FGF-2, and PDGF) for endothelial cell proliferation and microvessel formation. Most notably, the phenotype of TAMs is greatly similar to M2; once the phenotype deviates from M2, tumor growth would be suppressed [77]. Therefore, the enhanced proangiogenic activity is generally ascribed to polarized M2. However, M1 scarcely enhances the proangiogenic effect; inversely, it induces endothelial-to-mesenchymal transition and inhibits angiogenesis by releasing inflammatory factors such as TNF-α and IL-1β [89]. M2-dominated activation, hence, would lead to an amplification of angiogenic effects which plays a major role in some angiogenic diseases such as IH [14].
5. Macrophage Polarization in Vascular Dermatosis
Growing evidence supports that unbalanced macrophage polarization occurs in some vascular skin diseases, such as BD, psoriasis, SLE, and IH. As a group of typical vascular inflammatory dermatoses, BD, psoriasis, and SLE collectively exhibit same pathological mechanisms involving M1-mediated immune inflammation, T cell dysregulation, and OS damage. Meanwhile, they own the similar microenvironment which provides macrophage polarization with inflammatory cytokines that induce M1 polarization. In addition, M1-produced factors remarkably ascend in these diseases and positively correlate with disease activity. On the other hand, IH is an angiogenesis-related disease, in which polarized M2 and its cytokines substantially appear. Persistent activation of single polarized macrophages with their inflammatory mediators may closely implicate in the occurrence and development of those dermatoses; therefore, we propose that regulation of macrophage polarization direction would be a novel approach to treating these disorders.
5.1. M1 Polarization in Behcet's Disease (BD)
BD, a chronic recurrent multisystemic vascular inflammatory disease, is clinically characterized by oral aphthosis, genital ulcers, skin lesions, uveitis, and organ involvements [90, 91]. Patients with BD often suffer from relapsing painfully inflammatory attacks in involved organs. The histopathological feature of BD manifests as vasculitis with various inflammatory cell infiltrations and macrophages is a major one group of these inflammatory cells. Furthermore, the Th1-associated proinflammatory cytokines (e.g., TNF-α, INF-γ, IL-1β, IL-6) obviously emerged from the serum of active BD patients [92], which formed BD microenvironment that was regarded as a main inducing factor of BD. Later, Alpsoy et al. (2003) found in their experiment in vitro that the serum of BD patients could induce M1 macrophages [13]; meanwhile, study on BD-like mice model showed that the M1 highly expressed in BD-like mice compared with normal mice, followed by the increase of M1/M2 ratio in BD-like mice [43]. Besides, some M1-secreted factors, such as TNF-α, IL-1b, IL-6, IL-8, and IL-12, are closely associated with the disease activity [93, 94]. Up to now, it is thought that immune abnormality and oxidative damage are major players in BD pathogenesis. Relevant studies have demonstrated that T cell dysregulation, especially Th1 and Th17 expansions whereas Treg diminution, is partly responsible for BD [55, 95, 96]. Our previous studies, in the same way, confirmed that the OS-related parameters were significantly abnormal in BD patients, thereby considering that OS was one of the vital factors in BD pathogenesis. Furthermore, excessive ROS/RNS could promote the dysfunction of vascular endothelial cells in BD [62, 97]. These studies all indicate that there is a dominant M1 polarization in BD, which may further induce Th1/Th17 cell upregulation and cause OS damage. On the whole, M1 macrophages produce high-level proinflammatory cytokines and trigger the inflammatory events in BD [98, 99]; inflammatory factors from BD, in turn, recruit more M1 macrophages and aggravate inflammatory responses [13]. The current treatments for BD still display unsatisfactory efficacy; hence, macrophage polarization as a new therapeutic target would have important breakthroughs in its relief.
5.2. M1 Polarization in Psoriasis
Psoriasis is a chronic immune-mediated inflammatory disorder, histologically featuring as abnormal proliferation/differentiation of keratinocytes, excessive angiogenesis, and inflammatory cell infiltration in the dermis [100]. It has a great impact on the physical and psychological health of patients [101–103], but few therapeutic strategies are enough to satisfy. Thus, it is quite crucial to find the potential therapeutic targets for psoriasis. Emerging evidence supports that the recruitment and activation of macrophages in psoriatic skin lesions/blood vessels is a key pathogenic factor in the uncontrolled cutaneous/vascular inflammation [7, 104, 105]. A study from Lin et al. (2018) proved that a greatly high ratio of M1/M2 macrophages emerged from patients with severe psoriasis, and M1 in peripheral blood is absolutely superior to M2 [44]. Meanwhile, M1-related inflammatory cytokines obviously increase in psoriatic lesions, especially TNF-α. As a factor primarily derived from M1, TNF-α is considered as the master proinflammatory cytokine and is deemed to be a key candidate gene for the pathogenesis of psoriasis [106]. Apart from TNF-α, other M1-related proinflammatory cytokines (e.g., IL-6 and IL-1β) or chemokines (e.g., CXCL8) positively appear in psoriatic serum and facilitate inflammatory cell recruitment [107]. Although the exact mechanisms underlying initiation of psoriasis remain unclear, immune dysregulation and OS are mostly responsible for it. Th1/Th17 cell activation and Treg cell depletion could be triggered by M1, which in turn promote more M1 polarization and relatively suppress M2 activation via secreting IFN-γ, IL-17, and IL-23 [65]. The activation of IL-23/IL-17 axis due to dysregulation of Th1/Th17 cells is integral to the development of psoriasis, further to create a self-amplifying, feed-forward inflammatory response in keratinocytes [108]. On the one hand, the importance of ROS-induced OS in psoriasis was discussed in our previous document, namely, endogenous and exogenous factor-induced excessive ROS initiate OS that in turn promotes more ROS generation, further leading to immunological abnormality and the development of psoriasis [109]. Apart from that, ROS could induce the release of inflammatory factors that stimulate keratinocyte proliferation and angiogenesis, and directly damage the vascular endothelium as well as aggravate vascular inflammation. Above all indicate that M1 polarization is a key motivation to psoriasis; therefore, M1 polarization may be a potential therapeutic target for the treatment of psoriasis.
5.3. M1 Polarization in Systemic Lupus Erythematosus (SLE)
SLE, a multisystem autoimmune disease based on B cell immunity, involves the skin, connective tissue, and blood vessels [110]. It exhibits an unknown etiology with life-threatening manifestations. As a key part of the innate immune system, macrophages are probably responsible for SLE. Korman et al. (2014) favored that monocyte-to-macrophage differentiation could encourage the occurrence and development of SLE, possibly through polarizing macrophages towards M1 [111]. It has been long supposed that the imbalance between M1 and M2 is one of the possible causes of severe inflammation in SLE [18, 110, 112]. Evidence showed that several proinflammatory cytokines, e.g., TNF-α, GM-CSF, and IFN-γ, significantly elevated in the circulation of lupus patients, which formed a microenvironment to facilitate macrophage polarization towards M1 [113]. More importantly, many of the markers on M1 macrophages were found to remarkably ascend in SLE serum, which were implicated in the pathogenesis of SLE [114, 115]; for instance, BAFF, a member of TNF superfamily originating from M1 and an enhancement factor of M1 polarization [116, 117], primarily participates in stimulating B cell activation [118]; costimulatory molecules CD40 and CD86 also improve the ability of M1 to activate B cells and facilitate autoantibody production [119, 120]. Inflammatory cytokines, containing TNF-α, IL-1, IL-6, CXCL10, etc., obviously increase in SLE and positively stimulate M1 polarization [120, 121]. On the contrary, the failure in M2 polarization may be another crucial underlying mechanism in SLE. Studies show that macrophages in SLE patients exhibit low expression of CD163 (the marker molecule of M2), which indicates a defect of M2 polarization. It was confirmed in an experiment that adoptive transplantation of M2 not M1 macrophages significantly ameliorated the disease activity of SLE [112]. Although SLE is typically characterized by B cell hyperactivity, it may be aggravated in the presence of T cell dysregulation [122]. T cell dysregulation in SLE displays not only an abnormal activation of toxic cellular immunity due to Th1-Th17 cells elevation but also a failure of immune regulation from a decrease in Treg cells [123, 124]. Overactivated Th1/Th17 cells with their cytokines actively trigger a serial of pathological processes in SLE, including induction of vascular inflammation, recruitment of leukocytes, activation of B cells, and production of autoantibodies [125–127]; instead, the reduction of Tregs indicates the poor prognosis of SLE. In addition, ROS-induced OS damage is considered as another player in the pathogenesis of SLE and antioxidants targeting OS seem to be effective in SLE [128]. OS not only damages biomolecules to produce various autoantibodies in SLE [129, 130] but also contributes to vascular endothelial cell injury and triggers lupus vasculitis [63]. Anyhow, M1 overactivation with M2 absence would aggravate the imbalance between Th1/Th17 and Th2/Treg cells, and accelerate OS damage to cells/tissues, further accelerating the progression of SLE. Thus, regulation of M1/M2 polarization and enhancement of M2 anti-inflammatory molecules may slow the disorder progression in lupus models as well as in SLE patients [131, 132].
5.4. M2 Polarization in Infantile Hemangioma (IH)
As one of the most common benign tumors in infancy, IH is characterized by abnormal vascular endothelial cell proliferation and excessive angiogenesis; its pathogenesis involves genetic, environmental, and other factors [133]. This angiogenic disease is supposed to result from aberrant proliferation and differentiation of pluripotent progenitor cells. Generally, IH consists of three phases: proliferative phase, involuting phase, and involuted phase. The proliferating phase features as immature vascular mass [134, 135]. At present, it is believed that polarized M2 plays an important role in the proliferation period of hemangioma, which actively promotes the growth of vascular endothelial cell proliferation by secreting VEGF, FGF-2, and other proangiogenic factors [14, 15]. Hemangioma stem cells (HemSCs) are a class of stem cells with multiple differentiation potential in IH. Zhang et al. (2015) constructed a hemangioma mouse model in vitro through injecting HemSCs into the subcutaneous of mice, by which the differentiation potential of HemSCs was observed in a polarized M2 macrophage environment [136]. This finding indicates that M2 rather than M1 benefits the differentiation of HemSCs. Combined with previous research, a large number of CD163-positive cells, representing M2-polarized macrophages, were found surrounding the endothelial cells of proliferative IH and stimulating IH proliferation as well as VEGF/FGF signal transduction [14]. Accordingly, targeted regulation of M2 polarization during the proliferative phase of IH may be promising for management of IH.
6. Treatment Hotline
Considering the crucial role of dominant M1 or M2 in these dermatoses, various strategies are being explored to regulate the direction of their differentiation or block the transduction of downstream molecules, though most of them are still being in the pilot phase of cell or animal models. Here, we review the latest researches in the treatment of these vascular skin diseases by targeting macrophages, and more vehicles should be developed for modulating macrophage polarization in the future.
6.1. Progress in Therapies on Macrophage Polarization-Mediated Skin Diseases
Currently, many natural plant extracts or Chinese herbal ingredients have been widely studied on controlling macrophage polarization-mediated dermatosis owing to their multiple powerful activities, like anti-inflammation, immunoregulation, antioxidation, antiangiogenesis, and antitumor. It was reported that curcumin, a natural polyphenolic pigment derived from turmeric, could inhibit M1 polarization and decrease the level of IL-1β, IL-6, and TNF-α produced from M1 in BD patients [137]; what was more, curcumin induced anti-inflammatory M2 macrophage polarization and further mitigated the production of inflammatory cytokines [138]. Another study about curcumin treatment in a lupus mice model showed that curcumin markedly ameliorated lupus progression in mice, accompanied with a decrease of M1-derived BAFF production in the serum, spleen, and kidney as well as a reduction of activated B cells [139]. Similarly, a natural active compound from Astragalus membranaceus—cycloastragenol (CAG)—greatly ameliorated imiquimod- (IMQ-) induced psoriasiform dermatitis in mice by targeting regulation of proinflammatory macrophages (M1). CAG, on the one hand, obviously reduced the infiltration of M1 macrophages into psoriatic dermis; on the other hand, it in a dose-dependent mean lowered the level of proinflammatory cytokines (including IL-1β, TNF-α, and IL-6) in murine psoriatic skin and serum [140]. In addition, naringenin, a flavonoid compound, has been shown in a psoriasis-like mouse model to mitigate skin inflammation through accelerating the reprogramming of macrophages from M1 to M2 [141]. Although inflammatory cytokine antagonists have been gradually promoted in some autoimmune diseases recently, the application of exogenous anti-inflammatory factors is still limited to cell or animal models. For example, adalimumab as a TNF-α inhibitor could decrease the ratio of M1 to M2 macrophage in patients with psoriasis [44], while the cytokine IL-35 contributed to inhibiting the production of IL-6 and CXCL8 and alleviating the severe symptoms of IMQ-induced psoriasis-like mice via reducing the ratio of M1/M2 macrophages as well as the infiltration of M1 in local organs [142]. In the same way, Wu et al. (2017) suggested that local administration of TNF-α and IL-1β, produced from M1, would be a potential excellent option to IH through inducing endothelial-to-mesenchymal transition and accelerating hemangioma regression [89]. Moreover, some agents targeting the signaling pathways or receptors in macrophage polarization are undergoing an exploration in preliminary research. PAM3CSK4 (PAM3), a TLR2/1 agonist unlike other TLR agonists, could uniquely induce the conversion of lupus-patient monocytes to M2 in vitro; it was also shown that normal murine monocytes preferentially differentiated into M2 rather than M1 after weekly PAM3 treatment in the lupus-prone NZB/W mice (a mouse model of SLE whose symptoms and gender bias are similar to human) [132]. Thus, it raises the possibility of PAM3 being used to normalize the M1 : M2 ratio in SLE. Apart from above strategies, biomaterial application stands out. Recently, a promising study in vitro and in human psoriasis plaques has demonstrated that metallic polyphenol-enriched nanoparticles could not only reduce M1 but also suppress the production of proinflammatory cytokines, especially M1-derived TNF-α and IL-12; this new biological material, meanwhile, was found to powerfully repress NF-κB (an important signal pathway of M1 polarization) activation in macrophages in vitro [143].
6.2. Future Orientation towards Macrophage Polarization-Mediated Dermatosis
According to the different phenotype macrophages and their respective roles in the dermatosis, some critical targets are proposed and summarized for the treatment of skin diseases via regulating macrophage polarization (shown in Figure 3). In the M1-dominant skin disorders (e.g., BD, SLE, and psoriasis), if blocking the upstream signals of M1 polarization (like JAK-STAT1, IRF-STAT1, and MyD88-NF-κB), the level of M1 would decrease. Generally, polarized M1 can produce a lot of inflammatory mediators in serum (including TNF-α, IFN-γ, IL-1, IL-1β, IL-6, IL-10, IL-12, IL-18, IL-23, IL-27, NO, ROS). These inflammatory mediators from M1 are closely implicated in the occurrence and development of above dermatoses, so the exogenous intervention such as an addition of factor receptor antagonists may be a promising alternative. But in M2-dominant skin disorders, IH as the representative, the signal pathways involved in M2 polarization and the pathogenic molecules released from M2 are critical therapeutic targets for these diseases. Additional antagonists to hemangioma tissue contribute to the reversal of polarization state of M2 in IH via inhibition of M2 polarization-related signaling pathways (e.g., JAK/STAT6 and IL-10/STAT3) and downregulation of M2-related angiogenic factors (e.g., TNF-β, IL-10,VEGF-A, and FGF-2).
Figure 3.
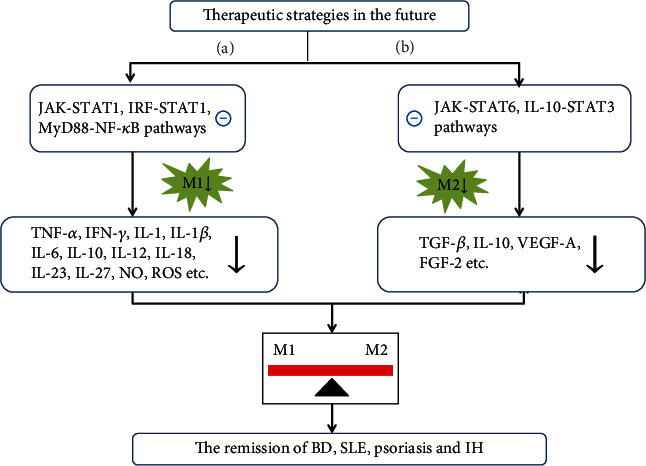
Future directions of treatment for macrophage polarization-mediated dermatosis. (a) Future treatment directions in M1 polarization-mediated skin diseases (BD, SLE, and psoriasis). (b) Future treatment directions in M2 polarization-mediated skin disease (IH). ㊀ indicates “inhibition” and ↓ indicates “downregulation”. Abbreviations: JAK: Janus kinase; STAT1/3/6: signal transducer and activator of transcription 1/3/6; IRF: interferon regulatory factor; IL-10: interleukin 10; MyD88: myeloid differentiation factor 88; NF-κB: nuclear factor kappa B; M1: classically activated macrophage; M2: alternatively activated macrophage; TNF-α: tumor necrosis factor alpha; IFN-γ: interferon gamma; IL-1/1β/6/10/12/18/23/27: interleukin 1/1β/6/10/12/18/23/27; ROS: reactive oxygen species; TGF-β: transforming growth factor-β; NO: nitric oxide; VEGF-A: vascular endothelial growth factor A; FGF-2: fibroblast growth factor 2; SLE: systemic lupus erythematosus; BD: Behcet's disease; IH: infantile hemangioma.
7. Conclusion
In conclusion, the effect of macrophage in different vascular dermatosis is variable owing to their M1/M2 phenotypes. Here, we present the evidence of macrophage polarization in M1/M2-mediated vascular skin diseases and discuss the possible pathological role of individual polarization types in these dermatoses. Typically in BD, psoriasis, and SLE, there is an enhancement in the ratio of M1/M2 and M1 plays a leading role in the continuous development and vicious cycle of inflammatory reactions, whereas in IH, the ratio of M1/M2 drops and the effect of M2 on angiogenesis are amplified. Targeting macrophage polarization to shift macrophages to M1 or M2 would be a novel strategy for control of vascular dermatosis; however, the mechanism of macrophage polarization in these diseases is not yet fully clarified and further studies in this field will be required. Besides, more therapeutic vehicles need to be developed and applied in clinical practice in the future.
Disclosure
Huiling Peng, Dehai Xian, and Jiexiong Liu are the co-first authors.
Conflicts of Interest
The authors declare that there is no conflict of interest regarding the publication of this paper.
References
- 1.Shirai T., Hilhorst M., Harrison D. G., Goronzy J. J., Weyand C. M. Macrophages in vascular inflammation – From atherosclerosis to vasculitis. Autoimmunity. 2015;48(3):139–151. doi: 10.3109/08916934.2015.1027815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lu C.-H., Lai C.-Y., Yeh D.-W., et al. Involvement of M1 macrophage polarization in endosomal toll-like receptors activated psoriatic inflammation. Mediators of Inflammation. 2018;2018:14. doi: 10.1155/2018/3523642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Murray P. J., Wynn T. A. Protective and pathogenic functions of macrophage subsets. Nature Reviews. Immunology. 2011;11(11):723–737. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bozec A., Soulat D. Latest perspectives on macrophages in bone homeostasis. Pflügers Archiv. 2017;469(3–4):517–525. doi: 10.1007/s00424-017-1952-8. [DOI] [PubMed] [Google Scholar]
- 5.da Rocha R. F., De Bastiani M. A., Klamt F. Bioinformatics approach to evaluate differential gene expression of M1/M2 macrophage phenotypes and antioxidant genes in atherosclerosis. Cell Biochemistry and Biophysics. 2014;70(2):831–839. doi: 10.1007/s12013-014-9987-3. [DOI] [PubMed] [Google Scholar]
- 6.Yang Y., Qin J., Lan L., et al. M-CSF cooperating with NF kappa B induces macrophage transformation from M1 to M2 by upregulating c-Jun. Cancer Biology & Therapy. 2013;15(1):99–107. doi: 10.4161/cbt.26718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fuentes-Duculan J., Suárez-Fariñas M., Zaba L. C., et al. A subpopulation of CD163-positive macrophages is classically activated in psoriasis. The Journal of Investigative Dermatology. 2010;130(10):2412–2422. doi: 10.1038/jid.2010.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Laria A., Lurati A., Marrazza M., Mazzocchi D., Re K. A., Scarpellini M. The macrophages in rheumatic diseases. Journal of Inflammation Research. 2016;9:1–11. doi: 10.2147/JIR.S82320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dunster J. L. The macrophage and its role in inflammation and tissue repair: mathematical and systems biology approaches. Wiley Interdisciplinary Reviews. Systems Biology and Medicine. 2016;8(1):87–99. doi: 10.1002/wsbm.1320. [DOI] [PubMed] [Google Scholar]
- 10.Gombozhapova A., Rogovskaya Y., Shurupov V., et al. Macrophage activation and polarization in post-infarction cardiac remodeling. Journal of Biomedical Science. 2017;24(1):p. 13. doi: 10.1186/s12929-017-0322-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hou Y., Zhu L., Tian H., et al. IL-23-induced macrophage polarization and its pathological roles in mice with imiquimod-induced psoriasis. Protein & Cell. 2018;9(12):1027–1038. doi: 10.1007/s13238-018-0505-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ma C., Xia Y., Yang Q. The contribution of macrophages to systemic lupus erythematosus. Clinical Immunology. 2019;207:1–9. doi: 10.1016/j.clim.2019.06.009. [DOI] [PubMed] [Google Scholar]
- 13.Alpsoy E., Kodelja V., Goerdt S., Orfanos C. E., Zouboulis C. C. Serum of patients with Behçet’s disease induces classical (pro-inflammatory) activation of human macrophages in vitro. Dermatology. 2003;206(3):225–232. doi: 10.1159/000068888. [DOI] [PubMed] [Google Scholar]
- 14.Wang F. Q., Chen G., Zhu J. Y., et al. M2-polarised macrophages in infantile haemangiomas: correlation with promoted angiogenesis. Journal of Clinical Pathology. 2013;66(12):1058–1064. doi: 10.1136/jclinpath-2012-201286. [DOI] [PubMed] [Google Scholar]
- 15.Tan E. M. S., Chudakova D. A., Davis P. F., Brasch H. D., Itinteang T., Tan S. T. Characterisation of subpopulations of myeloid cells in infantile haemangioma. Journal of Clinical Pathology. 2015;68(7):571–574. doi: 10.1136/jclinpath-2014-202846. [DOI] [PubMed] [Google Scholar]
- 16.Bhattacharya S. M2 macrophages and their role in rheumatic diseases. Rheumatology International. 2019;39(5):769–780. doi: 10.1007/s00296-018-4120-3. [DOI] [PubMed] [Google Scholar]
- 17.Fenyo I. M., Gafencu A. V. The involvement of the monocytes/macrophages in chronic inflammation associated with atherosclerosis. Immunobiology. 2013;218(11):1376–1384. doi: 10.1016/j.imbio.2013.06.005. [DOI] [PubMed] [Google Scholar]
- 18.Funes S. C., Rios M., Escobar-Vera J., Kalergis A. M. Implications of macrophage polarization in autoimmunity. Immunology. 2018;154(2):186–195. doi: 10.1111/imm.12910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Muraille E., Leo O., Moser M. Th1/Th2 paradigm extended: macrophage polarization as an unappreciated pathogen-driven escape mechanism? Frontiers in Immunology. 2014;5:p. 603. doi: 10.3389/fimmu.2014.00603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou D., Huang C., Lin Z., et al. Macrophage polarization and function with emphasis on the evolving roles of coordinated regulation of cellular signaling pathways. Cellular Signalling. 2014;26(2):192–197. doi: 10.1016/j.cellsig.2013.11.004. [DOI] [PubMed] [Google Scholar]
- 21.Decano J. L., Mattson P. C. Macrophages in vascular inflammation: origins and functions. Current Atherosclerosis Reports. 2016;18(6):p. 34. doi: 10.1007/s11883-016-0585-2. [DOI] [PubMed] [Google Scholar]
- 22.O’Neill L. A., Bowie A. G. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nature Reviews. Immunology. 2007;7(5):353–364. doi: 10.1038/nri2079. [DOI] [PubMed] [Google Scholar]
- 23.Ko H. J., Yang J. Y., Shim D. H., et al. Innate immunity mediated by myd88 signal is not essential for induction of lipopolysaccharide-specific B cell responses but is indispensable for protection against Salmonella enterica serovar Typhimurium infection. Journal of Immunology. 2009;182(4):2305–2312. doi: 10.4049/jimmunol.0801980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Donlin L. T., Jayatilleke A., Giannopoulou E. G., Kalliolias G. D., Ivashkiv L. B. Modulation of TNF-induced macrophage polarization by synovial fibroblasts. Journal of Immunology. 2014;193(5):2373–2383. doi: 10.4049/jimmunol.1400486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang N., Liang H. Molecular mechanisms that influence the macrophage m1-m2 polarization balance. Frontiers in Immunology. 2014;5:p. 614. doi: 10.3389/fimmu.2014.00614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sica A., Mantovani A. Macrophage plasticity and polarization: in vivo veritas. The Journal of Clinical Investigation. 2012;122(3):787–795. doi: 10.1172/JCI59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koh Y.-C., Yang G., Lai C.-S., Weerawatanakorn M., Pan M.-H. Chemopreventive effects of phytochemicals and medicines on M1/M2 polarized macrophage role in inflammation-related diseases. International Journal of Molecular Sciences. 2018;19(8):p. 2208. doi: 10.3390/ijms19082208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vergadi E., Ieronymaki E., Lyroni K., Vaporidi K., Tsatsanis C. Akt signaling pathway in macrophage activation and M1/M2 polarization. Journal of Immunology. 2017;198(3):1006–1014. doi: 10.4049/jimmunol.1601515. [DOI] [PubMed] [Google Scholar]
- 29.Park H. J., Lee S. J., Kim S. H., et al. IL-10 inhibits the starvation induced autophagy in macrophages via class I phosphatidylinositol 3-kinase (PI3K) pathway. Molecular Immunology. 2011;48(4):720–727. doi: 10.1016/j.molimm.2010.10.020. [DOI] [PubMed] [Google Scholar]
- 30.Covarrubias A. J., Aksoylar H. I., Horng T. Control of macrophage metabolism and activation by mTOR and Akt signaling. Seminars in Immunology. 2015;27(4):286–296. doi: 10.1016/j.smim.2015.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Biswas S. K., Chittezhath M., Shalova I. N., Lim J. Y. Macrophage polarization and plasticity in health and disease. Immunologic Research. 2012;53(1-3):11–24. doi: 10.1007/s12026-012-8291-9. [DOI] [PubMed] [Google Scholar]
- 32.Murray P. J. Macrophage polarization. Annual Review of Physiology. 2017;79(1):541–566. doi: 10.1146/annurev-physiol-022516-034339. [DOI] [PubMed] [Google Scholar]
- 33.Sica A., Erreni M., Allavena P., Porta C. Macrophage polarization in pathology. Cellular and Molecular Life Sciences. 2015;72(21):4111–4126. doi: 10.1007/s00018-015-1995-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Klimchenko O., di Stefano A., Geoerger B., et al. Monocytic cells derived from human embryonic stem cells and fetal liver share common differentiation pathways and homeostatic functions. Blood. 2011;117(11):3065–3075. doi: 10.1182/blood-2010-07-295246. [DOI] [PubMed] [Google Scholar]
- 35.Engel J. E., Chade A. R. Macrophage polarization in chronic kidney disease: a balancing act between renal recovery and decline? American Journal of Physiology. Renal Physiology. 2019;317(6):F1409–F1413. doi: 10.1152/ajprenal.00380.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ardura J. A., Rackov G., Izquierdo E., Alonso V., Gortazar A. R., Escribese M. M. Targeting macrophages: friends or foes in disease? Frontiers in Pharmacology. 2019;10:p. 1255. doi: 10.3389/fphar.2019.01255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hilhorst M., Shirai T., Berry G., Goronzy J. J., Weyand C. M. T Cell Macrophage interactions and granuloma formation in vasculitis. Frontiers in Immunology. 2014;5:p. 432. doi: 10.3389/fimmu.2014.00432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Biswas S. K., Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nature Immunology. 2010;11(10):889–896. doi: 10.1038/ni.1937. [DOI] [PubMed] [Google Scholar]
- 39.Potekhina A. V., Pylaeva E., Provatorov S., et al. Treg/Th17 balance in stable CAD patients with different stages of coronary atherosclerosis. Atherosclerosis. 2015;238(1):17–21. doi: 10.1016/j.atherosclerosis.2014.10.088. [DOI] [PubMed] [Google Scholar]
- 40.Virdis A., Dell'Agnello U., Taddei S. Impact of inflammation on vascular disease in hypertension. Maturitas. 2014;78(3):179–183. doi: 10.1016/j.maturitas.2014.04.012. [DOI] [PubMed] [Google Scholar]
- 41.Soleimani R., Mohammadi M., Saghebi S. A., Taghipour A., Khorsand Vakilzadeh A., Tavakkol Afshari J. Comparison of Th1/Th2 and Treg/Th17 ratios between wet and dry cupping therapies in Persian medicine. Avicenna Journal of Phytomedicine. 2020;10(1):24–34. [PMC free article] [PubMed] [Google Scholar]
- 42.Leitinger N., Schulman I. G. Phenotypic polarization of macrophages in atherosclerosis. Arteriosclerosis, Thrombosis, and Vascular Biology. 2013;33(6):1120–1126. doi: 10.1161/ATVBAHA.112.300173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Anower A. K. M. M., Shim J. A., Choi B., Kwon H. J., Sohn S. The role of classical and alternative macrophages in the immunopathogenesis of herpes simplex virus-induced inflammation in a mouse model. Journal of Dermatological Science. 2014;73(3):198–208. doi: 10.1016/j.jdermsci.2013.11.001. [DOI] [PubMed] [Google Scholar]
- 44.Lin S. H., Chuang H. Y., Ho J. C., Lee C. H., Hsiao C. C. Treatment with TNF-α inhibitor rectifies M1 macrophage polarization from blood CD14+ monocytes in patients with psoriasis independent of STAT1 and IRF-1 activation. Journal of Dermatological Science. 2018;91(3):276–284. doi: 10.1016/j.jdermsci.2018.05.009. [DOI] [PubMed] [Google Scholar]
- 45.Labonte A. C., Tosello-Trampont A. C., Hahn Y. S. The role of macrophage polarization in infectious and inflammatory diseases. Molecules and Cells. 2014;37(4):275–285. doi: 10.14348/molcells.2014.2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Magee C. N., Boenisch O., Najafian N. The role of costimulatory molecules in directing the functional differentiation of alloreactive T helper cells. American Journal of Transplantation. 2012;12(10):2588–2600. doi: 10.1111/j.1600-6143.2012.04180.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu Y., Lu H., Zhang C., Hu J., Xu D. Recent advances in understanding the roles of T cells in pressure overload- induced cardiac hypertrophy and remodeling. Journal of Molecular and Cellular Cardiology. 2019;129:293–302. doi: 10.1016/j.yjmcc.2019.01.005. [DOI] [PubMed] [Google Scholar]
- 48.Mantovani A., Sica A., Sozzani S., Allavena P., Vecchi A., Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends in Immunology. 2004;25(12):677–686. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 49.Wynn T. A. Fibrotic disease and the TH1/TH2 paradigm. Nature Reviews. Immunology. 2004;4(8):583–594. doi: 10.1038/nri1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Allen J. E., Wynn T. A. Evolution of Th2 immunity: a rapid repair response to tissue destructive pathogens. PLoS Pathogens. 2011;7(5, article 1002003) doi: 10.1371/journal.ppat.1002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Comte D., Karampetsou M. P., Tsokos G. C. T cells as a therapeutic target in SLE. Lupus. 2015;24(4-5):351–363. doi: 10.1177/0961203314556139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zheng Y., Danilenko D. M., Valdez P., et al. Interleukin-22, a TH17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature. 2007;445(7128):648–651. doi: 10.1038/nature05505. [DOI] [PubMed] [Google Scholar]
- 53.Emmi G., Silvestri E., Bella C. D., et al. Cytotoxic Th1 and Th17 cells infiltrate the intestinal mucosa of Behcet patients and exhibit high levels of TNF-α in early phases of the disease. Medicine. 2016;95(49):p. 5516. doi: 10.1097/MD.0000000000005516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shimizu J., Kaneko F. Helper T-cell responses to IL-12 family cytokines produced by antigen-presenting cells and the genetic background in Behcet’s disease. Genetics Research International. 2013;2013:11. doi: 10.1155/2013/363859.363859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chistiakov D. A., Kashirskikh D. A., Khotina V. A., Grechko A. V., Orekhov A. N. Immune-inflammatory responses in atherosclerosis: The role of myeloid cells. Journal of Clinical Medicine. 2019;8(11, article 1798) doi: 10.3390/jcm8111798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Samson M., Corbera-Bellalta M., Audia S., et al. Recent advances in our understanding of giant cell arteritis pathogenesis. Autoimmunity Reviews. 2017;16(8):833–844. doi: 10.1016/j.autrev.2017.05.014. [DOI] [PubMed] [Google Scholar]
- 57.Samson M. Physiopathologie des vascularites primitives des gros vaisseaux. La Revue de Médecine Interne. 2016;37(4):264–273. doi: 10.1016/j.revmed.2015.10.350. [DOI] [PubMed] [Google Scholar]
- 58.Liu G., Ma H., Qiu L., et al. Phenotypic and functional switch of macrophages induced by regulatory CD4+CD25+T cells in mice. Immunology and Cell Biology. 2011;89(1):130–142. doi: 10.1038/icb.2010.70. [DOI] [PubMed] [Google Scholar]
- 59.Sun S. W., Chen L., Zhou M., et al. BAMBI regulates macrophages inducing the differentiation of Treg through the TGF-β pathway in chronic obstructive pulmonary disease. Respiratory Research. 2019;20(1):p. 26. doi: 10.1186/s12931-019-0988-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vannini F., Kashfi K., Nath N. The dual role of iNOS in cancer. Redox Biology. 2015;6:334–343. doi: 10.1016/j.redox.2015.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tawakol A., Jaffer F. Imaging the intersection of oxidative stress, lipids, and inflammation: progress toward personalized care of atherosclerosis. Journal of the American College of Cardiology. 2018;71(3):336–338. doi: 10.1016/j.jacc.2017.11.031. [DOI] [PubMed] [Google Scholar]
- 62.Zhong J. Q., Xian D. H., Xiong X. Expression and correlation analysis of oxidative stress, Nrf2 and HO-1 in Behcet’s disease. Chinese Journal of Dermatology Venereol. 2016;30(1):6–9. [Google Scholar]
- 63.Tsai C. Y., Hsieh S. C., Lu C. S., et al. Cross-Talk between mitochondrial dysfunction-provoked oxidative stress and aberrant noncoding RNA expression in the pathogenesis and pathophysiology of SLE. International Journal of Molecular Sciences. 2019;20(20, article 5183) doi: 10.3390/ijms20205183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Franchini A. M., Hunt D., Melendez J. A., Drake J. R. FcγR-driven release of IL-6 by macrophages requires NOX2-dependent production of reactive oxygen species. The Journal of Biological Chemistry. 2013;288(35):25098–25108. doi: 10.1074/jbc.M113.474106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sun C., Sun L., Ma H., et al. The phenotype and functional alterations of macrophages in mice with hyperglycemia for long term. Journal of Cellular Physiology. 2012;227(4):1670–1679. doi: 10.1002/jcp.22891. [DOI] [PubMed] [Google Scholar]
- 66.Paik W., Alonzo F. Probiotic exopolysaccharide protects against systemic infection, inducing dual-functioning macrophages that restrict bacterial growth and limit inflammation. Infection and Immunity. 2019;87(1, article 00791) doi: 10.1128/IAI.00791-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Di Pietro M., Filardo S., De Santis F., Sessa R. Chlamydia pneumoniae infection in atherosclerotic lesion development through oxidative stress: A brief overview. International Journal of Molecular Sciences. 2013;14(7):15105–15120. doi: 10.3390/ijms140715105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Castaneda O. A., Lee S. C., Ho C. T. Macrophages in oxidative stress and models to evaluate the antioxidant function of dietary natural compounds. Journal of Food and Drug Analysis. 2017;25(1):111–118. doi: 10.1016/j.jfda.2016.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fuchs A. L., Schiller S. M., Keegan W. J., et al. Quantitative 1H NMR metabolomics reveal distinct metabolic adaptations in human macrophages following differential activation. Metabolites. 2019;9(11):p. 248. doi: 10.3390/metabo9110248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tan H. Y., Wang N., Li S., Hong M., Wang X., Feng Y. The reactive oxygen species in macrophage polarization: reflecting its dual role in progression and treatment of human diseases. Oxidative Medicine and Cellular Longevity. 2016;2016:16. doi: 10.1155/2016/2795090.2795090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li F., Zhu X., Yang Y., Huang L., Xu J. TIPE2 alleviates systemic lupus erythematosus through regulating macrophage polarization. Cellular Physiology and Biochemistry. 2016;38(1):330–339. doi: 10.1159/000438633. [DOI] [PubMed] [Google Scholar]
- 72.Liang L., Tan X., Zhou Q., et al. IL-1β triggered by peptidoglycan and lipopolysaccharide through TLR2/4 and ROS-NLRP3 inflammasome-dependent pathways is involved in ocular Behçet’s disease. Investigative Ophthalmology & Visual Science. 2013;54(1):402–414. doi: 10.1167/iovs.12-11047. [DOI] [PubMed] [Google Scholar]
- 73.Deng Z., Shi F., Zhou Z., et al. M1 macrophage mediated increased reactive oxygen species (ROS) influence wound healing via the MAPK signaling in vitro and in vivo. Toxicology and Applied Pharmacology. 2019;366:83–95. doi: 10.1016/j.taap.2019.01.022. [DOI] [PubMed] [Google Scholar]
- 74.Forstermann U., Xia N., Li H. Roles of vascular oxidative stress and nitric oxide in the pathogenesis of atherosclerosis. Circulation Research. 2017;120(4):713–735. doi: 10.1161/CIRCRESAHA.116.309326. [DOI] [PubMed] [Google Scholar]
- 75.Hucke S., Eschborn M., Liebmann M., et al. Sodium chloride promotes pro-inflammatory macrophage polarization thereby aggravating CNS autoimmunity. Journal of Autoimmunity. 2015;67:90–101. doi: 10.1016/j.jaut.2015.11.001. [DOI] [PubMed] [Google Scholar]
- 76.Zhou Y., Zhang T., Wang X., et al. Curcumin modulates macrophage polarization through the inhibition of the toll-like receptor 4 expression and its signaling pathways. Cellular Physiology and Biochemistry. 2015;36(2):631–641. doi: 10.1159/000430126. [DOI] [PubMed] [Google Scholar]
- 77.Rolny C., Mazzone M., Tugues S., et al. HRG inhibits tumor growth and metastasis by inducing macrophage polarization and vessel normalization through downregulation of PlGF. Cancer Cell. 2011;19(1):31–44. doi: 10.1016/j.ccr.2010.11.009. [DOI] [PubMed] [Google Scholar]
- 78.Qing L., Fu J., Wu P., Zhou Z., Yu F., Tang J. Metformin induces the M2 macrophage polarization to accelerate the wound healing via regulating AMPK/mTOR/NLRP3 inflammasome singling pathway. American Journal of Translational Research. 2019;11(2):655–668. [PMC free article] [PubMed] [Google Scholar]
- 79.De Palma M. Partners in crime: VEGF and IL-4 conscript tumour-promoting macrophages. The Journal of Pathology. 2012;227(1):4–7. doi: 10.1002/path.4008. [DOI] [PubMed] [Google Scholar]
- 80.Newman A. C., Hughes C. C. W. Macrophages and angiogenesis: a role for Wnt signaling. Vascular Cell. 2012;4(1):p. 13. doi: 10.1186/2045-824X-4-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kreimendahl F., Marquardt Y., Apel C., et al. Macrophages significantly enhance wound healing in a vascularized skin model. Journal of Biomedical Materials Research. Part A. 2019;107(6):1340–1350. doi: 10.1002/jbm.a.36648. [DOI] [PubMed] [Google Scholar]
- 82.Murdoch C., Muthana M., Coffelt S. B., Lewis C. E. The role of myeloid cells in the promotion of tumour angiogenesis. Nature Reviews. Cancer. 2008;8(8):618–631. doi: 10.1038/nrc2444. [DOI] [PubMed] [Google Scholar]
- 83.Lin E. Y., Li J. F., Gnatovskiy L., et al. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Research. 2006;66(23):11238–11246. doi: 10.1158/0008-5472.CAN-06-1278. [DOI] [PubMed] [Google Scholar]
- 84.Fantin A., Vieira J. M., Gestri G., et al. Tissue macrophages act as cellular chaperones for vascular anastomosis downstream of VEGF-mediated endothelial tip cell induction. Blood. 2010;116(5):829–840. doi: 10.1182/blood-2009-12-257832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Schmidt T., Carmeliet P. Bridges that guide and unite. Nature. 2010;465(7299):697–699. doi: 10.1038/465697a. [DOI] [PubMed] [Google Scholar]
- 86.Marrelli A., Cipriani P., Liakouli V., et al. Angiogenesis in rheumatoid arthritis: A disease specific process or a common response to chronic inflammation? Autoimmunity Reviews. 2011;10(10):595–598. doi: 10.1016/j.autrev.2011.04.020. [DOI] [PubMed] [Google Scholar]
- 87.Apte R. S., Richter J., Herndon J., Ferguson T. A. Macrophages inhibit neovascularization in a murine model of age-related macular degeneration. PLoS Medicine. 2006;3(8, article 3e310) doi: 10.1371/journal.pmed.0030310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jetten N., Verbruggen S., Gijbels M. J., Post M. J., de Winther M. P. J., Donners M. M. P. C. Anti-inflammatory M2, but not pro-inflammatory M1 macrophages promote angiogenesis in vivo. Angiogenesis. 2014;17(1):109–118. doi: 10.1007/s10456-013-9381-6. [DOI] [PubMed] [Google Scholar]
- 89.Wu K. Q., Muratore C. S., So E. Y., et al. M1 Macrophage–Induced Endothelial-to-Mesenchymal Transition Promotes Infantile Hemangioma Regression. The American Journal of Pathology. 2017;187(9):2102–2111. doi: 10.1016/j.ajpath.2017.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sakane T., Suzuki N., Nagafuchi H. Etiopathology of Behçet’s disease: immunological aspects. Yonsei Medical Journal. 1997;38(6):350–358. doi: 10.3349/ymj.1997.38.6.350. [DOI] [PubMed] [Google Scholar]
- 91.Mazzoccoli G., Matarangolo A., Rubino R., Inglese M., De Cata A. Behcet syndrome: from pathogenesis to novel therapies. Clinical and Experimental Medicine. 2016;16(1):10–12. doi: 10.1007/s10238-014-0328-z. [DOI] [PubMed] [Google Scholar]
- 92.Dalghous A. M., Freysdottir J., Fortune F. Expression of cytokines, chemokines, and chemokine receptors in oral ulcers of patients with Behcet’s disease (BD) and recurrent aphthous stomatitis is Th1-associated, although Th2-association is also observed in patients with BD. Scandinavian Journal of Rheumatology. 2009;35(6):472–475. doi: 10.1080/03009740600905380. [DOI] [PubMed] [Google Scholar]
- 93.Liu Y. C., Zou X. B., Chai Y. F., Yao Y. M. Macrophage polarization in inflammatory diseases. International Journal of Biological Sciences. 2014;10(5):520–529. doi: 10.7150/ijbs.8879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhou Z. Y., Chen S. L., Shen N., Lu Y. Cytokines and Behcet's Disease. Autoimmunity Reviews. 2012;11(10):699–704. doi: 10.1016/j.autrev.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 95.Houman H., Hamzaoui A., Ben Ghorbal I., Khanfir M., Feki M., Hamzaoui K. Abnormal expression of chemokine receptors in Behçet's disease: relationship to intracellular Th1/Th2 cytokines and to clinical manifestations. Journal of Autoimmunity. 2004;23(3):267–273. doi: 10.1016/j.jaut.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 96.Greco A., de Virgilio A., Ralli M., et al. Behçet's disease: New insights into pathophysiology, clinical features and treatment options. Autoimmunity Reviews. 2018;17(6):567–575. doi: 10.1016/j.autrev.2017.12.006. [DOI] [PubMed] [Google Scholar]
- 97.Zhong J., Xian D., Xiong X. Oral allicin in the treatment of Behcet’s disease through attenuating oxidative stress: a pilot study in 20 patients with mucocutaneous lesions. Dermatologic Therapy. 2017;30(1):1–4. doi: 10.1111/dth.12429. [DOI] [PubMed] [Google Scholar]
- 98.Gholijani N., Ataollahi M. R., Samiei A., Aflaki E., Shenavandeh S., Kamali-Sarvestani E. An elevated proinflammatory cytokines profile in Behcet’s disease: a multiplex analysis. Immunology Letters. 2016;186:46–51. doi: 10.1016/j.imlet.2016.12.001. [DOI] [PubMed] [Google Scholar]
- 99.Misumi M., Hagiwara E., Takeno M., et al. Cytokine production profile in patients with Behcet's disease treated with infliximab. Cytokine. 2003;24(5):210–218. doi: 10.1016/j.cyto.2003.09.003. [DOI] [PubMed] [Google Scholar]
- 100.Feng L., Song P., Xu F., et al. Cis-khellactone inhibited the proinflammatory macrophages via promoting autophagy to ameliorate imiquimod-induced psoriasis. Journal of Investigative Dermatology. 2019;139(9):1946–1956.e3. doi: 10.1016/j.jid.2019.02.021. [DOI] [PubMed] [Google Scholar]
- 101.Lowes M. A., Suarez-Farinas M., Krueger J. G. Immunology of psoriasis. Annual Review of Immunology. 2014;32(1):227–255. doi: 10.1146/annurev-immunol-032713-120225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Parisi R., Symmons D. P., Griffiths C. E., Ashcroft D. M., Identification and Management of Psoriasis and Associated ComorbidiTy (IMPACT) project team Global Epidemiology of Psoriasis: A Systematic Review of Incidence and Prevalence. The Journal of Investigative Dermatology. 2013;133(2):377–385. doi: 10.1038/jid.2012.339. [DOI] [PubMed] [Google Scholar]
- 103.Boehncke W. H., Schön M. P. Psoriasis. Lancet. 2015;386(9997):983–994. doi: 10.1016/S0140-6736(14)61909-7. [DOI] [PubMed] [Google Scholar]
- 104.Clark R. A., Kupper T. S. Misbehaving macrophages in the pathogenesis of psoriasis. Journal of Clinical Investigation. 2006;116(8):2084–2087. doi: 10.1172/JCI29441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wang H., Peters T., Sindrilaru A., Scharffetter-Kochanek K. Key role of macrophages in the pathogenesis of CD18 hypomorphic murine model of psoriasis. Journal of Investigative Dermatology. 2009;129(5):1100–1114. doi: 10.1038/jid.2009.43. [DOI] [PubMed] [Google Scholar]
- 106.Furue K., Ito T., Tsuji G., Kadono T., Furue M. Psoriasis and the TNF/IL23/IL17 axis. Giornale Italiano di Dermatologia e Venereologia. 2019;154(4):418–424. doi: 10.23736/S0392-0488.18.06202-8. [DOI] [PubMed] [Google Scholar]
- 107.Arango Duque G., Descoteaux A. Macrophage cytokines: involvement in immunity and infectious diseases. Frontiers in Immunology. 2014;5:p. 491. doi: 10.3389/fimmu.2014.00491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hawkes J. E., Yan B. Y., Chan T. C., Krueger J. G. Discovery of the IL-23/IL-17 signaling pathway and the treatment of psoriasis. Journal of Immunology. 2018;201(6):1605–1613. doi: 10.4049/jimmunol.1800013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lai R., Xian D., Xiong X., Yang L., Song J., Zhong J. Proanthocyanidins: novel treatment for psoriasis that reduces oxidative stress and modulates Th17 and Treg cells. Redox Report. 2018;23(1):130–135. doi: 10.1080/13510002.2018.1462027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chalmers S. A., Chitu V., Ramanujam M., Putterman C. Therapeutic targeting of macrophages in lupus nephritis. Discovery Medicine. 2015;20(108):43–49. [PubMed] [Google Scholar]
- 111.Korman B. D., Huang C.-C., Skamra C., et al. Inflammatory expression profiles in monocyte-to-macrophage differentiation in patients with systemic lupus erythematosus and relationship with atherosclerosis. Arthritis Research & Therapy. 2014;16(4):p. 4609. doi: 10.1186/ar4609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Li F., Yang Y., Zhu X., Huang L., Xu J. Macrophage polarization modulates development of systemic lupus erythematosus. Cellular Physiology and Biochemistry. 2015;37(4):1279–1288. doi: 10.1159/000430251. [DOI] [PubMed] [Google Scholar]
- 113.Lazzari E., Jefferies C. A. IRF5-mediated signaling and implications for SLE. Clinical Immunology. 2014;153(2):343–352. doi: 10.1016/j.clim.2014.06.001. [DOI] [PubMed] [Google Scholar]
- 114.Bennett L., Palucka A. K., Arce E., et al. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. The Journal of Experimental Medicine. 2003;197(6):711–723. doi: 10.1084/jem.20021553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Liu L., Allman W. R., Coleman A. S., Takeda K., Lin T. L., Akkoyunlu M. Delayed onset of autoreactive antibody production and M2-skewed macrophages contribute to improved survival of TACI deficient MRL-Fas/Lpr mouse. Scientific Reports. 2018;8(1):p. 1308. doi: 10.1038/s41598-018-19827-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mercado U. B lymphocyte stimulator in systemic lupus erythematosus. Revista Médica del Instituto Mexicano del Seguro Social. 2012;50(4):349–350. [PubMed] [Google Scholar]
- 117.Panchanathan R., Choubey D. Murine baff expression is up-regulated by estrogen and interferons: implications for sex bias in the development of autoimmunity. Molecular Immunology. 2013;53(1-2):15–23. doi: 10.1016/j.molimm.2012.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ding H., Wang L., Wu X., et al. Blockade of B-cell-activating factor suppresses lupus-like syndrome in autoimmune bxsb mice. Journal of Cellular and Molecular Medicine. 2010;14(6B):1717–1725. doi: 10.1111/j.1582-4934.2009.00817.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Katsiari C. G., Liossis S.-N. C., Souliotis V. L., Dimopoulos A. M., Manoussakis M. N., Sfikakis P. P. Aberrant expression of the costimulatory molecule CD40 ligand on monocytes from patients with systemic lupus erythematosus. Clinical Immunology. 2002;103(1):54–62. doi: 10.1006/clim.2001.5172. [DOI] [PubMed] [Google Scholar]
- 120.Li Y., Lee P. Y., Reeves W. H. Monocyte and macrophage abnormalities in systemic lupus erythematosus. Archivum Immunologiae et Therapiae Experimentalis. 2010;58(5):355–364. doi: 10.1007/s00005-010-0093-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Smallwood M. J., Nissim A., Knight A. R., Whiteman M., Haigh R., Winyard P. G. Oxidative stress in autoimmune rheumatic diseases. Free Radical Biology & Medicine. 2018;125:3–14. doi: 10.1016/j.freeradbiomed.2018.05.086. [DOI] [PubMed] [Google Scholar]
- 122.Mohan C., Putterman C. Genetics and pathogenesis of systemic lupus erythematosus and lupus nephritis. Nature Reviews. Nephrology. 2015;11(6):329–341. doi: 10.1038/nrneph.2015.33. [DOI] [PubMed] [Google Scholar]
- 123.Valencia X., Yarboro C., Illei G., Lipsky P. E. Deficient CD4+CD25highT regulatory cell function in patients with active systemic lupus erythematosus. Journal of Immunology. 2007;178(4):2579–2588. doi: 10.4049/jimmunol.178.4.2579. [DOI] [PubMed] [Google Scholar]
- 124.Xing Q., Wang B., Su H., Cui J., Li J. Elevated Th17 cells are accompanied by FoxP3+ Treg cells decrease in patients with lupus nephritis. Rheumatology International. 2012;32(4):949–958. doi: 10.1007/s00296-010-1771-0. [DOI] [PubMed] [Google Scholar]
- 125.Yang J., Yang X., Zou H., Li M. Oxidative stress and Treg and Th17 dysfunction in systemic lupus erythematosus. Oxidative Medicine and Cellular Longevity. 2016;2016:9. doi: 10.1155/2016/2526174.2526174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Li D., Guo B., Wu H., Tan L., Chang C., Lu Q. Interleukin-17 in systemic lupus erythematosus: a comprehensive review. Autoimmunity. 2015;48(6):353–361. doi: 10.3109/08916934.2015.1037441. [DOI] [PubMed] [Google Scholar]
- 127.Chen D. Y., Chen Y. M., Wen M. C., Hsieh T. Y., Hung W. T., Lan J. L. The potential role of Th17 cells and Th17-related cytokines in the pathogenesis of lupus nephritis. Lupus. 2012;21(13):1385–1396. doi: 10.1177/0961203312457718. [DOI] [PubMed] [Google Scholar]
- 128.Shah D., Mahajan N., Sah S., Nath S. K., Paudyal B. Oxidative stress and its biomarkers in systemic lupus erythematosus. Journal of Biomedical Science. 2014;21(1):23–35. doi: 10.1186/1423-0127-21-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Leishangthem B. D., Sharma A., Bhatnagar A. Role of altered mitochondria functions in the pathogenesis of systemic lupus erythematosus. Lupus. 2016;25(3):272–281. doi: 10.1177/0961203315605370. [DOI] [PubMed] [Google Scholar]
- 130.Kuren B. T., Scofield R. H. Lipid peroxidation in systemic lupus erythematosus. Indian Journal of Experimental Biology. 2006;44(5):349–356. [PubMed] [Google Scholar]
- 131.Krausgruber T., Blazek K., Smallie T., et al. IRF5 promotes inflammatory macrophage polarization and Th1-Th17 responses. Nature Immunology. 2011;12(3):231–238. doi: 10.1038/ni.1990. [DOI] [PubMed] [Google Scholar]
- 132.Horuluoglu B., Bayik D., Kayraklioglu N., Goguet E., Kaplan M. J., Klinman D. M. PAM3 supports the generation of M2-like macrophages from lupus patient monocytes and improves disease outcome in murine lupus. Journal of Autoimmunity. 2019;99:24–32. doi: 10.1016/j.jaut.2019.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Itinteang T., Tan S. T., Brasch H., Day D. J. Haemogenic endothelium in infantile haemangioma. Journal of Clinical Pathology. 2010;63(11):982–986. doi: 10.1136/jcp.2010.081257. [DOI] [PubMed] [Google Scholar]
- 134.Darrow D. H., Greene A. K., Mancini A. J., Nopper A. J. Diagnosis and management of infantile hemangioma. Pediatrics. 2015;136(4):1060–1104. doi: 10.1542/peds.2015-2485. [DOI] [PubMed] [Google Scholar]
- 135.Phung T. L., Hochman M. Pathogenesis of infantile hemangioma. Facial Plastic Surgery. 2012;28(6):554–562. doi: 10.1055/s-0032-1329930. [DOI] [PubMed] [Google Scholar]
- 136.Zhang W., Chen G., Wang F. Q., et al. Macrophages contribute to the progression of infantile hemangioma by regulating the proliferation and differentiation of hemangioma stem cells. The Journal of Investigative Dermatology. 2015;135(12):3163–3172. doi: 10.1038/jid.2015.321. [DOI] [PubMed] [Google Scholar]
- 137.Palizgir M. T., Akhtari M., Mahmoudi M., Mostafaei S., Rezaiemanesh A., Shahram F. Curcumin reduces the expression of interleukin 1βand the production of interleukin 6 and tumor necrosis factor alpha by M1 macrophages from patients with Behcet's disease. Immunopharmacology and Immunotoxicology. 2018;40(4):297–302. doi: 10.1080/08923973.2018.1474921. [DOI] [PubMed] [Google Scholar]
- 138.Gao S., Zhou J., Liu N., et al. Curcumin induces M2 macrophage polarization by secretion IL-4 and/or IL-13. Journal of Molecular and Cellular Cardiology. 2015;85:131–139. doi: 10.1016/j.yjmcc.2015.04.025. [DOI] [PubMed] [Google Scholar]
- 139.Li Q., Tan S., Xu K., et al. Curcumin attenuates lupus nephritis in MRL/lpr mice by suppressing macrophage-secreted B cell activating factor (BAFF) International Journal of Clinical and Experimental Pathology. 2019;12(6):2075–2083. [PMC free article] [PubMed] [Google Scholar]
- 140.Deng G., Chen W., Wang P., et al. Inhibition of NLRP3 inflammasome-mediated pyroptosis in macrophage by cycloastragenol contributes to amelioration of imiquimod-induced psoriasis- like skin inflammation in mice. International Immunopharmacology. 2019;74, article 105682 doi: 10.1016/j.intimp.2019.105682. [DOI] [PubMed] [Google Scholar]
- 141.Karuppagounder V., Arumugam S., Thandavarayan R. A., et al. Naringenin ameliorates skin inflammation and accelerates phenotypic reprogramming from M1 to M2 macrophage polarization in atopic dermatitis NC/Nga mouse model. Experimental Dermatology. 2016;25(5):404–407. doi: 10.1111/exd.12962. [DOI] [PubMed] [Google Scholar]
- 142.Zhang J., Lin Y., Li C., et al. IL-35 decelerates the inflammatory process by regulating inflammatory cytokine secretion and M1/M2 macrophage ratio in psoriasis. Journal of Immunology. 2016;197(6):2131–2144. doi: 10.4049/jimmunol.1600446. [DOI] [PubMed] [Google Scholar]
- 143.Crisan D., Scharffetter-Kochanek K., Crisan M., et al. Topical silver and gold nanoparticles complexed withCornus massuppress inflammation in human psoriasis plaques by inhibiting NF-κB activity. Experimental Dermatology. 2018;27(10):1166–1169. doi: 10.1111/exd.13707. [DOI] [PubMed] [Google Scholar]