

Abstract
The IL-7 receptor alpha chain (IL-7Rα or CD127) can be differentially expressed in memory CD8+ T cells. Here we investigated whether IL-7Rα could serve as a key molecule in defining a comprehensive landscape of heterogeneity in human effector memory (EM) CD8+ T cells using high-dimensional Cytometry by Time-Of-Flight (CyTOF) and single-cell RNA-seq (scRNA-seq). IL-7Rα had diverse, but organized, expressional relationship in EM CD8+ T cells with molecules related to cell function and gene regulation, which rendered an immune landscape defining heterogeneous cell subsets. The differential expression of these molecules likely has biological implications as we found in vivo signatures of transcription factors and homeostasis cytokine receptors, including T-bet and IL-7Rα. Our findings indicate the existence of heterogeneity in human EM CD8+ T cells as defined by distinct but organized expression patterns of multiple molecules in relationship to IL-7Rα and its possible biological significance in modulating downstream events.
Keywords: Effector memory CD8+ T cells, human, IL-7 receptor, high-dimensional single cell analysis
1. Introduction
Distinct subsets of CD4+ and CD8+ T cells are identified based on the expression of molecules including co-stimulatory molecules and receptors for cytokines and chemokines. CD4+ T helper (Th) 1, Th2 and Th17 differentially express CXCR3, CCR4 and CCR6 [1] while IL-1 receptor (R) I can define human Th17 cells [2]. In CD8+ T cells, the high affinity receptor alpha chain for IL-7 or IL-7Rα is differentially expressed by human effector memory (EM) CD8+ T cells, allowing the identification of IL-7Rα low and high EM CD8+ T cells with distinct cellular characteristics such as the expression of cytotoxic molecules, cytokines, co-stimulatory molecules, and DNA methylation [3–6].
The introduction of high-dimensional single cell analysis has revolutionized the understanding of cellular heterogeneity as determined by gene and protein expression. Single-cell RNA-seq (sc-RNA-seq) can be used to identify complex and rare cell populations and dissect the relationship among such cell populations [7]. cytometry or Cytometry by Time-Of-Flight (CyTOF) is a recently developed technology for multiparameter single cell analysis that permits unique high dimensional cell profiling at protein levels [8–10]. CyTOF utilizes heavy metal ions and mass spectrometry as labels and a readout, respectively, which allows the measuring of up to 40+ molecules in a single tube [11–13]. The high-dimensional CyTOF and scRNA-seq data can be analyzed to assess the multidimensional relationships of molecules expressed by single cells using computational methods such as dimensionality reduction and clustering algorithms [7–9]. The dimensionality reduction method of principal component analysis (PCA) was applied in analyzing human peripheral CD8+ T cells, showing diverse characteristics of total and viral specific CD8+ T cells [14–17]. Also, the relationships in multidimensional data can be visualized using the nonlinear dimensionality-reduction tool t-distributed stochastic neighbor embedding (t-SNE) that is used in combination with clustering analysis to robustly identify cellular subsets with distinct traits [18, 19]. Cellular development trajectories on multidimensional CyTOF data can be constructed using the method Wanderlust [20]. Also, the strength of the relationship among molecules expressed by cells can be quantified at the protein level via the application of the algorithm conditional-Density Resampled Estimate of Mutual Information (DREMI) on CyTOF data [21]. The function underlying the relationship can be visualized by conditional-Density Rescaled Visualization (DREVI) [21].
Human EM CD8+ T cells are divided into a few subsets based on the expression of a single or two molecules like CD27, CD28, CD45RA and IL-7Rα [3, 22, 23]. Previously, we showed the presence of human IL-7Rαhigh and low EM CD8+ T cell subsets with distinct cellular characteristics [3–6]. However, it is unknown whether IL-7Rα can serve as a key molecule in defining a comprehensive landscape heterogeneity in human EM CD8+ T cells as determined by high-dimensional single cell analysis. Here we addressed this question by analyzing CyTOF and scRNA-seq data of human EM CD8+ T cells using computational algorithms. The results of our study show heterogeneity in human EM CD8+ T cells as defined by distinct but organized expression patterns of multiple molecules, the unique position of IL-7Rα in defining such heterogeneity, and possible biological significance of differential expression of these molecules in modulating downstream events.
2. Materials and Methods
2.1. Human subjects
Peripheral blood was obtained after informed consent from healthy adult donors as done previously [2, 24, 25]. This work was approved by the institutional review committee of Yale University.
2.2. CyTOF Analysis
Peripheral blood mononuclear cells (PBMCs) were purified from blood using FicollPAQUE gradients. CyTOF analysis was done as previously done with some modifications [26]. All mass cytometry reagents were purchased from Fluidigm, Inc (South San Francisco, CA) unless otherwise noted. PBMCs (2 × 106 cells) were stained with a panel of metal-tagged antibodies (Supplemental Tables 1 and 2, including manufacturers) followed by Cisplatin staining. For intracellular staining, cells were fixed and permeabilized with Maxpar Fix 1 buffer and Maxpar Perm-S buffer, respectively. Stained cells were washed and kept overnight in the MaxPar Fix & Perm Buffer containing intercalator-Ir. Cells were resuspended with MaxPar Water containing EQ Four Element Calibration Beads and acquired on a CyTOF system Helios (Fluidigm). All FCS files were normalized and analyzed using the CYT [18], an open source analytic tool for CyTOF data, and FlowJo software (FlowJo, LLC). Also, FCS files from patients with SLE and their healthy controls were normalized using a CYTOF-preprocessing tool (https://github.com/KrishnaswamyLab/cytof-preprocessing). The FCS files were transformed using an inverse hyperbolic sine (arcsinh) function with a cofactor of 5 and pre-gated manually to exclude EQ beads, cell debris, cell doublets, and dead cells before additional analysis (Supplemental Fig. 5) [13]. PhenoGraph, t-SNE, Wanderlust, DREMI and DREMI were done on gated cells (2,500 cells) [19–21]. Also, we used a newly developed 3-dimensional (D) extension of DREMI and DREVI [27]. Here, the level of a third molecule was modeled as a function of the abundance of two molecules X and Y. The kernel density estimation used in 2D-DREMI [21] was extended to compute a 3D density estimate, which was then normalized by two parent dimensions rather than one dimension. The normalized, conditional distribution was visualized as a 2D surface that represents the “typical” behavior of molecule Z for each level of X and Y. Once the 3D-DREVI was computed, we computed 3D-DREMI by measuring the degree of information X and Y together which provided for the value of Z as in 2D-DREMI [21].
2.3. Single cell RNA-seq analysis
PBMCs were purified and stained with antibodies to APC-Cy7-CD16, Pacific Blue–CD8α, PE–Cy5–CD45RA and PE-Cy7-CCR7. Stained cells were into IL-7Rαlow and high EM (CCR7−) CD8+ T cells using a FACSAria® (BD Biosciences, San Jose, CA). The purity of cells was greater than 97%. For high-throughput single cell 3’ mRNA sequencing, we used a closed microwell array chip developed in the Rong Fan lab [28]. Briefly, single cells and uniquely barcoded mRNA capture beads were co-isolated in microwell array for single cell mRNA capture following lysis. Once the single cells and mRNA capture beads were loaded into the microwell, the microwell arrays were sealed using fluorinated oil (Fluorinert FC-40, Sigma Aldrich) to prevent cross-contamination after introducing freeze-thaw lysis buffer into the device. Three freeze-thaw cycles were used for the cell lysis, and then incubated at room temperature for 60 minutes for mRNA capture onto beads. After incubation, beads were retrieved from the device by flushing the beads out into an Eppendorf tube. Then, we followed previously reported protocols for reverse transcription, library construction, and sequencing [29, 30]. The Nextrera tagmentation (Nextera XT, Illumina) was used for the sequencing library constructions, and the finished libraries were sequenced on the HiSeq 2500 sequences (Illumina). Single-cell RNA-Seq data QA/QC was run on Partek Flow single cell module, and hg38_ensembl_release90_v2 was used for gene/feature annotation. STAR with default parameters was used to align sequence reads. Any cells with more than 1% of mitochondrial UMI counts were considered low quality. Cells with more than 2000 detected genes were checked to determine the rate of doublets. Any gene detected in less than three cells or a cell with less than 200 genes detected was excluded for downstream data analysis. A total of 1,732 cells from a donor were included in the final analysis. Using Seurat v2.0 (http://satijalab.org/seurat/), which is an R package for single cell genomics, the preprocessed data were log-normalized with a scale factor of 104. The clustering of the cells was done using only variable genes (2,037) selected by the FindVariableGenes (Seurat R package) with default parameters. The variable genes were then used to perform a PCA for dimensionality reduction. Significant PCs selected by a JackStraw test with 100 replicates and those with a p-value < 1e-5 were used to perform the clustering. After running the PCA, t-SNE analysis was run to visualize cells in a 2-D space. Clusters were identified using FindClusters (Seurat R package) using a 1.2. To identify differentially expressed genes in each of the clusters relative to the rest of the cells in the analysis, we used the function FindAllMarkers (Seurat R package). Only genes enriched and expressed at least in 25% of the cells in one of the two populations and with a log fold difference greater than 0.25 were considered. The scRNA-seq data set was submitted to Gene Expression Omnibus (GSE133167).
3. Results
3.1. High-dimensional CyTOF analysis shows heterogeneity in human EM CD8+ T cells expressing different levels of IL-7 receptor alpha chain.
We determined the expression of a set of 21 surface molecules, including IL-7Rα, in human EM CD8+ T cells using CyTOF to investigate the implication of IL-7Rα in defining cellular heterogeneity in the setting of high-dimensional profiling. These molecules included the ones encoded by differentially expressed genes (DEGs) in IL-7Rαlow and high EM CD8+ T cells [31] as well as those related to cell activation, co-stimulation, inhibition and migration. To characterize cell subsets in multiple donors, we ran the PhenoGraph algorithm on grouped EM CD8+ T cells from studied subjects based on the 21 molecules [19]. This revealed 185 cell subsets (Fig. 1A) that were landscaped on a t-SNE plot (Fig. 1B) where each subset and its size were indicated by a single point scaled to represent its proportion in the total 185 subsets. This landscape indicated that the subsets of EM CD8+ T cells from individual subjects could be grouped based on the similarities of the expression levels of the 21 molecules. Although the sizes of the subpopulations from each subject in individual clusters on the t-SNE plot were variable, each t-SNE cluster had subsets from most subjects (Fig. 1B). We took a metaclustering approach to merge the 185 subsets of EM CD8 into a set of secondary clusters or metaclusters [19, 32]. This analysis identified twelve metaclusters (Fig. 1C-D) with unique expression profiles of the analyzed molecules, including low and high levels of IL-7Rα expression (Fig 1D, t-SNE plot and heatmap). EM CD8+ T cell subsets in each cluster on the t-SNE plot belonged to the same metaclusters despite being from different subjects (Fig. 1B-C). The analyzed molecules could be grouped in relation to IL-7Rα expression in the metacluster analysis (Fig. 1D-E), showing the relationship of IL-7Rα expression with cell migration, co-stimulatory, and inhibitory molecules in EM CD8+ T cells. The proportions of individual metaclusters were variable, indicating quantitative heterogeneity of the EM CD8+ T cell subsets among subjects (Fig. 1F). Our findings support the existence of heterogeneous cell subsets with distinct levels of IL-7Rα expression in human EM CD8+ T cells.
Fig. 1. High-dimensional CyTOF analysis shows heterogeneity in human effector memory (EM) CD8+ T cells expressing different levels of IL-7 receptor alpha chain.

PBMCs of healthy human subjects were stained with antibodies to a set of molecules (Supplementary Table 1) and run on a Helios CyTOF instrument. The PhenoGraph clustering was performed on a group of EM CD8+ T cells from 13 individual donors based on the expression of 21 molecules including chemokine receptors, co-stimulatory and inhibitory molecules, which resulted in the division of the cells into 185 subpopulations as shown in (A). Subsequently, metaclustering (secondary clustering analysis) was done on these subpopulations (k parameter set at 15). t-SNE plots showing a landscape of the 185 subpopulations and their relationships to (B) individual donors and (C) metaclusters (MCs). Each subpopulation (subset) and its size were indicated by a single point scaled to represent its proportion in the total 185 subsets. (D) The MCs were named based on the expression profiles of individual MCs (heatmap) and indicated on a t-SNE plot generated on EM CD8+ T cells of the 13 donors. Each dot represents a single cell. Heatmap showing mean expression levels of 21 molecules by individual MCs. (E) t-SNE plot and bar graph showing expression levels of IL-7Rα by EM CD8+ T cells and individual MCs of the all 13 donors, respectively. (F) Graph showing the frequency of individual MCs (dots) in the 13 donors (bars and error bars indicate mean ± SEM, respectively).
3.2. The expression trajectories of chemokine receptors, co-stimulatory and inhibitory molecules have distinct but organized relationships with IL-7Rα expression in EM CD8+ T cells.
Given the expressional association of IL-7Rα with other molecules in EM CD8+ T cells, we explored the sequential relationship of such molecular expressions using the trajectory detection algorithm Wanderlust, which converts data from multi-parameter single-cell events into a one-dimensional linear trajectory [9, 20]. Here the starting point of a trajectory was designated by high levels of IL-7Rα and CD27 and low levels of CD57 because the former molecules are highly expressed by naïve CD8+ T cells while CD57 is considered a senescence marker of CD8+ T cells [23, 33]. The molecules differentially expressed by EM CD8+ T cells could be divided into several groups based on their Wanderlust trajectories. While the expression levels of IL-7Rα, CD27, CD28, CXCR4, CCR2 and CCR6 by EM CD8+ T cells gradually decreased in most studied subjects, the expression levels of CD57, 4–1BB, CXCR1 and CX3CR1 steadily increased (Fig. 2). This pattern became more apparent in all tested individuals as the graph smoothing level was increased to leave out noise (Supplemental Fig. 1A-B). Although the exact differentiation pathway(s) of EM CD8+ T cells is yet to be elucidated, our trajectory analysis infers that organized events occur sequentially across the expression of chemokine receptors and co-stimulatory molecules by human EM CD8+ T cells at the single cell level in relation to CD8+ T cell differentiation-associated molecules like IL-7Rα, CD27 and CD57.
Fig. 2. The expression trajectories of chemokine receptors, co-stimulatory and inhibitory molecules have distinct but organized relationships with the expression trajectory of IL-7 receptor alpha in human effector memory CD8+ T cells.

(A-B) The trajectory detection algorithm Wanderlust was applied to EM CD8+ T cells gated on the CyTOF data of PBMCs. EM CD8+ T cells that expressed high levels of IL-7Rα and CD27 and low levels of CD57 were designated as the youngest cells or the starting point of trajectories in the Wanderlust analysis. Graph smooth levels were set at 2 to allow matching differences in samples. Expression trajectories of (A) co-stimulatory and -inhibitory molecules and (B) chemokine receptors along with IL-7Rα.
3.3. High-dimensional single cell RNA-seq analysis shows heterogeneity in human EM CD8+ T cells expressing different levels of the IL7RA gene.
We next interrogated the relationship of IL7RA gene expression in defining heterogeneity of human EM CD8+ T cells at the gene level using high-dimensional scRNA-seq. By applying the PCA algorithms, we identified a set of genes that defined the gene expression profiles of individual EM CD8+ T cells. The genes which belonged to principal component 1 of the PCA included IL7RA, perforin (PRF1), and granzymes (Supplemental Fig. 2). Also, we analyzed the data using a combination of t-SNE and k-means clustering. This analysis revealed a set of genes, including IL7RA, that delineated the gene expression profiles of distinct cell subsets (Fig. 3A-C). In accordance with the results of CyTOF analysis, the identified cell clusters could divide into those with high and low levels of IL7RA expression, and heterogeneous cell subsets were found in cells expressing different levels of IL7RA. The characteristics of such cell subsets had some similarities to those of cell subsets detected by metaclustering on CyTOF data (Fig. 3C). Cells expressing low levels of IL7RA CX3CR1 expression but decreased CXCR4 expression compared to cells expressing high levels of IL7RA. Also, cells with low levels of IL7RA expression had high levels of PRF1, GZMB1, and TBX21 expression but low levels of CD27, CD28 and CD69 compared to the same cells expressing low levels of IL7RA (Fig 3D). We characterized the heterogeneity of EM CD8+ T cells using a supervised clustering approach based on the genes encoding molecules analyzed by CyTOF (Fig 3E). Although only some of these genes were detected at the single cell level, the clusters identified by this approach shared similarities with the cell clusters identified by CyTOF analysis. For instance, cells in cluster C1 detected by this analysis had high levels of IL7RA, CD69, CD27 and CXCR4 genes along with low levels of the SELL gene encoding CD62L as in cells belong to metacluster 1 (MC1) identified by CyTOF (Fig 3E and Fig 1D). We also determined the heterogeneity of EM CD8+ T cells based on a set of molecules related to CD8+ T cell functions (Fig 3F). Since we analyzed unstimulated EM CD8+ T cells, most cells hardly expressed cytokine genes except a small number of cells with the expression of the IFNG gene (Fig 3F, cluster F10). Of interest, a subset of IL7RAlow cells expressing GZMB and PRF1 had high expression levels of the LAMP1 gene which encodes lysosomal-associated membrane protein 1 (LAMP-1 or CD107a), a molecule associated with degranulation of cytotoxic molecules and cell migration (Fig 3F, cluster F3). This subset also expressed high levels of the fractalkine receptor CX3CR1, suggesting that highly cytotoxic EM CD8+ T cells may migrate to inflammatory sites through chemotaxis mediated by CX3CR1 and its ligand fractalkine. As previously reported [34], the T cell exhaustion-associated genes LAG3, CD244 (2B4) and CD160 were expressed by cells in the same cluster with low expression levels of the IL7RA gene (Fig 3F, cluster F2). These findings indicate that IL7RA has a unique place in defining distinct EM CD8+ T cell subsets at the gene expression level.
Fig. 3. Single-cell RNA-seq analysis shows heterogeneity in human effector memory CD8+ T cells expressing different levels of the IL-7 receptor alpha chain gene.
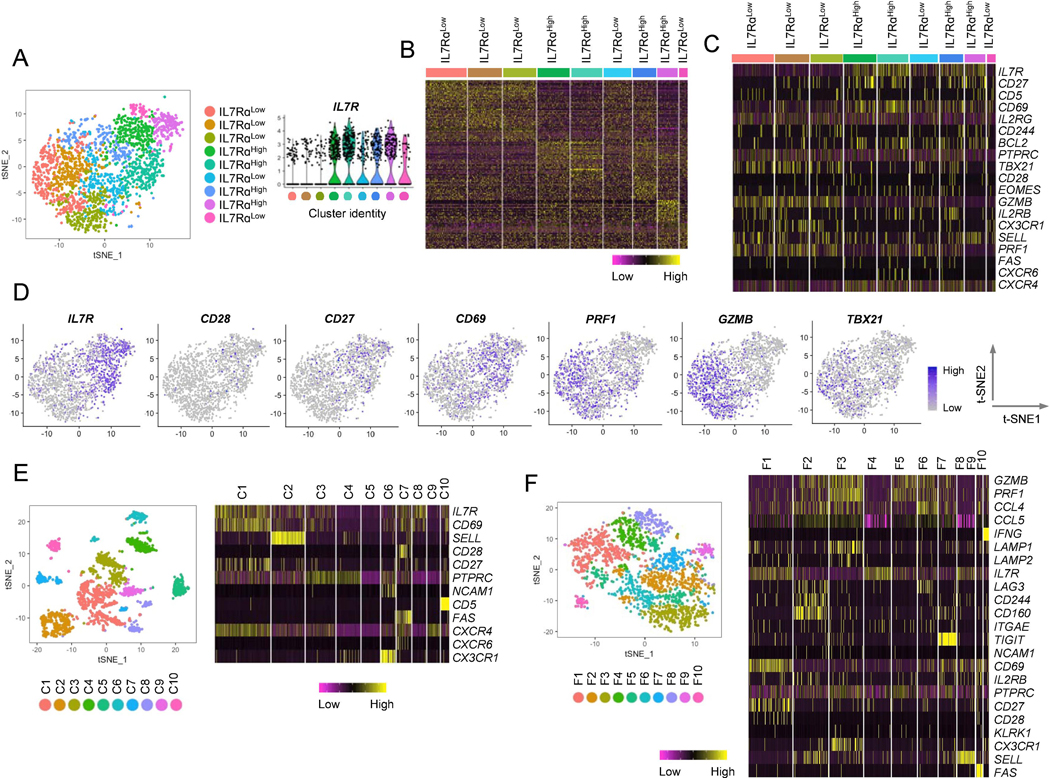
Single-cell RNA-seq analysis was performed on FACS-sorted EM CD8+T from PBMCs of a healthy donor. This analysis yielded data for 1,732 cells with variable expression levels of 2,037 genes. Unsupervised clustering revealed nine transcriptionally distinct subpopulations. (A) t-SNE plot showing nine transcriptionally distinct EM CD8+ T cells clusters with different levels of IL7RA gene expression. The expression of the IL7RA gene by the individual clusters were presented as violin plots. Individual cell clusters were indicated by different colors. Each dot represents a single cell. (B) Heatmap showing the gene expression profiles of individual clusters identified in (A). (C) Heatmap showing the expression values of a set of genes, including IL7RA and those related to IL7RA in Fig 1, in individual clusters. (D) t-SNE plots showing expression levels of the indicated molecules in EM CD8+ T cells. (E-F) t-SNE plots and heatmaps showing the results of supervised clustering analyses based on a set of genes employed for CyTOF (E) and genes associated with CD8+ T cell functions (F). C1–10 and F1-F10 indicate cell clusters.
3.4. Signatures of the transcription factor T-bet and T cell homeostasis cytokine receptors, including IL-7Rα, are present at different levels across EM CD8+ T cells.
The transcription factor T-bet (T-box transcription factor) encoded by the TBX21 plays a role in regulating perforin and granzyme B (GZMB) as well as in differentiating effector CD8+ T cells, including functionally exhausted ones expressing inhibitory molecules (e.g. 2B4) [35–37]. Our scRNA-seq analysis showed differential expression of TBX21, GZMB and PRF1 (perforin) in IL-7Rαlow and high EM CD8+ T cells (Fig. 3C-D). We further investigated this relationship at the protein level using DREMI and DREVI algorithms for CyTOF [21]. DREMI computes mutual information that describes how the state of Y varies with different states of X [21]. DREVI visualizes the function underlying this molecular interaction [21]. DREMI scores indicate the strength of the statistical dependency between two molecules. DREVI plots visualized the nature of their relationships. We noticed a reciprocal expressional relationship between IL-7Rα and T-bet at the single cell level (Fig. 4A) while T-bet had a positive expressional relationship with perforin, GZMB, 2B4 and CD57 (Fig 4A). In fact, the DREMI scores for perforin and GZMB were the highest ones in association with T-bet (Fig. 4A). The transcription factor eomesodermin (Eomes) can also regulate perforin and GZMB as well as the differentiation of CD8+ T cells [36, 38]. DREMI scores between Eomes and these molecules appeared to be less strong compared to T-bet (Supplemental Fig. 3). The expression trajectories of T-bet, perforin, and GZMB were closely related and had an inverse relationship with IL-7Rα as determined by Wanderlust (Fig. 4B and Supplemental Fig. 4A). We next explored the 3D relationships of these molecules using the recently developed 3D DREMI and DREVI [27]. EM CD8+ T cells expressing high levels of T-bet and Eomes had high levels of GZMB and perforin while IL-7Rα expression was low in those cells expressing high levels of T-bet and perforin (Fig. 4C).
Fig. 4. The signature of the transcription factor T-bet is present at different levels across human effector memory CD8+ T cells as determined by the expression of T-bet and its target molecules at the single cell level.

PBMCs of healthy human subjects were stained with antibodies to a set of molecules, including IL-7RA, T-bet, eomesodermin (Eomes), perforin, granzyme B, CD57, and 2B4, and run on a Helios CyTOF instrument. (A) 2-dimensional DREVI plots showing the expression relationship of IL-7Rα with T-bet as well as of T-bet with perforin, granzyme B, CD57, and 2B4 in EM CD8+ T cells of 5 donors (DO1-DO5). DREMI scores that indicate the strength of the statistical dependency between two molecules are shown above the DREVI plots. (B) Expression trajectories of T-bet, perforin, granzyme B, 2B4, CD57 and IL-7Rα in EM CD8+ T cells as determined by the trajectory detection algorithm Wanderlust. EM CD8+ T cells that expressed high levels of IL-7Rα and CD27 and low levels of CD57 were used as the youngest cells or the starting point of trajectories, in the Wanderlust analysis. (C) 3-dimensional (3D) DREVI plots showing the expression relationship of T-bet with perforin, granzyme B, IL-7Rα and Eomes CD8+ T cells. DREMI scores that indicate the strength of the statistical dependency in three molecules are shown above the DREVI plots. Representative data from 5 donors.
IL-7 and IL-15 are involved in CD8+ T cell homeostasis by enhancing anti-apoptotic molecule Bcl-2 expression [39, 40]. Both IL-7 and IL-15 receptor complexes have the common gamma chain (γC or CD132) in addition to IL-7Rα (CD127) and IL-2/15Rβ (CD122) chains, respectively [41]. The IL-2 receptor complex comprises γC, IL-2Rα (CD25) and IL-2/15Rβ [41]. Our Wanderlust trajectory analysis showed a gradual decrease in Bcl-2 along with IL-7Rα and γC in EM CD8+ T cells (Fig. 5A and Supplemental Fig. 4B). However, Bcl-2 and γC levels rebounded and IL-7Rα levels remained low while those of IL-15Rβ and CD25 re-rose. This interpretation may need to be taken carefully since Wanderlust trajectory analysis assumes that cells take a single differentiation pathway although more than one differentiation pathway can exist. We thus determined intermolecular quantitative relationships between the cytokine receptors and Bcl-2 using DREMI and DREVI algorithms (Fig. 5B), which showed modest intermolecular quantitative relationships. Since IL-7Rα and γC form the IL-7Rα complex to upregulate Bcl-2, we explored their relationships in EM CD8+ T cells using a 3D DREMI approach. Indeed, EMCD8+ T cells with high levels of Bcl-2 are those with high levels of IL-7Rα and C (Fig. 5C). A similar relationship was observed among Bcl-2, IL-7Rα, and CD122 as determined by the 3D DREMI and DREVI (Fig. 5C). Overall, our findings suggest that signatures of transcription factors and homeostasis cytokine receptors, including T-bet, IL-7Rα and γC, are present in EM CD8+ T cells in vivo at different levels as measured by the expressional levels of their downstream molecules including perforin, GZMB and Bcl-2 at the single cell level.
Fig. 5. The signature of T cell homeostasis cytokine receptors is present at different levels across human effector memory CD8+ T cells as determined by the expression of γC cytokine receptors and its target Bcl-2 at the single cell level.

PBMCs of healthy human subjects were stained with antibodies to a set of molecules, including the γC cytokine receptors IL-7Rα, CD122 (IL-15Rβ), CD25 (IL-2/15Rα), CD132 (γC) and the anti-apoptotic molecule Bcl-2, and run on a Helios CyTOF instrument as in Fig. 4. (A) Expression trajectories of IL-7Rα, CD122, CD25, CD132, and Bcl-2 in EM CD8+ T cells as determined by the trajectory detection algorithm Wanderlust. EM CD8+ T cells that expressed high levels of IL-7Rα and CD27 and low levels of CD57 were designated as the youngest cells or the starting point of trajectories, in the Wanderlust analysis. (B) 2-dimensional DREVI plots showing the expression relationship of IL-7Rα, CD122, CD25 and CD132 with Bcl-2 in EM CD8+ T cells of 5 donors (DO1-DO5). DREMI scores that indicate the strength of the statistical dependency between two molecules are shown above the DREVI plots. (C) 3-dimensional (3D) DREVI plots showing the expression relationship of IL-7Rα, CD122, CD132 and Bcl-2 in EM CD8+ T cells. DREMI scores that indicate the strength of the statistical dependency in three molecules are shown above the DREVI plots. Representative data from 5 donors.
4. Discussion
We investigated the role of IL-7Rα in defining heterogeneity of human EM CD8+ T cells and its expressional relationship with molecules implicated in such heterogeneity using high-dimensional CyTOF and scRNA-seq analyses. The results of our study showed that IL-7Rα has diverse but organized expressional relationship with molecules related to gene regulation, effector function, survival and migration in human EM CD8+ T cells, rendering a landscape of heterogeneous EM CD8+ T cell subsets. Such heterogeneity likely occurs in an orchestrated manner given the relationship in the expression trajectories and quantities of these molecules at the single cell level as assessed by the algorithms Wanderlust, DREMI and DREVI. The differential expression of the molecules may have functional implications as we found in vivosignatures of T-bet, IL-7Rα and γC at different levels across EM CD8+ T cells by determining the expression relationship of such molecules with their target molecules perforin, GZMB and Bcl-2 using 2D and novel 3D DREMI and DREVI. Our approach investigating the heterogeneity of EM CD8+ T cells at the single cell level based on a set of molecules, including IL-7Rα, can be a valuable tool in identifying altered CD8+ T cell subsets in health and disease. Overall, our findings indicate the existence of heterogeneous subsets in human EM CD8+ T cells as defined by the distinct but organized expressional relationship of IL-7Rα with multiple molecules and the possible biological significance of differential expression of such molecules in modulating downstream events.
Here we utilized CyTOF and scRNA-seq analyses in exploring heterogeneity of human EM CD8+ T cells, especially in relation to IL-7Rα expression. IL-7Rαlow and high EM CD8+ T cells could be divided into additional subsets based on the expression of these molecules. The phenotypic characteristics of subsets of EM CD8+ T cells were relatively similar among studied healthy subjects as determined by the metaclustering method although the proportions of such subsets appeared to be variable. It would be intriguing to investigate whether such heterogeneity changes at greater levels in pathologic conditions. Although the exact differentiation pathway(s) of IL-7Rαhigh and low EM CD8+ T cells is yet to be demonstrated, the results of our trajectory analysis are in line with previous studies showing expressional changes in co-stimulatory and inhibitory molecules along with IL-7Rα upon T cell stimulation [23, 33]. With antigenic CD8+ T cell stimulation, especially in the setting of repetitive and/or chronic stimulatory conditions, IL-7Rα and the co-stimulatory molecules CD27 and CD28 become down regulated on CD8+ T cells while the co-inhibitory molecule PD-1 and the cell senescence-associated molecule CD57 increase [23, 33]. Such changes could be related to genetic and epigenetic changes as suggested by the role of DNA methylation in regulating IL-7Rα and CX3CR1 expression in human CD8+ T cells [4, 6, 42]. Overall, the results of our Wanderlust trajectory analysis suggest that distinct sets of chemokine receptors, co-stimulatory and effector molecules tend to change in an organized manner in relation to IL-7Rα expression in EM CD8+ T cells at the single cell level.
Although perforin and GZMB are known to be transcriptionally regulated by T-bet and Eomes [38, 43], the in vivo quantitative relationship between these molecules in human CD8+ T cells at the single cell level was largely unknown. Here we first show a robust quantitative relationship of T-bet with perforin and GZMB in human CD8+ T cells using the recently developed algorithms DREMI and DREVI. The latter analysis indicates a linear type quantitative relationship between T-bet and perforin in EM CD8+ T cells. The expression levels of GZMB appeared to rise somewhat abruptly a certain level of T-bet expression in EM CD8+ T cells. These findings suggest that the dose-response relationship between T-bet and its target cytotoxic molecules perforin and GZMB may not be similar at the single cell level in human EM CD8+ T cells. Eomes had a similar but less robust expression relationship with perforin and GZMB compared to T-bet. In fact, our 3D DREMI and DREVI analysis supports a combinational role of T-bet and Eomes in regulating perforin and GZMB expression in EM CD8+ T cells at the single cell level.
Animal and in vitro studies showed the role of γC cytokines, including IL-7 and IL-15, in promoting memory T cell survival by up-regulating Bcl-2 [3, 40, 44, 45]. However, the quantitative relationship of Bcl-2 expression with γC cytokine receptor expression is not fully defined in human CD8+ T cells at the single cell level. The results of our analysis infer the existence of a quantitative relationship between Bcl-2 and the receptor subunits for IL-7 and IL-15 in human EM CD8+ T cells. This finding is further supported by our Wanderlust trajectory analysis showing the relationship in the expression of the same molecules at the single level. These results indicate that the expression levels of T cell homeostasis cytokine receptors change in EM CD8+ T cells, contributing to Bcl-2 expression, a possible signature of these cytokine receptors.
In summary, we believe our study is unique in that it has investigated heterogeneity of human EM CD8+ T cells in relation to the T cell homeostasis cytokine receptor IL-7Rα using high-dimensional CyTOF and scRNA-seq analyses. The results of our study showed the existence of heterogeneity in human EM CD8+ as defined by distinct but organized expression patterns of multiple molecules, including IL-7Rα, biological implications of differential expression of such molecules in controlling downstream events.
Supplementary Material
Highlights.
IL-7Rα has unique relationship with a set of molecules in EM CD8+ T cells.
Such a relationship defines heterogeneous cell subsets in EM CD8+ T cells.
Molecules expressed by these subsets may confer unique biological significance.
IL-7Rα has unique relationship with a set of molecules in EM CD8+ T cells.
Such a relationship defines heterogeneous cell subsets in EM CD8+ T cells.
Molecules expressed by these subsets may confer unique biological significance.
Acknowledgments
The authors thank Drs. Steven Kleinstein and Bram Gerritsen for their constructive discussions and critical review of the manuscript. We also thank Dr. Ala Nassar and Ms. Shelly Ren of the Yale CyTOF Core.
Funding sources
This work was supported in part by grants from the National Institutes of Health (2R56AG0280691, 1R01AG056728, and R21AI126604 to IK; K24AG042489, P30AG021342 (Yale Claude D. Pepper Older Americans Independence Center) to ACS; and R01AG055362 to IK and ACS).
Abbreviations:
- EM
effector memory
- IL-7Rα or CD127
IL-7 receptor alpha chain
- CyTOF
Cytometry by Time-Of-Flight
- scRNA-seq
single-cell RNA-seq
- Th
T helper
- R
receptor
- PCA
principal component analysis
- t-SNE
t-distributed stochastic neighbor embedding
- DREMI
conditional-Density Resampled Estimate of Mutual Information
- DREVI
conditional-Density Rescaled Visualization
- PBMCs
peripheral blood mononuclear cells
- DEGs
differentially expressed genes
- GZMB
granzyme B
Footnotes
Declaration of Competing Interest
The authors declare no known competing financial interests or personal relationships that could influence the work reported in this paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Sallusto F, Lanzavecchia A, Heterogeneity of CD4+ memory T cells: functional modules for tailored immunity, Eur J Immunol, 39 (2009) 2076–2082. [DOI] [PubMed] [Google Scholar]
- [2].Lee WW, Kang SW, Choi J, Lee SH, Shah K, Eynon EE, Flavell RA, Kang I, Regulating human Th17 cells via differential expression of IL-1 receptor, Blood, 115 (2010) 530–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Kim HR, Hong MS, Dan JM, Kang I, Altered IL-7R{alpha} expression with aging and the potential implications of IL-7 therapy on CD8+ T-cell immune responses, Blood, 107 (2006) 2855–2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Shin MS, You S, Kang Y, Lee N, Yoo SA, Park K, Kang KS, Kim SH, Mohanty S, Shaw AC, Montgomery RR, Hwang D, Kang I, DNA Methylation Regulates the Differential Expression of CX3CR1 on Human IL-7Ralphalow and IL-7Ralphahigh Effector Memory CD8+ T Cells with Distinct Migratory Capacities to the Fractalkine, J Immunol, 195 (2015) 2861–2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kim HR, Hwang KA, Kang I, Dual roles of IL-15 in maintaining IL-7RalphalowCCR7-memory CD8+ T cells in humans via recovering the phosphatidylinositol 3-kinase/AKT pathway, J Immunol, 179 (2007) 6734–6740. [DOI] [PubMed] [Google Scholar]
- [6].Kim HR, Hwang KA, Kim KC, Kang I, Down-regulation of IL-7Ralpha expression in human T cells via DNA methylation, J Immunol, 178 (2007) 5473–5479. [DOI] [PubMed] [Google Scholar]
- [7].Hwang B, Lee JH, Bang D, Single-cell RNA sequencing technologies and bioinformatics pipelines, Exp Mol Med, 50 (2018) 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Saeys Y, Gassen SV, Lambrecht BN, Computational flow cytometry: helping to make sense of high-dimensional immunology data, Nat Rev Immunol, 16 (2016) 449–462. [DOI] [PubMed] [Google Scholar]
- [9].Chester C, Maecker HT, Algorithmic Tools for Mining High-Dimensional Cytometry Data, J Immunol, 195 (2015) 773–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Spitzer MH, Nolan GP, Mass Cytometry: Single Cells, Many Features, Cell, 165 (2016) 780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Bandura DR, Baranov VI, Ornatsky OI, Antonov A, Kinach R, Lou X, Pavlov S, Vorobiev S, Dick JE, Tanner SD, Mass cytometry: technique for real time single cell multitarget immunoassay based on inductively coupled plasma time-of-flight mass spectrometry, Anal Chem, 81 (2009) 6813–6822. [DOI] [PubMed] [Google Scholar]
- [12].Ornatsky O, Bandura D, Baranov V, Nitz M, Winnik MA, Tanner S, Highly multiparametric analysis by mass cytometry, J Immunol Methods, 361 (2010) 1–20. [DOI] [PubMed] [Google Scholar]
- [13].Bendall SC, Simonds EF, Qiu P, Amir el AD, Krutzik PO, Finck R, Bruggner RV, Melamed R, Trejo A, Ornatsky OI, Balderas RS, Plevritis SK, Sachs K, Pe’er D, Tanner SD, Nolan GP, Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum, Science, 332 (2011) 687–696.21551058 [Google Scholar]
- [14].Newell EW, Sigal N, Bendall SC, Nolan GP, Davis MM, Cytometry by time-of-flight shows combinatorial cytokine expression and virus-specific cell niches within a continuum of CD8+ T cell phenotypes, Immunity, 36 (2012) 142–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Cheng Y, Newell EW, Deep Profiling Human T Cell Heterogeneity by Mass Cytometry, Adv Immunol, 131 (2016) 101–134. [DOI] [PubMed] [Google Scholar]
- [16].Newell EW, Sigal N, Nair N, Kidd BA, Greenberg HB, Davis MM, Combinatorial tetramer staining and mass cytometry analysis facilitate T-cell epitope mapping and characterization, Nat Biotechnol, 31 (2013) 623–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Swadling L, Capone S, Antrobus RD, Brown A, Richardson R, Newell EW, Halliday J, Kelly C, Bowen D, Fergusson J, Kurioka A, Ammendola V, Del Sorbo M, Grazioli F, Esposito ML, Siani L, Traboni C, Hill A, Colloca S, Davis M, Nicosia A, Cortese R, Folgori A, Klenerman P, Barnes E, A human vaccine strategy based on chimpanzee adenoviral and MVA vectors that primes, boosts, and sustains functional HCV-specific T cell memory, Sci Transl Med, 6 (2014) 261ra153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Amir el AD, Davis KL, Tadmor MD, Simonds EF, Levine JH, Bendall SC, Shenfeld DK, Krishnaswamy S, Nolan GP, Pe’er D, viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia, Nat Biotechnol, 31 (2013) 545–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Levine JH, Simonds EF, Bendall SC, Davis KL, Amir el AD, Tadmor MD, Litvin O, Fienberg HG, Jager A, Zunder ER, Finck R, Gedman AL, Radtke I, Downing JR, Pe’er D, Nolan GP, Data-Driven Phenotypic Dissection of AML Reveals Progenitor-like Cells that Correlate with Prognosis, Cell, 162 (2015) 184–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Bendall SC, Davis KL, Amir el AD, Tadmor MD, Simonds EF, Chen TJ, Shenfeld DK, Nolan GP, Pe’er D, Single-cell trajectory detection uncovers progression and regulatory coordination in human B cell development, Cell, 157 (2014) 714–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Krishnaswamy S, Spitzer MH, Mingueneau M, Bendall SC, Litvin O, Stone E, Pe’er D, Nolan GP, Systems biology. Conditional density-based analysis of T cell signaling in single-cell data, Science, 346 (2014) 1250689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Hamann D, Baars PA, Rep MH, Hooibrink B, Kerkhof-Garde SR, Klein MR, van Lier RA, Phenotypic and functional separation of memory and effector human CD8+ T cells, J Exp Med, 186 (1997) 1407–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Mahnke YD, Brodie TM, Sallusto F, Roederer M, Lugli E, The who’s who of T-cell differentiation: human memory T-cell subsets, Eur J Immunol, 43 (2013) 2797–2809. [DOI] [PubMed] [Google Scholar]
- [24].Kang KS, Lee N, Shin MS, Kim S, Yu Y, Mohanty S, Belshe RB, Montgomery RR, Shaw AC, Kang I, An altered relationship of influenza vaccine-specific IgG responses with T cell immunity occurs with aging in humans, Clin Immunol, 147 (2013) 79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Shah K, Lee WW, Lee SH, Kim SH, Kang SW, Craft J, Kang I, Dysregulated balance of Th17 and Th1 cells in systemic lupus erythematosus, Arthritis Res Ther, 12 (2010) R53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Shin MS, Yim K, Moon K, Park HJ, Mohanty S, Kim JW, Montgomery RR, Shaw AC, Krishnaswamy S, Kang I, Dissecting alterations in human CD8+ T cells with aging by high-dimensional single cell mass cytometry, Clin Immunol, 200 (2019) 24–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Krishnaswamy S, Zivanovic N, Sharma R, Pe’er D, Bodenmiller B, Learning Edge Rewiring in EMT from Single Cell Data, bioRxiv, (2017). [Google Scholar]
- [28].Dura B, Choi JY, Zhang K, Damsky W, Thakral D, Bosenberg M, Craft J, Fan R, scFTD-seq: freeze-thaw lysis based, portable approach toward highly distributed single-cell 3’ mRNA profiling, Nucleic Acids Res, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM, Trombetta JJ, Weitz DA, Sanes JR, Shalek AK, Regev A, McCarroll SA, Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets, Cell, 161 (2015) 1202–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Gierahn TM, Wadsworth MH 2nd, Hughes TK, Bryson BD, Butler A, Satija R, Fortune S, Love JC, Shalek AK, Seq-Well: portable, low-cost RNA sequencing of single cells at high throughput, Nat Methods, 14 (2017) 395–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Park HJ, Shin MS, Kim M, Bilsborrow JB, Mohanty S, Montgomery RR, Shaw AC, You S, Kang I, Transcriptomic analysis of human IL-7 receptor alpha (low) and (high) effector memory CD8(+) T cells reveals an age-associated signature linked to influenza vaccine response in older adults, Aging Cell, (2019) e12960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Pyne S, Hu X, Wang K, Rossin E, Lin TI, Maier LM, Baecher-Allan C, McLachlan GJ, Tamayo P, Hafler DA, De Jager PL, Mesirov JP, Automated high-dimensional flow cytometric data analysis, Proc Natl Acad Sci U S A, 106 (2009) 8519–8524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lee N, Shin MS, Kang I, T-cell biology in aging, with a focus on lung disease, J Gerontol A Biol Sci Med Sci, 67 (2012) 254–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Agresta L, Hoebe KHN, Janssen EM, The Emerging Role of CD244 Signaling in Immune Cells of the Tumor Microenvironment, Front Immunol, 9 (2018) 2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Glimcher LH, Townsend MJ, Sullivan BM, Lord GM, Recent developments in the transcriptional regulation of cytolytic effector cells, Nat Rev Immunol, 4 (2004) 900–911. [DOI] [PubMed] [Google Scholar]
- [36].Kaech SM, Cui W, Transcriptional control of effector and memory CD8+ T cell differentiation, Nat Rev Immunol, 12 (2012) 749–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Buggert M, Tauriainen J, Yamamoto T, Frederiksen J, Ivarsson MA, Michaelsson J, Lund O, Hejdeman B, Jansson M, Sonnerborg A, Koup RA, Betts MR, Karlsson AC, T-bet and Eomes are differentially linked to the exhausted phenotype of CD8+ T cells in HIV infection, PLoS Pathog, 10 (2014) e1004251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Pearce EL, Mullen AC, Martins GA, Krawczyk CM, Hutchins AS, Zediak VP, Banica M, DiCioccio CB, Gross DA, Mao CA, Shen H, Cereb N, Yang SY, Lindsten T, Rossant J, Hunter CA, Reiner SL, Control of effector CD8+ T cell function by the transcription factor Eomesodermin, Science, 302 (2003) 1041–1043. [DOI] [PubMed] [Google Scholar]
- [39].Boyman O, Krieg C, Homann D, Sprent J, Homeostatic maintenance of T cells and natural killer cells, Cell Mol Life Sci, 69 (2012) 1597–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kim HR, Hwang KA, H Park S, Kang I, IL-7 and IL-15: biology and roles in T-Cell immunity in health and disease, Crit Rev Immunol, 28 (2008) 325–339. [DOI] [PubMed] [Google Scholar]
- [41].Rochman Y, Spolski R, Leonard WJ New insights into the regulation of T cells by gamma(c) family cytokines, Nat Rev Immunol, 9 (2009) 480–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Chen Y, Zander R, Khatun A, Schauder DM, Cui W, Transcriptional and Epigenetic Regulation of Effector and Memory CD8 T Cell Differentiation, Front Immunol, 9 (2018) 2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Townsend MJ, Weinmann AS, Matsuda JL, Salomon R, Farnham PJ, Biron CA, Gapin L, Glimcher LH, T-bet regulates the terminal maturation and homeostasis of NK and Valpha14i NKT cells, Immunity, 20 (2004) 477–494. [DOI] [PubMed] [Google Scholar]
- [44].Graninger WB, Steiner CW, Graninger MT, Aringer M, Smolen JS, Cytokine regulation of apoptosis and Bcl-2 expression in lymphocytes of patients with systemic lupus erythematosus, Cell Death Differ, 7 (2000) 966–972. [DOI] [PubMed] [Google Scholar]
- [45].Wojciechowski S, Tripathi P, Bourdeau T, Acero L, Grimes HL, Katz JD, Finkelman FD, Hildeman DA, Bim/Bcl-2 balance is critical for maintaining naive and memory T cell homeostasis, J Exp Med, 204 (2007) 1665–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.