

Abstract
PURPOSE
The diagnosis of cancer predisposition in pediatric patients with cancer is vital for treatment decisions, surveillance, and management of at-risk family members. Somatic tumor testing can identify potential underlying constitutional variants that confer increased cancer risk. Here, we report the characteristics of constitutional variants identified through tumor testing.
MATERIALS AND METHODS
Data were abstracted from medical record review of 1,023 patients who received in-house somatic tumor testing over a 28-month period. Patients were identified for testing using referral criteria developed as a collaboration between genomic diagnostics, pathology, and oncology. Characteristics of patients who underwent constitutional testing, including family history and variant loss of heterozygosity, were tracked.
RESULTS
From 1,023 patients who underwent somatic tumor sequencing in a 28-month period, 210 variants were identified in 141 patients (13.8%) that were concerning for cancer predisposition syndromes requiring intervention. A total of 73 variants in 41 patients have undergone clinical confirmatory testing thus far. Of these, 26 variants were confirmed to be constitutionally present (35.6%). Among patients tested, 23 (56.1%) of 41 total patients were diagnosed with a cancer predisposition syndrome.
CONCLUSION
Our data demonstrate that more than one third of variants in tumor somatic sequencing that were concerning for underlying cancer predisposition were constitutionally confirmed. Overall, somatic tumor testing identified potential cancer predisposition syndromes in pediatric patients, and some would not have been identified on the basis of clinical history alone.
INTRODUCTION
The incidence of constitutional genetic aberrations leading to cancer predisposition in pediatric patients with cancer is estimated at 8% to 12%.1-4 Results from large somatic sequencing studies confirm that this rate of constitutional aberrations can be inferred from somatic sequencing results.5,6 The timely diagnosis of a cancer predisposition syndrome can have significant implications not only for the patient, but also for affected and unaffected family members. Follow-up of any features of a cancer predisposition syndrome is essential for appropriate management, surveillance, and family planning for these individuals.
Clinical and historical features that suggest a pediatric patient with cancer may have a predisposition syndrome include a family history of the same cancer or related cancers, clinical features of a predisposition syndrome, bilateral/multifocal primary cancer or multiple cancers, certain types of cancer that rarely occur except in a predisposed patient, and a much earlier than expected age at diagnosis. Indeed, certain tumors should lead to referral for constitutional testing in all cases,7 and referral criteria that are based on family history of cancer in first- and second-degree relatives have been established.1,8,9 Specific recommendations to test for a constitutional mutation in certain cancer types and/or predisposition syndromes, such as retinoblastoma, neuroblastoma, and Li-Fraumeni syndrome (LFS), also exist.10-12 Studies have shown, however, that clinical features and family history alone are not reliable predictors of underlying constitutional mutations leading to cancer predisposition syndromes.2,3 Furthermore, suspicion of cancer predisposition on the basis of genomic findings within the tumor has been shown to be a more powerful tool in uncovering potential constitutional change than tumor type and clinical history, and pediatric patients with cancer may have constitutional mutations in cancer predisposition genes in the absence of known risk factors.3
Somatic genetic testing of tumor tissue is used increasingly in the evaluation of children and adolescents with cancer for diagnosis, risk stratification, and treatment decisions.13 Results of tumor sequencing can suggest the presence of an underlying cancer predisposition syndrome through a variety of indicators, including a variant allele fraction (VAF) of 0.4 to 1.0 or identification of pathogenic or founder mutations in known cancer predisposition genes. As tumor sequencing becomes more common and comprehensive, it is important for both patients and family members that appropriate measures are in place for genetic counseling and subsequent constitutional testing. In this report, we describe our experience in conducting cancer predisposition evaluation on the basis of somatic sequencing findings in cancer samples identified using in-house next-generation sequencing testing panels. Appropriate referral is vital not only for conducting constitutional genetic testing, but also for identifying other at-risk family members.
Context
Key Objective
When a child is diagnosed with cancer, most families are uncertain why their child developed a rare and devastating disease. For an important minority, an underlying cancer predisposition syndrome as a result of a constitutional pathogenic variant may underlie malignancy; however, these can be difficult to identify in patients who lack syndromic features and/or a family history of cancer.
Knowledge Generated
This study demonstrates that pathogenic variants identified through somatic next-generation sequencing can indicate the presence of a constitutional cancer-predisposing variant. In a cohort of more than 1,000 pediatric patients with cancer who underwent somatic panel sequencing, nearly 14% had findings requiring referral for constitutional testing. Of those who underwent testing, more than one half were diagnosed with a cancer predisposition syndrome, some of whom would not have been identified on the basis of clinical history alone.
Relevance
In conducting somatic tumor sequencing, it is necessary to carefully evaluate for indicators of constitutional cancer predisposition syndromes.
MATERIALS AND METHODS
Data were abstracted from the medical records of all patients who underwent in-house somatic tumor genetic testing using multigene panels over a 28-month period. Potential constitutional likely pathogenic and pathogenic variants were identified during clinical analysis of somatic panels offered through the Children’s Hospital of Philadelphia Genomic Diagnostics Laboratory. These targeted panels provided comprehensive analysis of hematologic and solid tumors for fusion genes, single-nucleotide variants, insertion/deletion variants, and copy number variants for 117 to 238 genes13,14 (Appendix Table A1). These panels are performed on the majority of diagnostic samples obtained from Children’s Hospital of Philadelphia pediatric patients with cancer at initial diagnosis or relapse. Of note, retinoblastoma tumors are not routinely tested via this mechanism.
Variants were identified for potential follow-up constitutional testing if they met any of the following criteria: pathogenic or likely pathogenic variants in genes known to be associated with cancer predisposition15 with VAF between 0.4 and 1.0; large indels or exonic deletions/duplications in genes known to be associated with cancer predisposition regardless of VAF; and/or suspected constitutional pathogenic variants regardless of VAF if the variant was a known founder mutation, or if clinical features, such as tumor type, suggested constitutional predisposition related to the involved variant. These reasons for referral were divided into the following categories: VAF, Clinical Concern/Tumor Type, and Known Founder. The panels also provided copy number variation and loss of heterozygosity information that aided in the interpretation of the VAF for a given variant. Once identified in tumor tissue, the potential for suspicious variants to be of constitutional origin was described in the somatic tumor report issued to the ordering physician, usually an oncologist.
These suspected constitutional variants were tagged by the Genomic Diagnostics Laboratory for referral to the Cancer Predisposition Program (CPP). The CPP and ordering physician then discussed the need for cancer predisposition consultation, and pretest genetic counseling was arranged via the CPP as appropriate. If patients chose to pursue constitutional testing, targeted Sanger sequencing analysis or other appropriate genomic testing was performed using a constitutional—nontumor—sample, either blood or skin fibroblasts. Results were reported back to the CPP, which then contacted the patient or parents, and post-test genetic counseling was offered. Constitutional results were also reported to and reviewed with the oncologist. Somatic panel testing was not used as a substitute for clinical judgment, and a patient with concerning features of cancer predisposition would always be referred to the CPP regardless of somatic panel results. Clinical features and family history were abstracted from charts when available, but a complete family history review was not conducted by an oncologist, geneticist, or genetic counselor for patients not seen by the CPP. Factors included in the analysis of patients seen by the CPP included family history of cancer, presence of multifocal or bilateral disease, loss of heterozygosity in the tumor, and clinical features of diagnosis, such as clinical diagnostic criteria for neurofibromatosis type 1 (NF1) or tuberous sclerosis, in addition to the aforementioned referral criteria. Tumor type association with the presence of a constitutional variant—for example, rhabdoid tumor and SMARCB1 pathogenic variant—was also tracked.7 Data also included age at diagnosis and turnaround time of constitutional testing (days between the result report of somatic testing and the date of constitutional testing). As detailed in Results, some variants had been previously constitutionally confirmed, and some were confirmed on the basis of clinical features alone and did not have testing completed. Neither were included in the statistical analysis. Constitutional testing was listed as incomplete in all cases in which the CPP did not meet with the family to discuss the variant, even if testing may not ultimately have been suggested. Finally, results were deemed conclusive when the genetic testing was interpretable and inconclusive when additional sample would be required to make a definitive diagnosis.
Data were analyzed using STATA and R statistical programming languages, and statistical significance was determined using Wilcoxon rank-sum test and χ2 test, when appropriate. Visualizations were generated using the ggplot2 package.16
The human investigations performed in this study were completed after approval by the Children’s Hospital of Philadelphia Institutional Review Board and in accordance with the requirements of the Department of Health and Human Services, where appropriate.
RESULTS
Population Demographics
Records were reviewed for 1,023 patients who underwent somatic panel testing between February 2016 and June 2018. A total of 210 somatic variants in 141 patients were suspected to be constitutional pathogenic or likely pathogenic variants, which is 13.8% of patients who underwent somatic panel testing during this time period. Of 141 patients, 51 had CNS tumors, 63 had non-CNS solid tumors, and 27 had leukemia or lymphoma. Demographic features of these patients are listed in Table 1. VAF met criteria for referral (≥ 0.4) in 78% of cases (n = 164), including those with copy number variant in a potential cancer predisposition gene. The remainder of variants were referred for the presence of a founder mutation (2.9%; n = 6) or a tumor type–specific variant concerning for cancer predisposition (55.2%; n = 116), and some patients were referred for multiple reasons. VAF in all cases ranged from 0.05 to 0.98 (Table 2).
TABLE 1.
Demographics
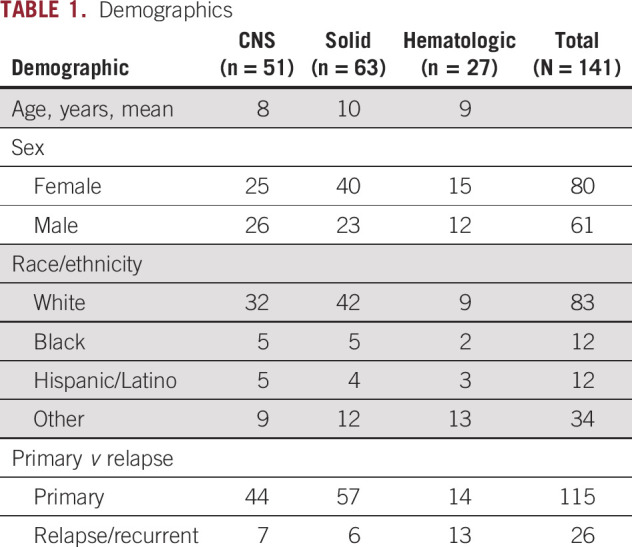
TABLE 2.
Variant Allele Frequency Distribution

Somatic Findings in Predisposition Genes
A total of 210 variants were identified in a total of 66 genes. A heatmap of genes with a suspected constitutional variant identified in 2 or more individuals is shown in Figure 1 (full variant list, including tumor type, in Appendix Table A2). As expected, potential constitutional variants were most frequently identified in the TP53 gene, occurring in 35% of individuals referred (n = 46). The second most frequent gene with potential constitutional variants was NF1 (17%; n = 27, including 6 individuals with an identified TP53 variant), followed by SMARCB1, MUTYH, APC, ATM, MSH6, WT1, and DICER1. Founder mutations were identified in 6 patients—for example, 4 of the 7 variants identified in APC were I1307K, a known founder risk allele in the Ashkenazi Jewish population that does not lead to familial adenomatous polyposis, but confers increased cancer risk in adulthood.17 Some variants identified had implications for immediate treatment decisions, such as TP53 variants that were identified in patients with anticipated radiation therapy, and an attempt was made to prioritize these patients for testing and counseling. Other individuals required the initiation of childhood screening for other tumors—for example, DICER1, WT1, 11p loss/aberrations, and non-I1307K APC variants—and in these cases screening was initiated during ongoing cancer treatment after a constitutional diagnosis was made.
FIG 1.
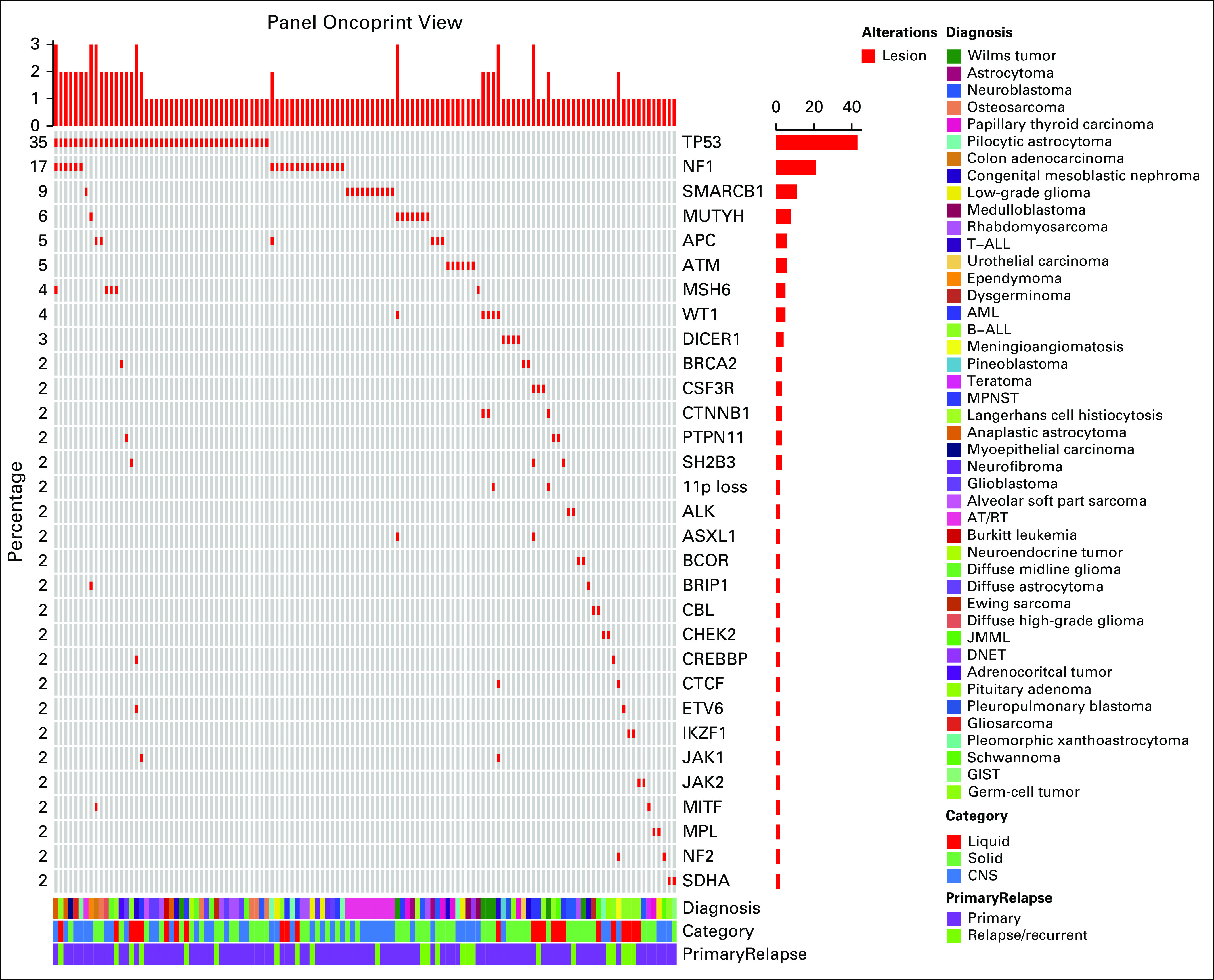
Somatic findings. Frequency of concerning somatic variants identified, as described by frequency, tumor type (liquid, non-CNS solid, and CNS), primary versus relapsed/refractory specimen, and specific cancer diagnosis. Not included are variants occurring in fewer than 2 individuals (Appendix Table A1). AML, acute myeloblastic leukemia; AT/RT, atypical teratoid/rhabdoid tumor; B-ALL, B-acute lymphoblastic leukemia; DNET, dysembryoplastic neuroepithelial tumor; GIST, GI stromal tumor; JMML, juvenile myelomonocytic leukemia; MPNST, malignant peripheral nerve sheath tumor; T-ALL, T-acute lymphoblastic leukemia.
Of note, several heterozygous variants identified had implications for adult cancer risk, but did not change management in the pediatric age range (MSH6, MUTYH, ATM, BRCA1, and BRCA2). These were still referred for genetic counseling, as they could have implications for family members and for the individual patient later in life. Testing decisions on these variants were made on a case-by-case basis, in discussion with the family and the oncologist, and in most cases, it was recommended that constitutional testing of the proband be deferred until age 18 years or older. Occasionally in adolescent patients, the patient preferred to complete testing.
Constitutional Testing
A total of 210 variants were determined to warrant follow-up for potential constitutional alterations on the basis of initial somatic panel review (Fig 2; Appendix Table A2). At the time of this study, no individuals were found to have a constitutional variant that was not indicated on panel testing.
FIG 2.

Schematic representation of patients referred for constitutional testing. Includes patients who did not have testing performed for clinical reasons, patients who still require referral and genetic testing, and results of testing for the patients for whom testing was conducted. Patients were presumed positive if they met clinical criteria for a diagnosis without the need for genetic testing, and were presumed negative when they did not meet clinical criteria for a diagnosis. In all cases, the individual was evaluated clinically.
Previously tested.
A total of 10 patients with 11 variants had undergone testing previously on the basis of clinical suggestion of a cancer predisposition syndrome before somatic sequencing. Of the variants previously tested, eight were constitutionally present and three were somatic only. These were not included in the analysis of characteristics of tested patients done below, as these patients had already been seen and/or were being observed by the CPP and thus were identified before somatic testing.
Constitutional testing completed.
Of 38 patients with constitutional testing that led to conclusive results, at least one potentially predisposing variant identified in tumor tissue was confirmed to be constitutional in 23 patients (60.5%) and in 26 total variants of 73 variants tested (35.6%; Fig 2). TP53 was the most frequently identified mutated gene by the laboratory referral pipeline. Of 21 cases in which constitutional TP53 testing was performed, the variant was constitutionally identified in 6 cases (28.6%). Of these individuals, three would not have otherwise been referred for LFS testing. Of the total number of patients with a somatic TP53 variant (n = 46), 19.6% were constitutionally affected (n = 9; includes those patients who previously tested positive). A full list of variants tested is included in Appendix Table A2.
A total of 3 variants in 3 individuals had inconclusive testing. In these cases, the sample was either contaminated with leukemia cells or the constitutional material obtained was insufficient. In all three cases, the patient died before the completion of additional testing.
In some cases, the finding was presumed positive without genetic testing. In 13 variants in 11 individuals, the finding was presumed positive. This was based on known familial mutations in APC I1307L (n = 2), BRCA1 (n = 1), and BRCA2 (n = 1), or on the basis of an existing clinical diagnosis in seven NF1 patients already being observed by the neurofibromatosis clinic. In six variants in five individuals, testing was presumed negative, as individuals did not meet criteria for a syndrome that was not present clinically. This included NF1 (n = 4), tuberous sclerosis (n = 1), and Kabuki syndrome (n = 1). These patients were clinically evaluated before this determination and the variant was discussed with the family. In these cases, variants were not included in the tested category in subsequent analyses; however, it is important to note that constitutional testing is not always required to confirm or rule out a diagnosis. In all cases, somatic mosaicism was considered and discussed with family when appropriate.
Testing not completed.
In some cases, constitutional testing was not performed after discussion with the family, per family preference and/or clinician recommendation. Constitutional testing was deferred in a total of 4 variants associated with adult-onset cancer risk until adulthood after discussion with family and review of family history (APC I1307L [n = 2], BLM [n = 1], MUTYH [n = 1]). This would likely be the recommendation for other somatic variants in adult-onset predisposition syndromes included in the genetic testing not done category; however, genetic counseling and family history review need to take place before constitutional testing is deferred completely. A total of 8 families—8 patients and 12 variants—preferred not to pursue constitutional testing, in one case because of a lack of insurance approval for testing.
Referral Still Required
Finally, a large number of patients—66 patients and 91 variants—still require referral to the CPP before constitutional testing can be completed. Our program is actively working to ensure that all families with a concerning somatic variant receive appropriate genetic counseling.
Characteristics of Tested Patients
Historically, the suspicion for constitutional cancer predisposition is based on clinical characteristics, including family history, physical features of a predisposition syndrome, presence of bilateral/multifocal disease, and/or a specific tumor type strongly associated with a predisposition syndrome. Characteristics of patients who were either confirmed or not confirmed constitutionally are included in Table 3. Using a cutoff of α = .05, patients who were identified to have an underlying tumor predisposition were younger (mean age, 6.15 years v 10.77 years; P = .0062). Testing turnaround time was not significantly faster in patients with an underlying predisposition (83 days v 108 days; P = .488). Patients with a concerning family history or bilateral or multifocal disease were more likely to have the variant confirmed constitutionally (P = .048 and .017, respectively; bilateral disease was present in three individuals with atypical teratoid/rhabdoid tumor and a SMARCB1 deletion and in one patient with a BARD1 mutation and a composite pheochromocytoma and neuroblastoma). Tumor type and variant association, such as rhabdoid tumor and SMARCB1 variant, was not predictive, with a near even frequency of being constitutional or not (P = .964). There was no significant difference between those confirmed and not confirmed in VAF ≥ 0.4 or in tumor loss of heterozygosity.
TABLE 3.
Characteristics of All Patients Who Underwent Clinical Testing

DISCUSSION
Somatic Findings Concerning for Cancer Predisposition
Of the more than 1,000 children, adolescents, and young adults who underwent somatic tumor testing in the 28 months included within this study period, 13.9% had somatic results that were concerning for a constitutional cancer predisposition. Of note, this does not include tumor types that routinely do not undergo somatic testing at our institution—retinoblastoma or Hodgkin lymphoma—or for which tissue was not available and thus is not a comprehensive evaluation of all patients with cancer at our institution. Referrals were made in most cases for variants with VAF > 0.4, although in 22.2% of variants VAF was lower than this threshold, and clinical suspicion based on the specific variant prompted CPP referral.
As expected, given the frequency of both constitutional and somatic mutations,2,18 TP53 was the most frequently mutated gene that prompted referral to the CPP. Of the 21 patients with a TP53 mutation in tumor tissue who were referred for testing, 6 (28.6%) were diagnosed with LFS on the basis of a constitutional finding of a known TP53 pathogenic/likely pathogenic variant, three of whom would not have been referred based on accepted LFS testing guidelines.15,19 Even including all somatic variants, at least 19.6% of patients with somatic LFS variants are constitutionally affected, a proportion higher than that observed in the adult population. All of these patients are now being observed by the CPP and are receiving recommended surveillance.20 The next most frequent mutation found was in NF1, but decisions about constitutional testing were sometimes made on the basis of the presence or absence of characteristic clinical features of NF1. These patients were all referred to our multidisciplinary neurofibromatosis clinic and observed there if clinical features of NF1 were present, and constitutional testing was not conducted routinely. For other variants, patients were tested and recommendations were made in accordance with accepted practices, when available (Appendix Table A2). Of note, for some variants clinical guidelines are not yet established, and in these cases families received genetic counseling regarding the lack of accepted tumor surveillance practices and/or additional study needed regarding the constitutional presence of a variant. In cases in which constitutional testing would not affect clinical management—for example, no additional childhood surveillance would be required—families were informed of the result and adult relatives were counseled that they could consider testing for themselves, but testing was usually not sent for the child. Finally, a discussion of somatic mosaicism occurred in the setting of inconclusive or negative results, or in cases in which families decided not to pursue genetic testing because of lack of clinical features of a disorder.
Additional study is required to analyze the power of somatic panel testing to identify pediatric patients with constitutional cancer predisposition, with and without the incorporation of clinical data, such as multifocality, tumor type, syndromic features, and family history. In addition, many patients have not yet been referred to the CPP to discuss constitutional testing, and qualitative review would suggest a variety of reasons for this, including emotional stress on the family at the time of cancer diagnosis and the additional stress of predisposition evaluation during treatment, although this has not been systematically studied at our institution. We plan to continue offering genetic testing and counseling to patients not yet referred to ensure an eventual 100% referral rate.
Interpretation of Somatic Results Concerning for Cancer Predisposition
The most significant differences in those constitutionally confirmed were known clinical risk factors, namely, a significant/concerning family history of cancer and bilateral or multifocal disease. In fact, all bilateral/multifocal tumors were associated with underlying cancer predisposition. Of interest, tumor type, VAF, and loss of heterozygosity (LOH) were not significant indicators of the constitutional presence of a variant. This suggests that VAF and LOH alone are not sufficient to rule out a cancer predisposition syndrome. Although it may seem surprising that tumor type was not predictive, this is consistent with the known prevalence of a cancer predisposition syndrome in these tumor types. Thus, clinical predictors of cancer predisposition remain important in the interpretation of somatic results.
More than one half of patients who received constitutional testing were found to have an underlying cancer predisposition syndrome, often requiring changes in treatment plan, ongoing surveillance, and familial testing. Some of these patients could have been referred for testing on the basis of patient age, type of cancer, and/or family history of cancer; however, others would not have been suspected as a result of a lack of these features. Thus, the incorporation of somatic testing results proves to be an important supplement to clinical judgment in identifying cancer predisposition syndromes.
In conclusion, somatic tumor sequencing is a powerful tool in pediatric cancer to risk-stratify patients and identify appropriate therapy, and its use is becoming increasingly widespread. This testing can also lead to the identification of variants that raise concern for an underlying constitutional cancer predisposition. Identification of these variants is vital for the treatment and ongoing tumor surveillance for both patients and family members. Collaboration between the genomic diagnostics laboratory, pathology, and oncology can assist in identifying and coordinating appropriate testing for cancer predisposition.
Appendix
TABLE A1.
Somatic Cancer Panels

TABLE A2.
All Identified Variants

Footnotes
Presented at the American College of Medical Genetics Annual Meeting, April 6, 2019, Seattle, WA.
Supported by National Institutes of Health Grant No. K12CA076931 (S.P.M.), the Precious Jules Foundation (S.P.M.), the Audrey E. Evans Endowed Chair in Molecular Oncology (G.M.B.), the Children's Hospital of Philadelphia Chair's Initiative (G.M.B.), and the Department of Pathology and Laboratory Medicine (L.F.S., D.G., and M.M.L.).
AUTHOR CONTRIBUTIONS
Conception and design: Suzanne P. MacFarland, Kristin Zelley, Stephen P. Hunger, Marilyn M. Li, Garrett M. Brodeur
Administrative support: Suzanne P. MacFarland, Garrett M. Brodeur
Collection and assembly of data: Suzanne P. MacFarland, Kristin Zelley, Lea F. Surrey, Daniel Gallo, Gerald Wertheim
Data analysis and interpretation: Suzanne P. MacFarland, Kristin Zelley, Minjie Luo, Pichai Raman, Stephen P. Hunger, Garrett M. Brodeur
Manuscript writing: All authors
Final approval of manuscript: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Pichai Raman
Consulting or Advisory Role: Scholar Rock
Travel, Accommodations, Expenses: Scholar Rock
Gerald Wertheim
Employment: Johnson & Johnson (I)
Stock and Other Ownership Interests: Johnson & Johnson (I)
Stephen P. Hunger
Stock and Other Ownership Interests: Amgen, Merck (I), Amgen (I), Pfizer (I)
Honoraria: Amgen
Consulting or Advisory Role: Novartis
No other potential conflicts of interest were reported.
REFERENCES
- 1.Brodeur GM, Nichols KE, Plon SE, et al. Pediatric cancer predisposition and surveillance: An overview, and a tribute to Alfred G. Knudson Jr. Clin Cancer Res. 2017;23:e1–e5. doi: 10.1158/1078-0432.CCR-17-0702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang J, Walsh MF, Wu G, et al. Germline mutations in predisposition genes in pediatric cancer. N Engl J Med. 2015;373:2336–2346. doi: 10.1056/NEJMoa1508054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Parsons DW, Roy A, Yang Y, et al. Diagnostic yield of clinical tumor and germline whole-exome sequencing for children with solid tumors. JAMA Oncol. 2016;2:616–624. doi: 10.1001/jamaoncol.2015.5699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mody RJ, Wu YM, Lonigro RJ, et al. Integrative clinical sequencing in the management of refractory or relapsed cancer in youth. JAMA. 2015;314:913–925. doi: 10.1001/jama.2015.10080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gröbner SN, Worst BC, Weischenfeldt J, et al. The landscape of genomic alterations across childhood cancers. Nature. 2018;555:321–327. doi: 10.1038/nature25480. Erratum: Nature 559:E10, 2018. [DOI] [PubMed] [Google Scholar]
- 6.Ma X, Liu Y, Liu Y, et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature. 2018;555:371–376. doi: 10.1038/nature25795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Postema FAM, Hopman SMJ, Aalfs CM, et al. Childhood tumours with a high probability of being part of a tumour predisposition syndrome; reason for referral for genetic consultation. Eur J Cancer. 2017;80:48–54. doi: 10.1016/j.ejca.2017.04.021. [DOI] [PubMed] [Google Scholar]
- 8.Malkin D, Li FP, Strong LC, et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science. 1990;250:1233–1238. doi: 10.1126/science.1978757. [DOI] [PubMed] [Google Scholar]
- 9.Jongmans MC, Loeffen JL, Waanders E, et al. Recognition of genetic predisposition in pediatric cancer patients: An easy-to-use selection tool. Eur J Med Genet. 2016;59:116–125. doi: 10.1016/j.ejmg.2016.01.008. [DOI] [PubMed] [Google Scholar]
- 10.Goudie C, Cullinan N, Villani A, et al. Retrospective evaluation of a decision-support algorithm (MIPOGG) for genetic referrals for children with neuroblastic tumors. Pediatr Blood Cancer. 2018;65:e27390. doi: 10.1002/pbc.27390. [DOI] [PubMed] [Google Scholar]
- 11.Kamihara J, Bourdeaut F, Foulkes WD, et al. Retinoblastoma and neuroblastoma predisposition and surveillance. Clin Cancer Res. 2017;23:e98–e106. doi: 10.1158/1078-0432.CCR-17-0652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kratz CP, Achatz MI, Brugières L, et al. Cancer screening recommendations for individuals with Li-Fraumeni syndrome. Clin Cancer Res. 2017;23:e38–e45. doi: 10.1158/1078-0432.CCR-17-0408. [DOI] [PubMed] [Google Scholar]
- 13.Surrey LF, MacFarland SP, Chang F, et al. Clinical utility of custom-designed NGS panel testing in pediatric tumors. Genome Med. 2019;11:32. doi: 10.1186/s13073-019-0644-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jain P, Surrey LF, Straka J, et al. Novel FGFR2-INA fusion identified in two low-grade mixed neuronal-glial tumors drives oncogenesis via MAPK and PI3K/mTOR pathway activation. Acta Neuropathol. 2018;136:167–169. doi: 10.1007/s00401-018-1864-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wickham H. ggplot2. Comput Stat. 2011;3:180–185. [Google Scholar]
- 17.Boursi B, Sella T, Liberman E, et al. The APC p.I1307K polymorphism is a significant risk factor for CRC in average risk Ashkenazi Jews. Eur J Cancer. 2013;49:3680–3685. doi: 10.1016/j.ejca.2013.06.040. [DOI] [PubMed] [Google Scholar]
- 18.Bouaoun L, Sonkin D, Ardin M, et al. TP53 variations in human cancers: New lessons from the IARC TP53 database and genomics data. Hum Mutat. 2016;37:865–876. doi: 10.1002/humu.23035. [DOI] [PubMed] [Google Scholar]
- 19.Leroy B, Ballinger ML, Baran-Marszak F, et al. Recommended guidelines for validation, quality control, and reporting of TP53 variants in clinical practice. Cancer Res. 2017;77:1250–1260. doi: 10.1158/0008-5472.CAN-16-2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Achatz MI, Porter CC, Brugières L, et al. Cancer screening recommendations and clinical management of inherited gastrointestinal cancer syndromes in childhood. Clin Cancer Res. 2017;23:e107–e114. doi: 10.1158/1078-0432.CCR-17-0790. [DOI] [PubMed] [Google Scholar]