

Abstract
Large copy-number variants (CNVs) are strongly associated with both developmental delay and cancer, but the type of disease depends strongly on when and where the mutation occurred, i.e., germline versus somatic. We used microarray data from UK Biobank to investigate the prevalence and penetrance of large autosomal CNVs and chromosomal aneuploidies using a standard CNV detection algorithm not designed for detecting mosaic variants. We found 160 individuals that carry >10 Mb copy number changes, including 56 with whole chromosome aneuploidies. Nineteen (12%) individuals had a diagnosis of Down syndrome or other developmental disorder, while 84 (52.5%) individuals had a diagnosis of hematological malignancies or chronic myeloproliferative disorders. Notably, there was no evidence of mosaicism in the blood for many of these large CNVs, so they could easily be mistaken for germline alleles even when caused by somatic mutations. We therefore suggest that somatic mutations associated with blood cancers may result in false estimates of rare variant penetrance from population biobanks.
Keywords: penetrance, cnv, aneuploidy, biobank, mosaic, somatic, germline
Main Text
Copy number variants (CNVs) are deletions or duplications of DNA that can vary in size from 50 basepairs to several hundred megabases,1 i.e., entire chromosomes. Individuals typically carry several thousand CNVs, most of which are small (<1 Mb) and rare (allele frequency < 1%).2, 3, 4 Large, rare pathogenic CNVs have historically been identified through clinical microarray testing of two distinct clinical cohorts: first, children with developmental disorders caused predominantly by germline mutations,5, 6, 7 and second, individuals with hematological and other cancers associated with somatically acquired mutations.8, 9, 10
The availability of large, well-genotyped population biobanks offers an opportunity to investigate the prevalence and penetrance of monogenic disease-causing variants.11 Several studies have already been published evaluating known developmental CNVs in ∼500,000 adults in the UK and Estonian Biobanks12, 13, 14, 15, 16, 17, 18, 19 and the penetrance of X chromosome aneuploidy has been investigated in UK Biobank.20 However, given the relatively advanced age of UK Biobank participants, ranging from 40 to 70 years (mean = 56.5 years) at recruitment, it is likely that some variants will be due to somatic mutation and age-related clonal hematopoiesis21,22 as has previously been observed in genome-wide association study cohorts.23,24 Importantly, somatic variants in adult population cohorts should not be used to evaluate the penetrance of germline CNVs known to cause developmental disorders, as this will result in spurious associations.
We sought to investigate large (>10 Mb) autosomal CNVs present in population datasets with the aim of determining whether they were likely to be germline (and therefore potentially useful for penetrance studies) or somatic (and therefore caused by clonal expansions). We used microarray data from UK Biobank, which recruited 502,506 individuals from across the UK between 2006 and 2010.25 Hospital Episode Statistics (HES) and cancer registry data were available for the whole cohort up to 31 March 2017, and GP records were available for half the cohort; all participants also provided a range of information (e.g., demographics, health status, lifestyle) via questionnaires. Genotypes for SNVs and indels were generated from blood-extracted DNA using the Affymetrix Axiom UK Biobank array (∼450,000 individuals) and the UKBiLEVE array (∼50,000 individuals) in 106 batches of ∼4,700 samples. This dataset underwent extensive central quality control.25
We called CNVs genome-wide in 488,377 individuals with array genotyping data in UK Biobank25 using PennCNV version 1.0.426 with log R ratio and B-allele frequency values for 805,426 genome-wide probe sets provided by UK Biobank. Very large PennCNV calls (i.e., multiple megabases) can sometimes be fragmented into many smaller calls, so we additionally calculated the sum of bases either deleted or duplicated on each chromosome per individual according to the PennCNV calls. We carried out visual inspection of each event in everyone with >10 Mb deleted and/or duplicated on a single chromosome to confirm breakpoints, event type and level of mosaicism (see examples in Figure 1). Around a third of the events showed no evidence of mosaicism in blood (based on a deviation of the B-allele frequencies from 0, 0.5, or 1 with a co-located increase/decrease in log R ratio) while two-thirds of events were consistent with the presence of a large CNV in a proportion of cells. Based on previous work investigating mosaicism in UK Biobank,20,27 we estimate that we were able to detect copy number changes present in >20%–25% of cells. There was a good correlation between the log R ratio and visual inspection of mosaicism28 (Figure 2).
Figure 1.

Example Mean Log R Ratio (LRR) and B-Allele Frequency Plots
Shown are (A) constitutive deletion of half the q-arm of chromosome 20, (B) four small non-mosaic deletions on chromosome 4, (C) mosaic deletion of the end of the q-arm of chromosome 6, (D) constitutive duplication of the whole of chromosome 21, (E) mosaic duplication of the whole of chromosome 19, and (F) triplication of chromosome 9p. Alleles (A and B) corresponding to each of B-allele frequencies are indicated. Red, copy number change; blue, normal copy number.
Figure 2.
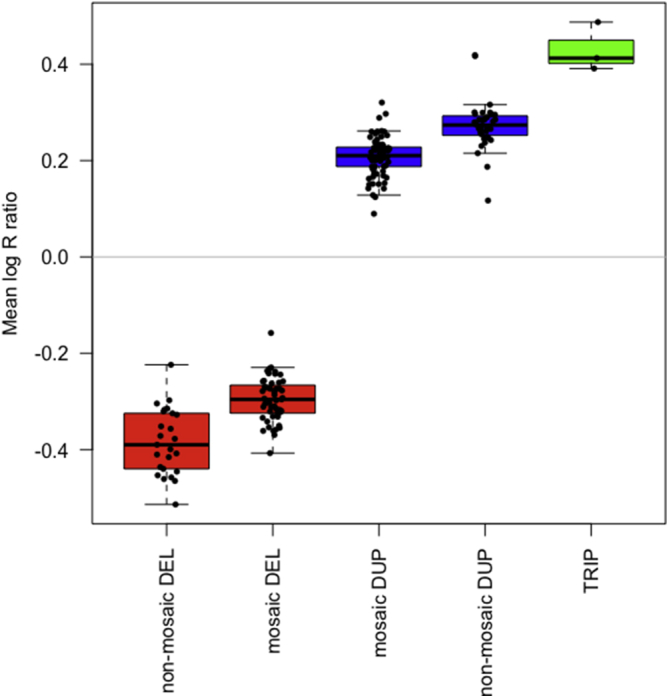
Mean Log R Ratio of CNVs
Boxplot of mean log R ratios of large CNVs, grouped by whether there was evidence of mosaicism based on visual inspection of the data. Red, deletion (DEL); blue, duplication (DUP); green, triplication (TRIP); median and interquartile ranges shown.
We identified 160 individuals in UK Biobank (61% male versus 46% in the whole of UK Biobank; Pearson’s chi-square p = 0.025) with >10 Mb involved in copy number events on a single autosome (Figure 3 and Table S1). This male bias has been observed previously and is thought to be due to higher male-specific rates of certain hematological malignancies.24 In the majority (134/160) of individuals, this was caused by a single large CNV; 19 individuals had 2 separate events (17 on 2 different chromosomes); 5 individuals had 3 separate events (all involving at least 2 different chromosomes); and 2 individuals had 4 or 5 separate events on the same chromosome. Individual events ranged in size from 0.9 Mb to 198 Mb (mean = 56 Mb, stdev = 51 Mb) and included both unique events and recurrent events. There were 64 whole chromosome duplications of chromosomes 3, 8, 9, 12, 14, 18, 19, and 21, including four individuals with two trisomies and three individuals with three trisomies.
Figure 3.

Summary of Large Autosomal CNVs Identified
Circos plot of all large autosomal CNVs in UK Biobank; chromosomes 1–22 are indicated, and CNVs on the same chromosome in the same person are shown on the same track. Red, deletion; blue, duplication; green, triplication.
Of the autosomal aneuploidies, only trisomy 21 is compatible with adult life when present constitutively and causes Down syndrome.29 Twelve individuals had a duplication of chromosome 21, of whom 11 had a diagnosis of Down syndrome in their HES or GP records (GP records were not available for the remaining individual). A further six individuals in UK Biobank had Down syndrome recorded in their HES records, but their microarray data showed no evidence of trisomy 21. It is unclear whether these discrepancies are caused by sample mix-ups, errors in HES records, or misdiagnoses. Of those with large CNVs, a further eight individuals had ICD-10 codes or GP records consistent with various developmental disorders, including congenital malformations, developmental disorder (intellectual disability/handicap or epilepsy), and bipolar affective disorder (Table S1).
We suggest that the rest of the whole chromosome duplications and the majority of large CNVs are likely to be somatic mutations caused by clonal expansions, some of which are compatible with being present in (apparently) healthy individuals. Several lines of evidence suggest that the majority of the large CNVs were likely caused by somatic mutations associated with either cancer10,30 or age-related hematopoietic clonal expansions.22,31 First, 79/160 (50%) individuals had a recurrent duplication of chromosome 8, 9, 12, or 19 or large deletions on chromosome 11q, 13q, 17p, and 20q that are consistent with those observed previously in lymphocytic and myeloid leukemias10,30,32,33 and JAK2-related myeloproliferative neoplasms.34 Second, 98/160 (61%) individuals had neoplasms recorded in their HES records or cancer registry data compared with 80,046 (17%) across the whole of UK Biobank (p < 2 × 10−16). Sixty-four (40%) were malignant neoplasms of lymphoid, hematopoietic, and related tissues (ICD-10 codes: C81-96), a significant enrichment above the whole of UK Biobank (n = 3,869, 0.8%, p < 2 × 10−16); and a further 20 were polycythemia vera, myelodysplastic syndrome, and chronic myeloproliferative diseases (ICD-10 codes: D45-47), again a significant enrichment above the whole of UK Biobank (n = 646, 0.1% p < 2 × 10−16). Third, individuals with large CNVs and neoplasms were older and taller versus the others with large CNVs but no record of neoplasms, as expected in cancer; in contrast, individuals with large CNVs and a developmental disorder were younger and shorter versus the others, consistent with many developmental disorders (Figure 4).
Figure 4.

Characteristics of Individuals in UK Biobank with >10 MB Copy Number Changes
Boxplot of age in years (A) at recruitment and height of individuals (B) grouped by whether they had a neoplasm, developmental disorder, or neither coded in their HES or cancer registry records. Red, developmental disorder; green, neoplasm; blue, neither neoplasm nor developmental disorder recorded; dotted red line, average for UK Biobank; median and interquartile ranges shown; p values from paired samples Wilcoxon test in R.
Nonetheless, 43/160 (27%) individuals with a large CNV in UK Biobank have neither a developmental disorder nor a neoplasm (of any sort) recorded to date. This observation has a range of explanations, including record error, lack of hospitalization, absence of GP records (currently available for only around half the cohort), benign hematopoietic clonal expansions, or neoplasms that have not yet developed or been diagnosed. Given the prognostic link between chromosomal instability and tumorigenesis,30,35 unfortunately the latter explanation is likely to be true in many cases.
Mosaic chromosomal alterations27 and Y chromosome loss36 in UK Biobank have previously been linked to age-related clonal hematopoiesis, both of which can be easily excluded from studies seeking to investigate penetrance of germline CNVs. Indeed, although some mosaic variants can be detected by PennCNV, low-level mosaic variants are often not detected using standard variant calling algorithms. However, the presence of very rare autosomal aneuploidies, some of which do not appear to be mosaic based on intensity data from microarrays, suggest that caution should be used when interpreting rare variants (of any size) in population biobanks. For example, we note that six individuals in UK Biobank have complete or partial trisomy of chromosome 3, of whom 5/6 have non-Hodgkin’s lymphoma. Presumed germline duplications of 3q29 have previously been causally linked with early death (OR = 27.8) and cancer (OR = 37.5) in UK Biobank,12 but we suggest that these associations more likely reflect reverse causality, with cancer causing chromosome 3 duplication and early death. A similar issue has previously been highlighted for sequence variants in cancer driver genes where rare mutations are also a cause of developmental disorders, such as ASXL1 and DNMT3A.37 Based on our analysis, the issue of somatic mutations confounding analyses of variants presumed to be germline can only be partially addressed by assigning the mosaic status of each variant; critically evaluating the validity of a variant (such as the breakpoints of a CNV), the plausibility of a finding (such as presence of constitutive autosomal aneuploidy), and likelihood of different mutational mechanisms (such as clonal expansion) are also important. As genome-wide sequencing becomes widely available in aging cohorts such as UK Biobank, researchers should be aware of potential confounding caused by somatic mutations present in high proportions of cells.
Acknowledgments
This research has been conducted using the UK Biobank Resource under Application Number 49847. The authors would like to acknowledge the use of the University of Exeter High-Performance Computing facility in carrying out this work. The authors declare no competing interests.
Published: June 22, 2020
Footnotes
Supplemental Data can be found online at https://doi.org/10.1016/j.ajhg.2020.06.001.
Data and Code Availability
This study did not generate new datasets or code. The code used during this study is available at https://github.com/WGLab/PennCNV. All bona fide researchers can apply to use the UK Biobank resource for health-related research that is in the public interest, https://www.ukbiobank.ac.uk/.
Supplemental Data
List of All >10 Mb Copy Number Changes in UK Biobank Detected via SNP-Genotyping Arrays DUP, duplication; DEL, deletion; LRR, log R ratio; St Dev LRR, standard deviation of the log R ratio; N SNPs, number of SNPs in the call.
References
- 1.Zarrei M., MacDonald J.R., Merico D., Scherer S.W. A copy number variation map of the human genome. Nat. Rev. Genet. 2015;16:172–183. doi: 10.1038/nrg3871. [DOI] [PubMed] [Google Scholar]
- 2.Conrad D.F., Pinto D., Redon R., Feuk L., Gokcumen O., Zhang Y., Aerts J., Andrews T.D., Barnes C., Campbell P., Wellcome Trust Case Control Consortium Origins and functional impact of copy number variation in the human genome. Nature. 2010;464:704–712. doi: 10.1038/nature08516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Craddock N., Hurles M.E., Cardin N., Pearson R.D., Plagnol V., Robson S., Vukcevic D., Barnes C., Conrad D.F., Giannoulatou E., Wellcome Trust Case Control Consortium Genome-wide association study of CNVs in 16,000 cases of eight common diseases and 3,000 shared controls. Nature. 2010;464:713–720. doi: 10.1038/nature08979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Collins R.L., Brand H., Karczewski K.J., Zhao X., Alföldi J., Francioli L.C., Khera A.V., Lowther C., Gauthier L.D., Wang H., Genome Aggregation Database Production Team. Genome Aggregation Database Consortium A structural variation reference for medical and population genetics. Nature. 2020;581:444–451. doi: 10.1038/s41586-020-2287-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cooper G.M., Coe B.P., Girirajan S., Rosenfeld J.A., Vu T.H., Baker C., Williams C., Stalker H., Hamid R., Hannig V. A copy number variation morbidity map of developmental delay. Nat. Genet. 2011;43:838–846. doi: 10.1038/ng.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coe B.P., Witherspoon K., Rosenfeld J.A., van Bon B.W.M., Vulto-van Silfhout A.T., Bosco P., Friend K.L., Baker C., Buono S., Vissers L.E.L.M. Refining analyses of copy number variation identifies specific genes associated with developmental delay. Nat. Genet. 2014;46:1063–1071. doi: 10.1038/ng.3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Veltman J.A. Genomic microarrays in clinical diagnosis. Curr. Opin. Pediatr. 2006;18:598–603. doi: 10.1097/MOP.0b013e3280105417. [DOI] [PubMed] [Google Scholar]
- 8.Shlien A., Malkin D. Copy number variations and cancer. Genome Med. 2009;1:62. doi: 10.1186/gm62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Campbell P.J., Stephens P.J., Pleasance E.D., O’Meara S., Li H., Santarius T., Stebbings L.A., Leroy C., Edkins S., Hardy C. Identification of somatically acquired rearrangements in cancer using genome-wide massively parallel paired-end sequencing. Nat. Genet. 2008;40:722–729. doi: 10.1038/ng.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ozery-Flato M., Linhart C., Trakhtenbrot L., Izraeli S., Shamir R. Large-scale analysis of chromosomal aberrations in cancer karyotypes reveals two distinct paths to aneuploidy. Genome Biol. 2011;12:R61. doi: 10.1186/gb-2011-12-6-r61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wright C.F., West B., Tuke M., Jones S.E., Patel K., Laver T.W., Beaumont R.N., Tyrrell J., Wood A.R., Frayling T.M. Assessing the Pathogenicity, Penetrance, and Expressivity of Putative Disease-Causing Variants in a Population Setting. Am. J. Hum. Genet. 2019;104:275–286. doi: 10.1016/j.ajhg.2018.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crawford K., Bracher-Smith M., Owen D., Kendall K.M., Rees E., Pardiñas A.F., Einon M., Escott-Price V., Walters J.T.R., O’Donovan M.C. Medical consequences of pathogenic CNVs in adults: analysis of the UK Biobank. J. Med. Genet. 2019;56:131–138. doi: 10.1136/jmedgenet-2018-105477. [DOI] [PubMed] [Google Scholar]
- 13.Kendall K.M., Bracher-Smith M., Fitzpatrick H., Lynham A., Rees E., Escott-Price V., Owen M.J., O’Donovan M.C., Walters J.T.R., Kirov G. Cognitive performance and functional outcomes of carriers of pathogenic copy number variants: analysis of the UK Biobank. Br. J. Psychiatry. 2019;214:297–304. doi: 10.1192/bjp.2018.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Owen D., Bracher-Smith M., Kendall K.M., Rees E., Einon M., Escott-Price V., Owen M.J., O’Donovan M.C., Kirov G. Effects of pathogenic CNVs on physical traits in participants of the UK Biobank. BMC Genomics. 2018;19:867. doi: 10.1186/s12864-018-5292-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kendall K.M., Rees E., Escott-Price V., Einon M., Thomas R., Hewitt J., O’Donovan M.C., Owen M.J., Walters J.T.R., Kirov G. Cognitive performance among carriers of pathogenic copy number variants: analysis of 152,000 UK biobank subjects. Biol. Psychiatry. 2017;82:103–110. doi: 10.1016/j.biopsych.2016.08.014. [DOI] [PubMed] [Google Scholar]
- 16.Kendall K.M., Rees E., Bracher-Smith M., Legge S., Riglin L., Zammit S., O’Donovan M.C., Owen M.J., Jones I., Kirov G., Walters J.T.R. Association of rare copy number variants with risk of depression. JAMA Psychiatry. 2019;76:818–825. doi: 10.1001/jamapsychiatry.2019.0566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Macé A., Tuke M.A., Deelen P., Kristiansson K., Mattsson H., Nõukas M., Sapkota Y., Schick U., Porcu E., Rüeger S. CNV-association meta-analysis in 191,161 European adults reveals new loci associated with anthropometric traits. Nat. Commun. 2017;8:744. doi: 10.1038/s41467-017-00556-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Männik K., Mägi R., Macé A., Cole B., Guyatt A.L., Shihab H.A., Maillard A.M., Alavere H., Kolk A., Reigo A. Copy number variations and cognitive phenotypes in unselected populations. JAMA. 2015;313:2044–2054. doi: 10.1001/jama.2015.4845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aguirre M., Rivas M.A., Priest J. Phenome-wide Burden of Copy-Number Variation in the UK Biobank. Am. J. Hum. Genet. 2019;105:373–383. doi: 10.1016/j.ajhg.2019.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tuke M.A., Ruth K.S., Wood A.R., Beaumont R.N., Tyrrell J., Jones S.E., Yaghootkar H., Turner C.L.S., Donohoe M.E., Brooke A.M. Mosaic Turner syndrome shows reduced penetrance in an adult population study. Genet. Med. 2019;21:877–886. doi: 10.1038/s41436-018-0271-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Genovese G., Kähler A.K., Handsaker R.E., Lindberg J., Rose S.A., Bakhoum S.F., Chambert K., Mick E., Neale B.M., Fromer M. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 2014;371:2477–2487. doi: 10.1056/NEJMoa1409405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jaiswal S., Ebert B.L. Clonal hematopoiesis in human aging and disease. Science. 2019;366:366. doi: 10.1126/science.aan4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vattathil S., Scheet P. Extensive hidden genomic mosaicism revealed in normal tissue. Am. J. Hum. Genet. 2016;98:571–578. doi: 10.1016/j.ajhg.2016.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Machiela M.J., Zhou W., Sampson J.N., Dean M.C., Jacobs K.B., Black A., Brinton L.A., Chang I.-S., Chen C., Chen C. Characterization of large structural genetic mosaicism in human autosomes. Am. J. Hum. Genet. 2015;96:487–497. doi: 10.1016/j.ajhg.2015.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bycroft C., Freeman C., Petkova D., Band G., Elliott L.T., Sharp K., Motyer A., Vukcevic D., Delaneau O., O’Connell J. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562:203–209. doi: 10.1038/s41586-018-0579-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang K., Li M., Hadley D., Liu R., Glessner J., Grant S.F.A., Hakonarson H., Bucan M. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007;17:1665–1674. doi: 10.1101/gr.6861907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Loh P.-R., Genovese G., Handsaker R.E., Finucane H.K., Reshef Y.A., Palamara P.F., Birmann B.M., Talkowski M.E., Bakhoum S.F., McCarroll S.A., Price A.L. Insights into clonal haematopoiesis from 8,342 mosaic chromosomal alterations. Nature. 2018;559:350–355. doi: 10.1038/s41586-018-0321-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Conlin L.K., Thiel B.D., Bonnemann C.G., Medne L., Ernst L.M., Zackai E.H., Deardorff M.A., Krantz I.D., Hakonarson H., Spinner N.B. Mechanisms of mosaicism, chimerism and uniparental disomy identified by single nucleotide polymorphism array analysis. Hum. Mol. Genet. 2010;19:1263–1275. doi: 10.1093/hmg/ddq003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hassold T., Hunt P. To err (meiotically) is human: the genesis of human aneuploidy. Nat. Rev. Genet. 2001;2:280–291. doi: 10.1038/35066065. [DOI] [PubMed] [Google Scholar]
- 30.Ben-David U., Amon A. Context is everything: aneuploidy in cancer. Nat. Rev. Genet. 2020;21:44–62. doi: 10.1038/s41576-019-0171-x. [DOI] [PubMed] [Google Scholar]
- 31.Shlush L.I. Age-related clonal hematopoiesis. Blood. 2018;131:496–504. doi: 10.1182/blood-2017-07-746453. [DOI] [PubMed] [Google Scholar]
- 32.Landau D.A., Tausch E., Taylor-Weiner A.N., Stewart C., Reiter J.G., Bahlo J., Kluth S., Bozic I., Lawrence M., Böttcher S. Mutations driving CLL and their evolution in progression and relapse. Nature. 2015;526:525–530. doi: 10.1038/nature15395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marasca R., Maffei R., Martinelli S., Fiorcari S., Bulgarelli J., Debbia G., Rossi D., Rossi F.M., Rigolin G.M., Martinelli S. Clinical heterogeneity of de novo 11q deletion chronic lymphocytic leukaemia: prognostic relevance of extent of 11q deleted nuclei inside leukemic clone. Hematol. Oncol. 2013;31:88–95. doi: 10.1002/hon.2028. [DOI] [PubMed] [Google Scholar]
- 34.James C., Ugo V., Le Couédic J.-P., Staerk J., Delhommeau F., Lacout C., Garçon L., Raslova H., Berger R., Bennaceur-Griscelli A. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434:1144–1148. doi: 10.1038/nature03546. [DOI] [PubMed] [Google Scholar]
- 35.Danielsen H.E., Pradhan M., Novelli M. Revisiting tumour aneuploidy - the place of ploidy assessment in the molecular era. Nat. Rev. Clin. Oncol. 2016;13:291–304. doi: 10.1038/nrclinonc.2015.208. [DOI] [PubMed] [Google Scholar]
- 36.Thompson D.J., Genovese G., Halvardson J., Ulirsch J.C., Wright D.J., Terao C., Davidsson O.B., Day F.R., Sulem P., Jiang Y., International Lung Cancer Consortium (INTEGRAL-ILCCO) Breast Cancer Association Consortium. Consortium of Investigators of Modifiers of BRCA1/2. Endometrial Cancer Association Consortium. Ovarian Cancer Association Consortium. Prostate Cancer Association Group to Investigate Cancer Associated Alterations in the Genome (PRACTICAL) Consortium. Kidney Cancer GWAS Meta-Analysis Project. eQTLGen Consortium. Biobank-based Integrative Omics Study (BIOS) Consortium. 23andMe Research Team Genetic predisposition to mosaic Y chromosome loss in blood. Nature. 2019;575:652–657. doi: 10.1038/s41586-019-1765-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carlston C.M., O’Donnell-Luria A.H., Underhill H.R., Cummings B.B., Weisburd B., Minikel E.V., Birnbaum D.P., Tvrdik T., MacArthur D.G., Mao R., Exome Aggregation Consortium Pathogenic ASXL1 somatic variants in reference databases complicate germline variant interpretation for Bohring-Opitz Syndrome. Hum. Mutat. 2017;38:517–523. doi: 10.1002/humu.23203. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
List of All >10 Mb Copy Number Changes in UK Biobank Detected via SNP-Genotyping Arrays DUP, duplication; DEL, deletion; LRR, log R ratio; St Dev LRR, standard deviation of the log R ratio; N SNPs, number of SNPs in the call.
Data Availability Statement
This study did not generate new datasets or code. The code used during this study is available at https://github.com/WGLab/PennCNV. All bona fide researchers can apply to use the UK Biobank resource for health-related research that is in the public interest, https://www.ukbiobank.ac.uk/.