

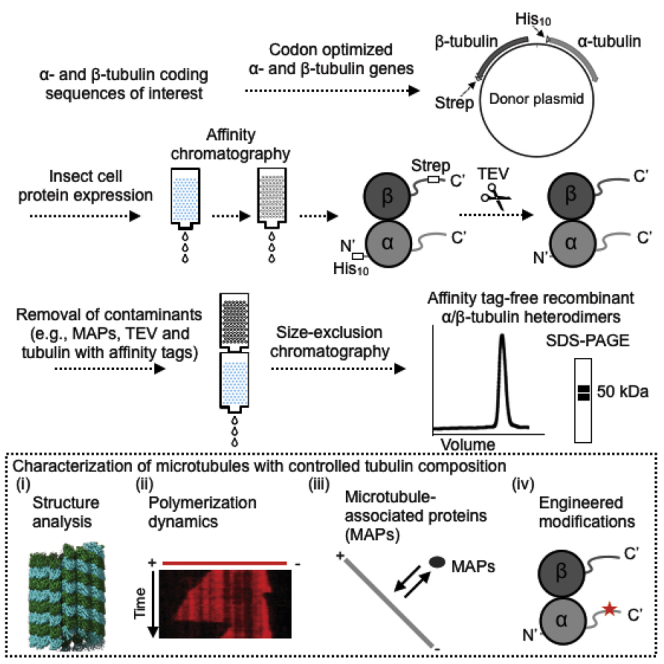
Summary
α/β-tubulin heterodimers, which can harbor diverse isotypes and post-translational modifications, polymerize into microtubules that are fundamental to many cellular processes. Due to long-standing challenges in generating recombinant tubulin, however, it has been difficult to examine the properties of specific tubulin isotypes. Here, we provide a protocol for purifying milligrams of affinity tag-free, isotypically pure recombinant tubulin. Our method can be applicable to any tubulin of interest, opening the door to dissecting how tubulin diversity regulates microtubule function.
For complete details on the use and execution of this protocol, please see Ti et al. (2018).
Graphical Abstract
Highlights
-
•
Strategy to generate isotypically pure affinity tag-free recombinant tubulin
-
•
Strategy is generally applicable to any desired tubulin sequence
-
•
Purified tubulin has no detectable post-translational modifications
-
•
Recombinant tubulin is of sufficient yield for biochemical and biophysical assays
α/β-tubulin heterodimers, which can harbor diverse isotypes and post-translational modifications, polymerize into microtubules that are fundamental to many cellular processes. Due to long-standing challenges in generating recombinant tubulin, however, it has been difficult to examine the properties of specific tubulin isotypes. Here, we provide a protocol for purifying milligrams of affinity tag-free, isotypically pure recombinant tubulin. Our method can be applicable to any tubulin of interest, opening the door to dissecting how tubulin diversity regulates microtubule function.
Before You Begin
Prepare a Baculovirus Donor Plasmid that Encodes α- and β-Tubulin Genes
As a proof-of-concept, we provide a detailed protocol that we used to purify milligram quantities of affinity tag-free human α1B/β2B- and α1B/β3-tubulin, which are the major tubulin isotypes expressed in neurons (Ludueña and Banerjee, 2008).
Note: The cDNA codons for the α-tubulin and β-tubulin genes of choice should initially be optimized for expression in High Five (Trichoplusia ni) cells.
-
1.
For the purification of human α1B/β3- and α1B/β2B-tubulin, use the molecular biology method of choice to assemble a codon-optimized α1B-tubulin gene fragment and one human β2B- or β3-tubulin codon-optimized gene fragment into a pFastBac dual vector such that a polyhedrin promoter and a p10 promoter drive the expression of α- and β-tubulin, respectively.
-
2.
Include an L21 enhancer (AACTCCTAAAAAACCGCCACC) (Sano et al., 2002) at the 5′ end of the start codon (ATG) for each sequence.
-
3.
We also include a nucleotide sequence (from 5′ to 3′) encoding a tobacco etch virus (TEV) protease site, a Gly-Gly-Ser-Gly-Gly linker and a Strep-tag II (Schmidt and Skerra, 2007) at the 3′ end of the β-tubulin cDNA sequence, and a sequence (from 5′ to 3′) encoding a decahistidine tag, a TEV protease site, and an Ala-Pro linker at the 5′ end of the α-tubulin cDNA sequence. Please see Figure 1 for the schematic showing the organization of the α-tubulin and β-tubulin transcription units that are cloned into a pFastBac Dual vector.
Figure 1.

Schematic of Constructs Encoding α- and β-tubulin transcription units
(A and B) The design of the construct of α-tubulin (A) and β-tubulin (B) genes in a pFastBac Dual donor plasmid.
CRITICAL: We found that an expression construct with N terminus tagged α1B-tubulin and C terminus tagged β2B- or β3-tubulin is essential to achieve optimal yield. Compared to the removal of the β-tubulin C-terminal affinity tag, the TEV digestion that removes the N-terminal decahistidine tag of α-tubulin is less efficient. A Ser-Gly linker between the TEV cleavage site and the α1B-tubulin N terminus increases the efficiency of the TEV digestion, but the tubulin heterodimers precipitate after tag cleavage. We found that an Ala-Pro linker is critical for the removal of the decahistidine tag of α1B-tubulin without reducing the solubility of tubulin heterodimers.
Alternatives: Because our affinity tag-based strategy does not depend on affinity for tubulin-binding domains (e.g. TOG domain (Widlund et al., 2012)) or cycles of microtubule polymerization and depolymerization (Gell et al., 2011), properties which depend on tubulin primary sequence (Pamula et al., 2016, Ti et al., 2016), we expect that researchers can adopt this strategy to any tubulin species, isotype, or post-translational modification of interest.
Alternatives: For generating other unique/modified tubulins, use the protein tagging strategy described above as a starting point. Alternatives include the choice and position (N- vs. C terminus) of affinity tags, as well as the length and composition of the linker, each of which may need to be modified for optimal protein expression and yield.
Purify and Reconstitute Tobacco Etch Virus (TEV) Protease in a Tubulin-Friendly Buffer
TIMING: ∼30 h from cell lysis to frozen aliquots of TEV stored at −80°C.
Express TEV in BL21(DE3) Rosetta Cells
-
4.
Transform BL21(DE3) Rosetta cells (Novagen) with plasmid pRK793 (Addgene #8827 (Kapust et al., 2001)) and grow single colonies on LB plates that contain 100 μg/mL ampicillin and 34 μg/mL chloramphenicol.
-
5.
Inoculate a single colony into 10 mL of LB medium with antibiotics. Grow the cell culture at 37°C and 220 rpm orbital rotation for 16-18 h.
-
6.
Dilute 10 mL of overnight culture into 1 L of LB and grow the culture at 37°C and 220 rpm orbital rotation until the OD600 is between 0.6 and 0.8.
-
7.
After cooling the culture down in a 4°C cold room for 1 h, add IPTG to 1 mM for the induction of protein expression at 19°C and 220 rpm orbital rotation for 16-18 h.
Cell Lysis and Clarification
-
8.
Harvest the cells by centrifugation at 4,000 rcf for 10 min at 4°C.
-
9.
Resuspend the cell pellet from every 1 L of culture with 25 mL of lysis buffer (50 mM sodium phosphate, 10% (w/v) glycerol, 10 mM imidazole, 100 mM NaCl, and 1 mM 2-mercaptoethanol, pH 8.0) supplemented with 3 μL of 25 U/μL Benzonase (EMD Millipore), 1 mM PMSF (freshly prepared in isopropanol), 0.2 mg/mL lysozyme and 1 tablet of complete EDTA-free protease inhibitor (Roche).
-
10.
Lyse the cells by six rounds of 1 min sonication (1 s per burst) at 30% power output with 1 min interval between each round (Fisherbrand Model 500 Sonic dismembrator with a probe that has a diameter of 2 mm).
-
11.
Centrifuge the cell lysate at 40,000 rpm (∼130,000 rcf) in a Type 45 Ti rotor (Beckman) for 45 min.
Nickel Affinity Chromatography
-
12.
For cell lysate from every 1 L of culture, equilibrate 3 mL of Ni-NTA resin (QIAGEN) with lysis buffer.
-
13.
Gently stir the clarified cell lysate with the equilibrated Ni-NTA resin for 30 min.
-
14.
Collect the TEV-bound Ni-NTA resin in a reusable gravity chromatography column.
-
15.
For purification from 6 L of cell culture (∼18 mL of resin bed), flow 100 mL of lysis buffer through the column, followed by 300 mL of wash buffer (50 mM sodium phosphate, 10% (w/v) glycerol, 10 mM imidazole, 300 mM NaCl, and 0.5 mM 2-mercaptoethanol, pH 8.0).
-
16.
Elute TEV with elution buffer (50 mM sodium phosphate, 10% (w/v) glycerol, 200 mM imidazole, 100 mM NaCl, and 2 mM 2-mercaptoethanol, pH 8.0).
-
17.
Detect the fractions that contain protein by Bradford assay and pool together (∼60 mL for a purification from 6 L of cell culture).
Size Exclusion Chromatography
-
18.
Use a 10 kDa MWCO centrifugal filter unit to reduce the volume of pooled Ni-NTA eluate to ∼10 mL.
-
19.
Load the concentrated protein solution onto a Superdex 75 HiLoad 16/60 column (GE Healthcare) equilibrated in gel-filtration buffer (50 mM Phosphate, 10% (w/v) glycerol, 300 mM KCl, and 10 mM 2-mercaptoethanol, pH 8.0) at 1 mL/min.
-
20.
Keep flowing the gel filtration buffer through the column at 1 mL/min, and collect the flow-through in 2 mL fractions. TEV elutes at ∼70 mL on this column.
Characterization and Storage
-
21.
Analyze the purity of the TEV elution fractions by SDS-PAGE.
-
22.
Pool the fractions that contain at least 95% pure TEV (MW = 27 kDa) and dialyze against 2 L of storage buffer (40 mM HEPES, 30% (w/v) glycerol, 150 mM KCl, 1 mM MgCl2, and 3 mM 2-mercaptoethanol, pH 7.5) for ∼16 h.
-
23.
Measure the protein concentration by Bradford assay using bovine serum albumin (BSA) as a standard.
-
24.
Snap freeze small (∼100-200 μL) aliquots of TEV in liquid nitrogen and store the protein at −80°C. Ideally, for a purification from 6 L of cell culture, the final protein concentration should be at least 7 mg/mL with a total yield of ∼100 mg. In our lab, a 2.5% molar ratio of TEV purified in this manner is sufficiently active to cleave > 95% of its substrate within ∼16 h at 4°C.
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-α-tubulin (clone DM1A) | Sigma-Aldrich | T9026; RRID: AB_477593 |
| Mouse monoclonal anti-β-tubulin (clone G-8) | Santa Cruz | sc-55529; RRID: AB_2210962 |
| Bacterial and Insect Cell Strains | ||
| Rosetta (DE3) pLysS competent cells | Millipore | 70956 |
| High Five cells | Thermo Fisher | B85502 |
| Sf9 cells | Thermo Fisher | 11496015 |
| Critical Commercial Columns and Consumables | ||
| HisTrap HP | GE life science | 17-5247-01 |
| Ni-NTA agarose | QIAGEN | 30230 |
| StrepTrap HP | GE life science | 29-0486-53 |
| HiTrap SP Sepharose FF | GE life science | 17-5054-01 |
| Superdex 200 16/600 | GE life science | 17-1069-01 |
| Superdex 75 16/600 | GE life science | 28-9893-33 |
| 0.22 μm Millex-GP PES membrane | Millipore | SLGP033RS |
| 0.45 μm Acrodisc Supor Membrane | Pall | 4614 |
| Amicon Ultra 50K MWCO centrifugal filter unit | Millipore | UFC901024 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Sf-900 II SFM | Thermo Fisher | 10902096 |
| Express Five SFM | Thermo Fisher | 10486025 |
| Fetal Bovine Serum (heat inactivated) | Sigma Aldrich | F7678 |
| CellFectin II | Thermo Fisher | 10362100 |
| antibiotic-antimyocotic | Thermo Fisher | 15240-062 |
| 200 mM L-glutamine | Thermo Fisher | 25030-081 |
| HEPES | Sigma-Aldrich | H4034 |
| Imidazole | Sigma-Aldrich | I202 |
| Potassium Chloride | Fisher | P217-3 |
| Lithium Chloride | Sigma-Aldrich | 62476 |
| Magnesium Chloride | Fisher | BP214 |
| β-mercaptoethanol | Sigma-Aldrich | M6250 |
| Tris(2-carboxyethyl)phosphine hydrochloride | Thermo Fisher | PI77720 |
| 10X Strep-Tactin Regeneration buffer with 10 mM 2-[4 -hydroxy-benzeneazo]benzoic acid (HABA) | Iba | 2-1002-100 |
| EGTA | Sigma-Aldrich | E3889 |
| GTP | Sigma-Aldrich | G8877 |
| ATP | Sigma-Aldrich | A2383 |
| GMPCPP | Jena Bioscience | NU-405S |
| Benzonase | Millipore | 70746-3 |
| Complete EDTA-free protease inhibitor | Roche | 11873580001 |
| PIPES | Sigma-Aldrich | P1851 |
| Paclitaxel (Taxol) | Sigma-Aldrich | T7191-5mg |
| TEV protease | Kapust et al., 2001 | Addgene #8827 |
| Bovine serum albumin (BSA) | bioWORLD | 22070008-3 |
| Phenol:Chloroform:Isoamyl Alcohol 25:24:1 Saturated with 10 mM Tris, pH8.0, 1 mM EDTA | Sigma-Aldrich | P2069 |
| Phenylmethylsulfonyl fluoride (PMSF) | Goldbio | P-470 |
| Ampicillin | Goldbio | A-301 |
| Kanamycin | Goldbio | K-120 |
| Gentamicin | Goldbio | G-400 |
| Tetracyclin | Goldbio | T-101 |
| 5-Bromo-4-chloro-3-indolyl β-D-galactopytanoside (X-gal) | Santa Cruz Biotechnology | Sc-280488 |
| d-desthiobiotin | Sigma-Aldrich | D1411 |
| Glycerol | Fisher Scientific | G33500 |
| Oligonucleotides | ||
| Cloning primer at the 5′ end of tubulin alpha1B 5′GAGCTCACTAGTAACTCCTAAAAAA CCGCCACCATGCATCATCATCATCAC CATCACCACCACCACGAGAACCTGT ACTTCCAAGGCGCTCCTATGAGGGA ATGTATCTCTATTCATGTGG3′ |
This paper | N/A |
| Cloning primer at the 3′ end of tubulin alpha1B 5′CTCGACAAGCTTTTAGTACTCTTCG CCTTCTTCCTCACCC3′ |
This paper | N/A |
| Cloning primer at the 5′ end of tubulin beta3 CGGGATCTCGAGAACTCCTAAAAA ACCGCCACCATGAGGGAAATCGTT CACATTCAGGC |
This paper | N/A |
| Cloning primer at the 3′ end of tubulin beta3 TCTCCCGGTACCTTACTTCTCGAAC TGAGGGTGGGACCAGCCACCAGAA CCACCGCCTTGGAAGTACAGGTTC TCCTTTGGACCTTGAGCTTCGCTTTCC |
This paper | N/A |
| Recombinant DNA | ||
| pFastBac Dual | Fisher | 10712024 |
| Codon optimized Human tubulin alpha1B (NP_006073.2) in pCloneEZ-NRS-Blunt-Amp | Epoch Life Science | N/A |
| Codon optimized Human tubulin beta2B (NM_178012.4) in pFastBac Dual | Epoch Life Science | N/A |
| Codon optimized Human tubulin beta3 (NP_006077.2) in pCloneEZ-NRS-Blunt-Amp | Epoch Life Science | N/A |
| Deposited Data | ||
| Raw mass spectrometry data of recombinant human α1B/β2B-tubulin | This paper | https://doi.org/10.17632/s8vgfy9xz5.1 |
| Raw mass spectrometry data of recombinant human α1B/β3-tubulin | This paper | https://doi.org/10.17632/64yp27r788.1 |
Materials and Equipment
Prepare DH10Bac LB Agar Plates
Prepare LB/Agar containing 50 μg/mL kanamycin, 7 μg/mL gentamycin, 10 μg/mL tetracycline, 100 μg/mL X-gal, and 40 μg/mL IPTG in disposable Petri dishes. The plates should be wrapped in aluminum foil and stored at 4°C for less than 2 weeks.
Prepare the Buffers for Bacmid Preparation
Optional: For the preparation of all the buffers, use ultrapure water from other water purifying systems that purify water to the same quality as the Milli-Q systems.
P1 Buffer
P1 buffer contains 50 mM Tris-HCl, 10 mM EDTA, 100 μg/mL RNase A, at pH 8.0. To prepare 1 L of P1 buffer, combine:
-
•
6.06 g of Tris-base
-
•
3.72 g of EDTA·2H2O
-
•
800 mL of room temperature Milli-Q water
Stir until all the powder has dissolved. Adjust the pH to 8.0 using 12.1 M HCl, and then bring the volume to 1 L with Milli-Q water. Add 100 mg of RNase A to the solution and store at 4°C.
P2 Buffer
P2 buffer contains 200 mM NaOH and 1% (w/v) sodium dodecyl sulfate (SDS). To prepare 1 L of P2 buffer, combine:
-
•
8.09 g of NaOH
-
•
50 mL of 20% (w/v) SDS solution
-
•
930 mL of room temperature Milli-Q water
Stir until all the NaOH has dissolved. Adjust the final volume to 1 L using Milli-Q water.
N3 Buffer
N3 buffer contains 4.2 M guanidine hydrochloride and 0.9 M potassium acetate, at pH 4.8. To prepare 500 mL of N3 buffer, combine:
-
•
200.6 g of guanidine hydrochloride
-
•
44.16 g of potassium acetate
-
•
200 mL of room temperature Milli-Q water
Bring the volume to 450 mL using Milli-Q water. Adjust the pH to 4.8 using glacial acetic acid. Adjust the final volume to 500 mL using Milli-Q water.
Maintain and Expand Insect Cell Stocks
Grow Sf9 cells in Sf-900 II medium (Thermo Fisher Scientific) supplemented with 10% (v/v) fetal bovine serum (FBS) and 1% (v/v) Antibiotic-Antimycotic (GIBCO) at 27°C and 110 rpm orbital rotation. To maintain a stock, we typically split the cells every day to 20-40 mL at 1-2 × 106 cells/mL in a 250 mL sterile Erlenmeyer flask. The doubling time under these conditions is roughly 24 h.
Grow High Five cells in Express Five medium (Thermo Fisher Scientific) supplemented with 18 mM L-glutamine and 1% (v/v) Antibiotic-Antimycotic at 27°C and 110 rpm orbital rotation. To maintain a stock, we typically split the cells every day to 20-40 mL at 1-2 × 106 cells/mL in a 250 mL sterile Erlenmeyer flask. For protein expression, we typically expand the cells to a volume of 1.2 L at a density of 3 × 106 cells/mL. One 2.8 L Erlenmeyer flask can hold a maximum 600 mL of cell culture. Under these conditions, High Five cells triple in density every 24 h.
Prepare a 5-Fold Concentrated Stock Solution of BRB80 Buffer (5X BRB80)
5X BRB80 buffer contains 400 mM PIPES, 5 mM MgCl2, and 5 mM EGTA, at pH 6.8. To prepare 1 L of 5X BRB80 buffer, combine:
-
•
120.8 g of PIPES
-
•
1.02 g of MgCl2
-
•
1.9 g of EGTA
-
•
27 g of KOH
-
•
800 mL of room temperature Milli-Q water
Stir until all the PIPES has dissolved. Adjust the pH to 6.8 using 5 M KOH, and then bring the volume to 1 L with Milli-Q water. After filtering through a 0.22 μm filter, prepare 50-mL aliquots that can be stored at −20°C.
Clean Purification Columns
We typically use the following procedures to clean the reusable 1 mL columns after each purification. The columns are left in filtered Milli-Q water for another purification within 2 weeks. Otherwise, the columns are stored in 20% (v/v) ethanol.
Connect 4 X 1 mL HisTrap HP columns in series and clean with:
-
•
10 mL of 1 M imidazole, pH 7.0
-
•
10 mL of 6 M guanidine hydrochloride
-
•
10 mL of Milli-Q water
Connect 2 X 1 mL StrepTrap HP columns in series and clean with:
-
•
5 mL of 0.5 M NaOH
-
•
5 mL of Milli-Q water
Optional: The StrepTrap HP columns can also be regenerated with 10 mL of 1 mM 2-[4 -hydroxy-benzeneazo]benzoic acid (HABA) (100 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, and 1 mM HABA, pH 8.0), followed by 10 mL of wash buffer (100 mM Tris-HCl, 150 mM NaCl, and 1 mM EDTA, pH 8.0), and 10 mL of Milli-Q water.
Connect 2 X 1 mL HiTrap SP columns in series and clean with:
-
•
5 mL of 6 M guanidine hydrochloride
-
•
5 mL of Milli-Q water
Lysis Buffer
Lysis buffer contains 50 mM HEPES, 20 mM imidazole, 100 mM KCl, 1 mM MgCl2, 0.5 mM 2-mercaptoethanol, and 0.1 mM GTP, at pH 7.2. To prepare 300 mL of lysis buffer for the purification of recombinant tubulin from 1.2 L of High Five cell culture, combine in a beaker:
-
•
3.57 g of HEPES
-
•
6 mL of 1M imidazole (pH 7.0)
-
•
2.24 g of KCl
-
•
300 μL of 1M MgCl2
-
•
293 mL of pre-chilled Milli-Q water.
The buffer should be chilled on ice before adjusting the pH to 7.2 with KOH. Right before use, add 10.5 μL of 14.3 M 2-mercaptoethanol and 15.6 mg of GTP powder to the lysis buffer.
HisTrap Elution Buffer
HisTrap elution buffer contains 1X BRB80, 500 mM imidazole, 2 mM 2-mercaptoethanol, and 0.2 mM GTP, at pH 7.2. To prepare 50 mL of HisTrap elution buffer, combine in a 50 mL conical centrifuge tube:
-
•
10 mL of 5X BRB80
-
•
25 mL of 1M imidazole (pH 7.0)
-
•
14.8 mL of pre-chilled Milli-Q water.
After mixing by inverting the tube, the buffer should be chilled on ice before adjusting the pH to 7.2 with HCl. Right before use, add 7 μL of 14.3 M 2-mercaptoethanol and 5.2 mg of GTP powder to the HisTrap elution buffer.
Strep Wash Buffer A
Strep wash buffer A contains 1X BRB80, 10% (w/v) glycerol, 0.1% (w/v) Tween-20, 1 mM 2-mercaptoethanol, and 0.1 mM GTP, at pH 7.2. To prepare 50 mL of strep wash buffer A, combine in a 50 mL conical centrifuge tube:
-
•
10 mL of 5X BRB80
-
•
5 g of glycerol (directly weighted in the 50 mL tube)
-
•
250 μl of 20% (w/v) Tween-20
-
•
37 mL of pre-chilled Milli-Q water
After mixing by inverting the tube, the buffer should be chilled on ice before adjusting the pH to 7.2 with KOH. Right before use, add 3.5 μL of 14.3 M 2-mercaptoethanol and 2.6 mg of GTP powder to the buffer.
Strep Wash Buffer B
Strep wash buffer B contains 1X BRB80, 10 mM MgCl2, 5 mM ATP, 1 mM 2-mercaptoethanol, and 0.1 mM GTP, at pH 7.2. To prepare 50 mL of strep wash buffer B, combine in a 50 mL conical centrifuge tube:
-
•
10 mL of 5X BRB80
-
•
0.5 mL of 1 M MgCl2
-
•
137.8 mg of ATP
-
•
39 mL of pre-chilled Milli-Q water
After mixing by inverting the tube, the buffer should be chilled on ice before adjusting the pH to 7.2 with KOH. Right before use, add 3.5 μL of 14.3 M 2-mercaptoethanol and 2.6 mg of GTP powder to the buffer.
Strep Elution Buffer
Strep elution buffer contains 1X BRB80, 20 mM imidazole, 3 mM desthiobiotin, 2 mM 2-mercaptoethanol, and 0.2 mM GTP, at pH 7.2. To prepare 150 mL of strep elution buffer, combine in a beaker:
-
•
30 mL 5X BRB80
-
•
3 mL 1M imidazole (pH 7.0)
-
•
116.1 mL of pre-chilled Milli-Q water
The buffer should be chilled on ice before adjusting the pH to 7.2 with KOH. Right before use, add 21 μL of 14.3 M 2-mercaptoethanol, 15.6 mg of GTP powder, and 0.9 mL of 0.5 M desthiobiotin (prepared in dimethyl sulfoxide (DMSO)) to the buffer. The addition of desthiobiotin does not change the pH of the buffer.
Gel Filtration Buffer
Gel filtration buffer contains 1X BRB80, 5% (w/v) glycerol, 2 mM 2-mercaptoethanol, and 0.2 mM GTP, at pH 6.8. To prepare 300 mL of gel filtration buffer, combine in a beaker:
-
•
60 mL 5X BRB80
-
•
15 g of glycerol (directly weighed in the beaker)
-
•
225 mL of pre-chilled Milli-Q water.
The buffer should be chilled on ice before adjusting the pH to 6.8 with HCl. Right before use, add 42 μL of 14.3 M 2-mercaptoethanol and 31.5 mg of GTP powder to the buffer, and then filter the buffer through a 0.22 μm filter.
Step-By-Step Method Details
Note:Figure 2 shows an overall schematic of the workflow from the generation of bacmid to the frozen aliquots of purified recombinant human tubulin.
Figure 2.

Overall Schematic of the Workflow in This Protcol
For each stage (dotted box), the expected finish time and the corresponding steps are annotated.
Optional: Prepare baculovirus following Invitrogen’s Bac-to-Bac manual (Invitrogen MAN0000414 A.0) and skip to “Infection of High Five Insect Cells.” However, we use a slightly modified version of the Bac-to-Bac protocol, described below. In theory, both methods should produce similar protein expression results.
DH10Bac Transformation
Generate the DH10Bac strains that contain the expression bacmid with the α- and β-tubulin genes of interest.
-
1.
Transform 3 μL of a pFastBac Dual donor plasmid (∼500 ng/μL) that encodes the codon-optimized α- and β-tubulin genes into 100 μL of DH10Bac competent cells. Plate the cells onto DH10Bac LB agar plates. Incubate the plate at 37°C for 1 day.
-
2.
Pick one white colony to streak onto another DH10Bac LB agar plate. Incubate the plate at 37°C for 1 day.
-
3.
Inoculate 6 mL of LB medium containing 50 μg/mL kanamycin, 7 μg/mL gentamycin, and 10 μg/mL tetracycline with cells from step 2 (no need to isolate a single colony at this step). Incubate the culture at 37°C and 230 rpm orbital rotation for about 18 h.
Optional: Pick two white colonies in Step 2 in case there is a problem downstream (e.g., transfected cell culture obtains microbial contamination). In Step 3, inoculate one 6 mL LB culture with cells from each individual white colony.
Optional: For Step 1 and Step 2, an extra day of incubation at 37°C can be helpful to distinguish the blue colonies from the white colonies.
Bacmid Preparation
Isolate the expression bacmid from the DH10Bac strains.
Optional: Use the P1, P2, and N3 buffers from the Qiagen plasmid miniprep kit. In our hands, using either the home-made buffer or the Qiagen buffer generates bacmid with comparable quality.
-
4.
Harvest the cells from Step 3 at 4,000 rcf for 10 min at 4°C.
-
5.
Resuspend the cell pellet in 250 μL of P1 buffer. Transfer the cells to a new 1.7 mL microcentrifuge tube.
-
6.
Add 250 μL of P2 buffer into the cell suspension and mix by inverting the tube 4-6 times.
-
7.
Add 350 μL of N3 buffer into the cell suspension and mix by inverting the tube 4-6 times.
-
8.
Centrifuge the cell lysate at 21,000 rcf for 10 min at room temperature.
-
9.
Transfer the supernatant to a 2 mL microcentrifuge tube and add 1 mL of phenol:chloroform:isoamyl alcohol (25:24:1, v/v). Mix by inverting the tube 4-6 times.
-
10.
Centrifuge at 21,000 rcf for 2 min at room temperature.
-
11.
Transfer the upper aqueous phase to a new 2 mL microcentrifuge tube.
-
12.
Repeat Step 9 to 11 twice. Transfer the aqueous phase to a new 2 mL microcentrifuge tube and add 1 mL of anhydrous chloroform. Mix by inverting the tube 4-6 times.
-
13.
Centrifuge at 21,000 rcf for 2 min at room temperature.
-
14.
Transfer the final aqueous phase (∼750 μl) to a 15 mL conical centrifuge tube. Add 10% volume of 8 M lithium chloride. Mix by tapping the bottom of the tube.
-
15.
Add 2.5 mL of 100% ethanol. Incubate on ice for 20 min.
-
16.
Centrifuge at 4,000 rcf for 20 min at 4°C. Aspirate the supernatant.
-
17.
Gently resuspend the pellet in 1 mL of ice-cold 80% (v/v) ethanol. Transfer the solution to a new 1.7 mL microcentrifuge tube. Centrifuge at 21,000 rcf for 10 min at 4°C.
-
18.
Aspirate the supernatant. Air dry the purified bacmid pellet at room temperature for at least 30 min. Cover the microcentrifuge tubes with a sheet of plastic wrap to minimize airborne contamination.
-
19.
Dissolve the dried pellet in 50 μL sterile Milli-Q water. Quantify the DNA concentration and estimate the quality by measuring the absorption at 260 nm (A260) and at 280 nm (A280). In general, the DNA concentration should be ∼6 μg/μL with an A260/A280 of ∼2.0.
Optional: Use ultrapure water from other water purifying systems that purify water to the same quality as the Milli-Q systems.
Optional: Use primers that flank the α- or β-tubulin genes to confirm the tubulin genes are in the bacmid via PCR. A protocol can be found in Invitrogen’s Bac-to-Bac manual. The PCR products can also be purified and sent for Sanger DNA sequencing.
PAUSE POINT: The purified bacmid DNA can be stored at 4°C for up to 1 week.
Generating the Recombinant Baculovirus
Transfect the expression bacmid into Sf9 cells to generate baculovirus.
For transfection and virus production, Sf9 cells should be in log phase (2∼2.5x106 cells/mL) at the time of plating. The following steps are for transfection of one construct:
Transfection
-
20.
Plate 9x105 Sf9 cells into two 9.5 cm2 flat-bottom wells of a 6-well plate. Incubate at 27°C for 1 h to allow cell attachment. During this time, continue to step 21.
-
21.
Dilute 12 μg of purified bacmid DNA into 200 μl of serum- and antibiotic-free Sf-900 II medium in a sterilized 1.7 mL microcentrifuge tube.
-
22.
Mix Cellfectin II Reagent (Thermo Fisher Scientific) by inverting the Cellfectin stock tube 6 times. In a sterilized 1.7 mL microcentrifuge tube, dilute 12 μl of Cellfectin II into 200 μl of serum- and antibiotic-free Sf-900 II medium.
-
23.
Combine the diluted bacmid and Cellfectin II reagent. Mix gently by tapping the microcentrifuge tube. Incubate at room temperature for 45 min.
-
24.
For the attached Sf9 cells from Step 20, aspirate the medium and then rinse each well with 2 mL of serum- and antibiotic-free Sf-900 II medium. After aspirating the washing medium, add 0.8 mL of serum- and antibiotic-free Sf-900 II medium per well.
-
25.
Add the bacmid:Cellfectin II mixture (∼206 μl per well) to the attached Sf9 cells. Incubate at 27°C for 5 h.
-
26.
Aspirate the medium and add 2 mL of Sf-900 II medium supplemented with 10% (v/v) FBS and 1% (v/v) Antibiotic-Antimycotic per well. Incubate at 27°C for 72 h.
Harvest P1 Virus
-
27.
Resuspend the cells with a 5 mL serological pipette. Combine the cell culture from both wells to a 15 mL conical centrifuge tube. Centrifuge at 1,000 rcf for 5 min at room temperature.
-
28.
Filter the supernatant through a sterilized 25 mm diameter syringe filter with a 0.45 μm pore size polyethersulfone membrane into another 15 mL conical centrifuge tube. This is the P1 virus stock.
Generate P2 Virus
-
29.
Plate 9x106 Sf9 cells into a 75 cm2 vented cap tissue culture flask. Incubate at 27°C for 1 h to allow cell attachment.
-
30.
Replace medium with 23 mL of Sf-900 II medium supplemented with 10% (v/v) FBS and 1% (v/v) Antibiotic-Antimycotic. Add 2 mL of the P1 virus and incubate at 27°C for 60 h.
-
31.
Resuspend the cells with a 10 mL serological pipette. Transfer the cell culture to a 50 mL conical centrifuge tube. Centrifuge at 1,000 rcf for 5 min at room temperature.
-
32.
Filter the supernatant through a sterilized 25 mm diameter syringe filter with a 0.45 μm pore size polyethersulfone membrane into another 50 mL conical centrifuge tube. This is the P2 virus stock.
Generate P3 Virus
-
33.
Plate 9x106 Sf9 cells into a 75 cm2 vented cap tissue culture flask. Incubate at 27°C for 1 h to allow cell attachment.
-
34.
Aspirate the medium, add 1 mL of P2 virus and 24 mL of Sf-900 II medium supplemented with 10% (v/v) FBS and 1% (v/v) Antibiotic-Antimycotic. Incubate at 27°C for 60 h.
-
35.
Resuspend the cells with a 10 mL serological pipette. Transfer the cell culture to a 50 mL conical centrifuge tube. Centrifuge at 1,000 rcf for 5 min at room temperature.
-
36.
Filter the supernatant through a sterilized 25 mm diameter syringe filter with a 0.45 μm pore size polyethersulfone membrane into another 50 mL conical centrifuge tube. This is the P3 virus stock.
Optional: To verify the expression of recombinant tubulin, we use liquid nitrogen to flash freeze the infected Sf9 cell pellets that produce the P3 virus and then use the following protocol to check for protein expression in these cells by western blot.
-
a.
Resuspend the cell pellet in 1 mL of lysis buffer (1X BRB80, 0.1% (w/v) Tween-20, 1% (w/v) Triton X-100, 3 U/mL benzonase, and 1X Roche EDTA-free mini protease inhibitor). Transfer the cell suspension to a 1.7 mL microcentrifuge tube. Incubate on ice for 20 min.
-
b.
Centrifuge the cell lysate at 21,000 rcf for 10 min at 4°C.
-
c.
Use 15 μL of sample to run a western blot analysis using primary antibodies against α- or β-tubulin. After secondary antibody and image development, there should be two individual bands at ∼50 kD. By comparing to the western blot analysis of uninfected Sf9 cells, we have found that the lower band corresponds to the endogenous insect tubulin, and the upper band corresponds to the recombinant α-tubulin with the decahistidine tag or the recombinant β-tubulin with the Strep-tag II (Figure 3).
Figure 3.

Immunoblot Analysis of Tubulin Expression in Sf9 Cells
(A and B) The Sf9 cells harvested after generating human α1B/β3-tubulin P3 virus. Antibody against α-tubulin (A) or β-tubulin (B) was used for western blots (WB).
(C and D) The infected (lane 1) and the uninfected control Sf9 cells (lane 2). Antibody against α-tubulin (C) or β-tubulin (D) was used for western blots (WB).
Infecting High Five Cells for Protein Expression
Express the α- and β-tubulin of interest in High Five cells.
-
37.
When High Five cell density is at 2.5∼3.5x106 cells/mL, add 6 mL of P3 virus per 600 mL of cell culture. Incubate the infected insect cell culture at 27°C and 110 rpm orbital rotation for 60 h.
Optional: As with any untested protein purification strategy, generating recombinant tubulin other than human α1B/β2B- and α1B/β3-tubulin may require time-course experiments to determine the infection time that maximizes protein yield. However, the procedure outlined here can act as a good starting point.
Purification of Recombinant Tubulin
Isolate affinity tag-free recombinant α/β-tubulin heterodimers.
Cell Lysis
-
38.
Harvest cells by centrifugation at 1,000 rcf for 10 min at 4°C
Figure 4.

Representative Images of the Harvested High Five Cell
(A) Harvested cell pellets and supernatant (600 mL of cell culture for each bottle). (B) The supernatant from 1.2 L of cell culture.
-
39.
For cells harvested from 1.2 L of culture, add 5 μL of 25 U/μL benzonase and 2 tablets of Roche complete EDTA-free protease inhibitor to 40 mL of lysis buffer.
-
40.
Use a 10 mL serological pipette to resuspend the cell pellet with the lysis buffer supplemented with benzonase and protease inhibitor.
-
41.
Lyse cells by 20 strokes of dounce homogenization on ice. We use a serrated Teflon pestle (Thomas C 4812).
Clarification
-
42.
Centrifuge the homogenized cell lysate at 55,000 rpm (∼222,000 rcf) in a Type 70 Ti rotor for 1 h at 4°C.
-
43.
Collect the supernatant with a 10 mL serological pipette. Avoid pipetting the bottom 25% of the supernatant (∼5 mL per centrifuge tube).
-
44.
Separately collect the turbid bottom fraction of the supernatant into 1.7 mL microcentrifuge tubes. Centrifuge in a benchtop microcentrifuge at 21,000 rcf for 10 min at 4°C, then pool this supernatant with the rest from step 43.
-
45.
Use a 10 mL syringe to filter the supernatant through 33 mm diameter syringe filters with a 0.22 μm pore size polyethersulfone membrane. In general, one filter can clarify ∼10 mL of the supernatant.
Optional: Take 20 μl of sample for SDS-PAGE and western blot analysis.
Nickel Affinity Chromatography
-
46.
Equilibrate two 1 mL HisTrap HP columns (GE Healthcare) connected in series with 4 mL of lysis buffer at 1 mL/min.
-
47.
Load the 0.22 μm filtered cell lysate onto the columns at 1 mL/min (e.g., using a Superloop).
Optional: Take 20 μl of sample from the flow-through from this step for SDS-PAGE and western blot analysis.
-
48.
Wash the columns with at least 35 mL of lysis buffer, or until the UV absorption (A280) reaches baseline.
-
49.
Elute with HisTrap elution buffer at 1 mL/min in 0.25 mL fractions. The fractions are collected in a multi-well microplate (Greiner cat# 655101).
Optional: To minimize buffer consumption, leave pump A with the lysis buffer while pumping the HisTrap elution buffer through pump B.
Optional: Elute the two columns separately. Together with the western blot analysis (see Expected Outcomes), this strategy can help determine whether both HisTrap HP columns properly sequester decahistidine-tagged α-tubulin. This information can be useful for troubleshooting when the final yield is lower than expected (see Troubleshooting). Detach the second column and elute the first column until baseline, then attach the second column and continue the elution until baseline. See Figure 5A for a typical elution profile we observe from the two 1 mL HisTrap HP columns.
Figure 5.

Typical elution profiles from each chromatography step
(A and B) Elution profile of recombinant human α1B/β3-tubulin from two 1 mL HisTrap columns (A) and two 1 mL StrepTrap columns (B).
(C) The chromatogram of the flow-through from HiTrap SP/HisTrap HP columns.
(D and E) Elution profile of recombinant human α1B/β3-tubulin (D; peak volume, 81.1 mL) or bovine brain tubulin (E; peak volume, 80.4 mL) from size-exclusion chromatography. *, non-specific instrument singal. V0, void volume. 1CV, one column volume.
-
50.
Pool all the fractions that have an A280 value above baseline into a chilled 50 mL conical tube. Estimate the volume of this eluate (typically ∼9 mL). To maximize yield, use ∼300 μl of lysis buffer to rinse out the wells of the fraction collection plate and add this to the eluate.
-
51.
Dilute the pooled HisTrap eluate to ∼30 mL with lysis buffer.
Optional: Take 20 μl of sample from the HisTrap eluate for SDS-PAGE and western blot analysis.
Strep Tag II Affinity Chromatography
-
52.
Equilibrate two 1 mL StrepTrap HP columns (GE Healthcare) connected in series with 4 mL of buffer containing 66% lysis buffer (from pump A) and 34% HisTrap elution buffer (from pump B) at 1 mL/min.
-
53.
Filter the diluted HisTrap eluate from step 51 through a 33 mm diameter/0.22 μm pore size syringe polyethersulfone membrane filter.
-
54.
Load the filtered protein solution onto the equilibrated StrepTrap HP columns using a Superloop at 1 mL/min.
Optional: Take 20 μl of sample from the flow-through of the StrepTrap column for SDS-PAGE and western blot analysis.
-
55.
Wash the columns with 4 mL of buffer containing 66% lysis buffer (from pump A) and 34% HisTrap elution buffer (from pump B) at 1 mL/min.
-
56.
Wash the columns with 25 mL of strep wash buffer A (from pump B), followed by 25 mL of strep wash buffer B (from pump B) at 1 mL/min.
-
57.
Wash the columns with 10 mL of lysis Buffer (from pump A) to get rid of baseline nucleotide signal at 1 mL/min.
-
58.
Elute with strep elution buffer (from pump A) at 0.8 mL/min in 0.25 mL fractions. The fractions are collected in a multi-well microplate.
Optional: Elute the two columns separately. Together with the western blot analysis (see Expected Outcomes), this strategy can be useful if the final yield is lower than expected (e.g., if one of the StrepTrap HP columns has lost binding affinity; see Troubleshooting). Detach the second column and elute the first column until baseline, then attach the second column and continue the elution until baseline. See Figure 5B for the elution profile from two 1 mL StrepTrap HP columns.
Optional: Take 20 μl of sample from the eluate of the StrepTrap column for SDS-PAGE and western blot analysis.
-
59.
Pool all the fractions that have an A280 value above the baseline into a chilled 50 mL conical centrifuge tube (typically ∼7 mL). To maximize yield, use ∼300 μl of strep elution buffer to rinse out the wells of the collection plate. Dilute this eluate to ∼25 mL with strep elution buffer to reduce the extent of changes in the buffer composition in the following TEV digestion step.
TEV Digestion
-
60.
Dilute ∼4 mg TEV (> 7 mg/mL stored in 40 mM HEPES, 150 mM KCl, 30% (w/v) glycerol, 1 mM MgCl2, and 3 mM 2-mercaptoethanol, pH 7.5) into 5 mL of strep elution buffer. Add this TEV solution to the eluate from Step 59. Incubate on ice for 2 h. Mix gently every 10 min by inverting the tube 4-6 times.
Optional: Take 20 μl of sample for SDS-PAGE and western blot analysis.
Cation Exchange-Nickel Affinity Chromatography
-
61.
Centrifuge the digested protein at 4,000 rcf for 30 sec to bring all the liquid to the bottom of the tube.
-
62.
Equilibrate two 1 mL HiTrap SP columns connected in series and upstream of two 1 mL HisTrap HP columns with 5 mL of Strep elution buffer at 1 mL/min.
-
63.
Filter the digested protein from Step 61 through a 33 mm diameter/0.22 μm pore size syringe polyethersulfone membrane filter.
-
64.
Load the filtered protein solution onto the equilibrated HiTrap SP/HisTrap HP columns setup at 1 mL/min using a Superloop. Immediately start collecting the flow-through, which contains the recombinant tubulin. As the TEV is stored in a nucleotide-free buffer, the nucleotide concentration of the digested protein is ∼5% lower than the nucleotide concentration of the strep elution buffer. The concentration of tubulin in the flow-through is generally not high enough to compensate for the loss of UV absorbance due to nucleotide dilution. Therefore, the flow-through will typically have slightly lower UV absorbance than the strep elution buffer. Flow and collect another 10 mL of strep elution buffer through the columns to maximize protein recovery.
Figure 5C shows the chromatogram of the flow-through from HiTrap SP/HisTrap HP columns of purification with an above average yield of tubulin (∼4 mg from 1.2 L cell culture). The absorbance of the flow-through at 280 nm is slightly higher than the background absorbance of the strep elution buffer.
Optional: Take 20 μl of sample from the flow-through for SDS-PAGE and western blot analysis.
Size Exclusion Chromatography
-
65.
Equilibrate a Superdex 200 HiLoad 16/60 column with 130 mL of gel filtration buffer at 1 mL/min.
-
66.
Pre-equilibrate a 15 mL 50 kDa MWCO centrifugal filter unit (Millipore UFC901024) with ice-cold gel filtration buffer.
-
67.
Concentrate the flow-through from Step 64 (∼40 mL) to less than 3 mL.
-
68.
Load the concentrated protein solution from Step 67 onto the equilibrated Superdex 200 HiLoad 16/60 column at 1 mL/min.
-
69.
Keep flowing the gel filtration buffer through the column at 1 mL/min for one column volume. Tubulin dimers elute at ∼80-85 mL on this column (Figures 5D and 5E). Pool all the fractions that have an A280 value above the baseline into a 50 mL conical centrifuge tube.
Optional: Take 20 μl of sample from the eluate of the size-exclusion column for SDS-PAGE and western blot analysis.
Measure the Protein Concentration and Storage
-
70.
Concentrate gel filtration eluate to less than 1 mL using another 50 kDa MWCO centrifugal filter unit pre-equilibrated in ice-cold gel filtration buffer. To determine the purity of the concentrated protein, take 1-5 μl sample for SDS-PAGE analysis. Typically, densitometric analysis of Coomassie stained SDS-PAGE gels indicates that the purity at this step is greater than 95%.
-
71.
Measure the protein concentration by Bradford assay using BSA as a standard, or by absorption at 280 nm (ϵ280 = 115,000 M-1cm-1 and MW = 100 kDa). On average, this protocol should generate ∼3 mg of recombinant tubulin from 1.2 L of insect cell culture.
-
72.
Snap freeze small aliquots (∼20 μl) of concentrated protein in liquid nitrogen and store at −80°C.
Expected Outcomes
Western Blot Analysis
We use primary antibodies against α- or β-tubulin for western blots to analyze the protein samples collected at different stages of the purification. The presence of affinity tags on both recombinant tubulin subunits gives them a slower electrophoretic mobility relative to endogenous insect tubulin subunits (Figure 3C and D). We use this band shift to judge the expression level of the recombinant tubulin in insect cells, and to determine the effectiveness of the affinity purification steps. As the removal of affinity tags increases the electrophoretic mobility, we employ the mobility shift to assess the efficiency of the tag cleavage. A typical purification western blot (Figure 6) gives the following band patterns, all located near the 50 kDa marker:
-
•
Cell lysate: two bands each for both α- and β-tubulin.
-
•
HisTrap flow-through: one lower band for α-tubulin and two bands for β-tubulin.
-
•
HisTrap eluate: one upper band for α-tubulin and two bands for β-tubulin.
-
•
Strep flow-through: one upper band for α-tubulin and one lower band for β-tubulin.
-
•
Strep eluate: one upper band for α-tubulin and one upper band for β-tubulin.
-
•
HiTrap SP/HisTrap tandem column flow-through: one lower band for α-tubulin and one lower band for β-tubulin.
-
•
Size-exclusion column eluate: one lower band for α-tubulin and one lower band for β-tubulin.
Figure 6.

SDS-PAGE and Immunoblot Analysis of Protein Samples Collected during the Purification of Recombinant Human α1B/β3-Tubulin
(A) Coomassie-stained SDS-PAGE gel. (B and C) Antibody against α-tubulin (B) or β-tubulin (C) was used for western blots (WB). 1, protein ladder; 2, clarified cell lysate; 3, flow-through from the HisTrap HP columns; 4, eluate from the HisTrap HP columns; 5, flow-through from the StrepTrap HP columns; 6, eluate from the StrepTrap HP columns; 7, flow-through from the tandem HiTrap SP/HisTrap HP columns; 8, eluate from the Superdex 200 16/60 size exclusion column.
Mass Spectrometry
In addition to analyzing a Coomassie-stained SDS-PAGE gel to determine the purity, the recombinant tubulin should be analyzed using mass spectrometry. We recommend running > 5 μg of tubulin on a 4%–20% Tris-Glycine SDS-PAGE gel (Novex). The tubulin band is excised, reduced, alkylated, trypsinized, and analyzed by tandem LC-MS/MS. Ideally, the sample should contain > 99% recombinant α- and β-tubulin, and there should be no detectable peptides corresponding to insect cell tubulin.
When analyzing the mass spectrometry data for our purified human α1B/β3- and α1B/β2B-tubulin, we do not find peptides containing common tubulin post-translational modifications (PTMs) (e.g., acetylation, ubiquitination, glycosylation, polyglycylation (polyG) and polyglutamylation (polyE)). However, as the identification of certain complex PTMs (e.g., polyG and polyE) can be challenging, we provide our raw mass spectrometry data for further independent processing and analysis (Supplemental Data). Additionally, we note that electron spray ionization analysis of purified recombinant tubulin generated in another study (Vemu et al., 2016) also did not detect any PTMs. Current evidence indicates that the baculovirus-driven insect cell expression system generates recombinant tubulin with no PTMs.
Gel Filtration of Mammalian Brain Tubulin as a Standard
We also advise confirming that the purified recombinant tubulin exists as a dimer with the correct hydrodynamic radius. A quick way to do this is to compare the size exclusion trace from Step 69 with a sample of mammalian brain tubulin diluted in size exclusion buffer and run under identical flow rates. In both cases, tubulin should elute as a single peak, and the peaks from both runs should appear almost identical in shape and retention volume position (Figures 5D and 5E).
Polymerizing the Recombinant Tubulin
For any tubulin purification, it is essential to confirm that the purified protein is polymerization-competent. A reliable way to do this is to assemble microtubules in the presence of a microtubule-stabilizing drug, such as Taxol, or a GTP analog, such as GMPCPP. As stated in the “Limitations” section, commonly used additives such as DMSO or glycerol may not be sufficient to promote spontaneous assembly of recombinant tubulin as it does for brain tubulin; indeed, this is the case for our human α1B/β3- and α1B/β2B-tubulin (Ti et al., 2018). We must stress that researchers should spend some time testing a variety of polymerization methods to find the one that best promotes formation of microtubules from their respective recombinant tubulin source. In any case, the resulting microtubule pellets can be quantified by SDS-PAGE for protein content, or directly visualized using fluorescence microscopy if stained with fluorescent antibodies or spiked with fluorescently labeled mammalian tubulin (Figure 7). An alternative approach would be to use label-less microscopy methods (differential interference contrast (DIC), dark-field (DF), interference reflection microscopy (IRM), or -electron microscopy (-EM) to visualize the microtubules polymerized from recombinant tubulin (Bormuth et al., 2007, Gittes et al., 1993, Horio and Hotani, 1986, Mahamdeh et al., 2018, Mandelkow and Mandelkow, 1985, Walker et al., 1988). Below, we describe how we polymerize recombinant human α1B/β2B- or α1B/β3-tubulin using Taxol or GMPCPP. These methods can be modified to use different microtubule-stabilizing drugs or nucleotide analogs, if necessary.
Figure 7.

Representative Total-Internal Reflection Fluorescence (TIRF) Microscopy Images of Microtubules
(A) taxol- and (B) GMPCPP-stabilized recombinant human α1B/β3 microtubules. X-rhodamine-labeled recombinant human α1B/β3 tubulin (~3.5%) was added to visualize filaments immobilized on kinesin-5 coated coverslips (Ti et al., 2018). Scale bar, 6 μm.
Prepare Buffers for Testing the Polymerization Activity of Purified Recombinant Tubulin
-
•
Tubulin dilution buffer: 1X BRB80 containing 5% (w/v) glycerol.
-
•
Taxol-microtubule resuspension buffer: 1X BRB80 containing 1 mM Tris(2-carboxyethyl)phosphine (TCEP) and 15 μM Taxol (10 mM paclitaxel stock solution prepared in DMSO).
-
•
GMPCPP-microtubule resuspension buffer: 1X BRB80 containing 1 mM TCEP.
Polymerization of Taxol-Stabilized Recombinant Microtubules
-
1.
Quickly thaw one aliquot of recombinant human α1B/β2B- or α1B/β3-tubulin between your fingers. Once thawed, place the protein on ice.
-
2.
In a 1.7 mL microcentrifuge tube, quickly mix recombinant tubulin, GTP from a 100 mM stock, and TCEP from a 500 mM stock with tubulin dilution buffer to prepare a mixture containing ∼1 mg/mL tubulin, 2 mM GTP and 1 mM TCEP in 50 μL. Incubate this mixture on ice for 5 min.
-
3.
Centrifuge the mixture 90,000 rpm (∼290,000 rcf) in a TLA120.1 rotor for 10 min at 4°C.
-
4.
Transfer the supernatant to a new pre-chilled 1.7 mL microcentrifuge tube. Incubate at 37°C for 2 min.
-
5.
Add 1 μM Taxol (prepared in DMSO) to the tubulin solution at a 1:10 dilution (final 0.1 μM Taxol). Incubate in a 37°C water bath for 10 min.
-
6.
Add 10 μM Taxol (prepared in DMSO) to the tubulin solution at a 1:10 dilution (final 1 μM Taxol). Incubate in a 37°C water bath for 10 min.
-
7.
Add 100 μM Taxol (prepared in DMSO) to the tubulin solution at a 1:10 dilution (final 10 μM Taxol). Incubate in a 37°C water bath for 15 min.
-
8.
Centrifuge the Taxol-stabilized microtubules at 90,000 rpm (∼290,000 rcf) in a TLA120.1 rotor for 10 min at 37°C.
-
9.
Remove the supernatant. Resuspend the microtubule pellet in 50 μL of warm (37°C) Taxol-microtubule resuspension buffer.
Polymerization of GMPCPP-Stabilized Recombinant Microtubules
-
1.
Quickly thaw one to two aliquots of recombinant human α1B/β2B- or α1B/β3-tubulin between your fingers. Leave the protein on ice
-
2.
In a 1.7 mL microcentrifuge tube, mix recombinant tubulin, GMPCPP from a 10 mM stock, and TCEP from a 500 mM stock with tubulin dilution buffer to prepare a mixture containing ∼1.5 mg/mL tubulin, 1 mM GMPCPP, and 1 mM TCEP in 100 μL. Incubate this mixture on ice for 10 min.
-
3.
Incubate mixture at 37°C for 1 h.
-
4.
Centrifuge at 90,000 rpm (∼290,000 rcf) in a TLA120.1 rotor for 10 min at 37°C.
-
5.
Remove the supernatant. Resuspend the microtubule pellet in 30 μL of chilled on ice GMPCPP-microtubule resuspension buffer. Incubate on ice for 1 h.
-
6.
Centrifuge at 90,000 rpm (∼290,000 rcf) in a TLA120.1 rotor for 10 min at 4°C.
-
7.
Transfer the supernatant to a new 1.7 mL microcentrifuge tube. Add GMPCPP from a 10 mM stock, TCEP from a 500 mM stock, and tubulin dilution buffer to prepare a mixture containing depolymerized tubulin, 1 mM GMPCPP, and 1 mM TCEP in 40 μL. Incubate on ice for 10 min.
-
8.
Incubate at 37°C for 1 h.
-
9.
Centrifuge at 90,000 rpm (∼290,000 rcf) in a TLA120.1 rotor for 10 min at 37°C.
-
10.
Remove the supernatant. Resuspend the microtubule pellet in 30 μL of warm (37°C) GMPCPP-microtubule resuspension buffer.
Limitations
Our current strategy relies on TEV to cleave off the affinity tags from recombinant α/β-tubulin heterodimers. The TEV digestion leads to remnant residues at the N terminus of α-tubulin (Gly-Ala-Pro) and at the C terminus of β-tubulin (Glu-Asn-Leu-Tyr-Phe-Gln). The effect of these additional residues on the biochemical properties of our purified tubulin is currently not clear. We do note that these appended peptides are not as highly charged as affinity tags that remain on recombinant tubulin generated by other methods (e.g., histidine tag or the FLAG tag; (Minoura et al., 2013, Vemu et al., 2016). More complex protein engineering techniques, such as subtiligase protein ligation (Henager et al., 2016), could be used in the future to generate recombinant human tubulin with its complete native peptide sequence.
Our current method generates enough recombinant tubulin for at least 10-20 typical in vitro assays (∼3 mg tubulin from 1.2 L of insect cell culture). In principle, the purification can be scaled up as needed, but we anticipate that cell lysis could be a major bottleneck. Lysing a significantly larger quantity of cells by dounce homogenization will lead to longer purification times, which reduces tubulin yield in our hands. Alternative lysis methods such as using mild detergents, sonication, or a microfluidizer would have to be used. We have not yet extensively tested these methods and thus do not know how they might affect the purity and integrity of the resulting tubulin. We advise researchers to proceed with caution when scaling up/changing the lysis method and to properly characterize the purified tubulin using, at the very least, the quality control procedures outlined in the “Expected Outcomes” section below.
From a biochemistry and structural biology standpoint, tubulin can be quite heterogeneous. For example, S. cerevisiae tubulin has a much lower critical concentration than bovine brain tubulin (Davis et al., 1993), yet it is not easily stabilized by paclitaxel (Taxol), a microtubule-stabilizing drug (Barnes et al., 1992, Schiff et al., 1979). In another example, glycerol and DMSO are commonly used to induce the polymerization of bovine brain tubulin in vitro. However, we find that recombinant human α1B/β2B- or α1B/β3-tubulin does not form microtubules in the presence of these additives. To assemble α1B/β2B or α1B/β3 microtubules in vitro, we need to instead use Taxol, guanosine-5′-[(α,β)-methyleno] triphosphonate (GMPCPP, a slowly hydrolyzing GTP analog), or GMPCPP-stabilized microtubule “seeds” to initiate polymerization. Even so, stabilized human microtubules tend to be much shorter-lived than their bovine brain counterparts – in our hands, Taxol-stabilized human α1B/β3-microtubules fully depolymerize within a day (Ti et al., 2018), but bovine brain microtubules prepared in the same way can last for weeks. Thus, traditionally used assays and microtubule polymerization methods may not always be appropriate for an uncharacterized recombinant tubulin. Conditions that favor microtubule polymerization may need to be empirically determined for each new recombinant tubulin generated.
Troubleshooting
Problem
The P3 virus does not express the recombinant tubulin (Step 36)
Possible Solution
Redo the transfection using freshly purified bacmid. The chloroform extraction is critical to generate bacmid that produces virus with a sufficiently high multiplicity of infection (MOI).
Problem
Precipitation appears after adding TEV protease (Step 60)
Possible Solutions
Make an at least 8-fold dilution from the concentrated (∼7 mg/mL) TEV stock solution using strep elution buffer.
We have noticed that mechanical agitation induces tubulin precipitation (e.g., with batch resin binding). To mix TEV with the tubulin solution, gently invert the conical tubes five times every 10 min. Do not use vortex or a rotary mixer.
Problem
The yield is much lower than expected. (Step 71)
Possible Solutions
Grow High Five cells in Express Five medium supplemented with 18 mM glutamine and 1% (v/v) Antibiotic-Antimycotic. We find that the yield of recombinant human tubulin can be significantly reduced if other insect cell lines (e.g., Sf9 or Sf21 cells) or culture media are used.
If the binding capacity of the HisTrap or the StrepTrap columns is compromised, the elution peaks from the affinity columns will have UV absorbance smaller than expected, and the western blot analysis will show significant amount of unbound recombinant tubulin in the flow-through. Using fresh or regenerated columns can solve this problem.
Add more TEV protease for digestion. We usually aim for 1 mg of TEV protease per 1 mg of recombinant tubulin heterodimer.
Make sure that the two HiTrap SP columns are upstream of the two HisTrap HP columns. We find that our TEV protease can associate with recombinant tubulin, likely through an interaction between a positively charged polyarginine tag on our TEV construct and the negatively charged tubulin C-terminal tails. We therefore use cation exchange (HiTrap SP) to dissociate and sequester TEV from the purified tubulin. The downstream HisTrap columns are mainly used to remove uncleaved tubulin from the prep. However, our TEV construct also contains a polyhistidine tag; with an incorrect chromatography setup in Step 59 (e.g., HisTrap HP columns followed by HiTrap SP columns), there is a risk of pulling down the TEV protease along with the TEV-associated tubulin heterodimers, significantly reducing the tubulin yield.
Problem
The tubulin does not polymerize using standard brain tubulin polymerization protocols.
Possible Solution
See “Limitations” and “Expected Outcomes” sections.
Acknowledgments
This research was supported by the NIH (GM65933, PI: T.M.K.). M.W. was supported by an HFSP Fellowship (LT000025/18-L1) and a Merck Postdoctoral Fellowship from Rockefeller University.
Author Contributions
Conceptualization, S.C.T. and T.M.K.; Methodology, S.C.T.; Validation, M.W.; Writing - Original Draft, S.C.T and M.W.; Writing - Review and Editing, S.C.T., M.W., and T.M.K.; Funding Acquisition, T.M.K.
Declaration of Interests
The authors declare no competing interests.
Contributor Information
Shih-Chieh Ti, Email: jeffti@hku.hk.
Tarun M. Kapoor, Email: kapoor@rockefeller.edu.
References
- Barnes G., Louie K.A., Botstein D. Yeast proteins associated with microtubules in vitro and in vivo. Mol. Biol. Cell. 1992;3:29–47. doi: 10.1091/mbc.3.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bormuth V., Howard J., Schäffer E. LED illumination for video-enhanced DIC imaging of single microtubules. J. Microsc. 2007;226:1–5. doi: 10.1111/j.1365-2818.2007.01756.x. [DOI] [PubMed] [Google Scholar]
- Davis A., Sage C.R., Wilson L., Farrell K.W. Purification and biochemical characterization of tubulin from the budding yeast Saccharomyces cerevisiae. Biochemistry. 1993;32:8823–8835. doi: 10.1021/bi00085a013. [DOI] [PubMed] [Google Scholar]
- Gell C., Friel C.T., Borgonovo B., Drechsel D.N., Hyman A.A., Howard J. Purification of tubulin from porcine brain. Methods Mol. Biol. 2011;777:15–28. doi: 10.1007/978-1-61779-252-6_2. [DOI] [PubMed] [Google Scholar]
- Gittes F., Mickey B., Nettleton J., Howard J. Flexural rigidity of microtubules and actin filaments measured from thermal fluctuations in shape. J. Cell Biol. 1993;120:923–934. doi: 10.1083/jcb.120.4.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henager S.H., Chu N., Chen Z., Bolduc D., Dempsey D.R., Hwang Y., Wells J., Cole P.A. Enzyme-catalyzed expressed protein ligation. Nat. Methods. 2016;13:925–927. doi: 10.1038/nmeth.4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horio T., Hotani H. Visualization of the dynamic instability of individual microtubules by dark-field microscopy. Nature. 1986;321:605–607. doi: 10.1038/321605a0. [DOI] [PubMed] [Google Scholar]
- Kapust R.B., Tözsér J., Fox J.D., Anderson D.E., Cherry S., Copeland T.D., Waugh D.S. Tobacco etch virus protease: mechanism of autolysis and rational design of stable mutants with wild-type catalytic proficiency. Protein Eng. 2001;14:993–1000. doi: 10.1093/protein/14.12.993. [DOI] [PubMed] [Google Scholar]
- Ludueña R.F., Banerjee A. The Isotypes of Tubulin. In: Fojo T., editor. The Role of Microtubules in Cell Biology, Neurobiology, and Oncology. Humana Press; 2008. pp. 123–175. [Google Scholar]
- Mahamdeh M., Simmert S., Luchniak A., Schäffer E., Howard J. Label-free high-speed wide-field imaging of single microtubules using interference reflection microscopy. J. Microsc. 2018;272:60–66. doi: 10.1111/jmi.12744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandelkow E.M., Mandelkow E. Unstained microtubules studied by cryo-electron microscopy. Substructure, supertwist and disassembly. J. Mol. Biol. 1985;181:123–135. doi: 10.1016/0022-2836(85)90330-4. [DOI] [PubMed] [Google Scholar]
- Minoura I., Hachikubo Y., Yamakita Y., Takazaki H., Ayukawa R., Uchimura S., Muto E. Overexpression, purification, and functional analysis of recombinant human tubulin dimer. FEBS Lett. 2013;587:3450–3455. doi: 10.1016/j.febslet.2013.08.032. [DOI] [PubMed] [Google Scholar]
- Olmsted J.B., Borisy G.G. Ionic and nucleotide requirements for microtubule polymerization in vitro. Biochemistry. 1975;14:2996–3005. doi: 10.1021/bi00684a032. [DOI] [PubMed] [Google Scholar]
- Pamula M.C., Ti S.C., Kapoor T.M. The structured core of human β tubulin confers isotype-specific polymerization properties. J. Cell Biol. 2016;213:425–433. doi: 10.1083/jcb.201603050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano K., Maeda K., Oki M., Maéda Y. Enhancement of protein expression in insect cells by a lobster tropomyosin cDNA leader sequence. FEBS Lett. 2002;532:143–146. doi: 10.1016/s0014-5793(02)03659-1. [DOI] [PubMed] [Google Scholar]
- Schiff P.B., Fant J., Horwitz S.B. Promotion of microtubule assembly in vitro by taxol. Nature. 1979;277:665–667. doi: 10.1038/277665a0. [DOI] [PubMed] [Google Scholar]
- Schmidt T.G., Skerra A. The Strep-tag system for one-step purification and high-affinity detection or capturing of proteins. Nat. Protoc. 2007;2:1528–1535. doi: 10.1038/nprot.2007.209. [DOI] [PubMed] [Google Scholar]
- Ti S.C., Pamula M.C., Howes S.C., Duellberg C., Cade N.I., Kleiner R.E., Forth S., Surrey T., Nogales E., Kapoor T.M. Mutations in Human Tubulin Proximal to the Kinesin-Binding Site Alter Dynamic Instability at Microtubule Plus- and Minus-Ends. Dev. Cell. 2016;37:72–84. doi: 10.1016/j.devcel.2016.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ti S.C., Alushin G.M., Kapoor T.M. Human beta-Tubulin Isotypes Can Regulate Microtubule Protofilament Number and Stability. Dev Cell. 2018;47:175–190.e5. doi: 10.1016/j.devcel.2018.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vemu A., Atherton J., Spector J.O., Szyk A., Moores C.A., Roll-Mecak A. Structure and Dynamics of Single-isoform Recombinant Neuronal Human Tubulin. J. Biol. Chem. 2016;291:12907–12915. doi: 10.1074/jbc.C116.731133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker R.A., O’Brien E.T., Pryer N.K., Soboeiro M.F., Voter W.A., Erickson H.P., Salmon E.D. Dynamic instability of individual microtubules analyzed by video light microscopy: rate constants and transition frequencies. J. Cell Biol. 1988;107:1437–1448. doi: 10.1083/jcb.107.4.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widlund P.O., Podolski M., Reber S., Alper J., Storch M., Hyman A.A., Howard J., Drechsel D.N. One-step purification of assembly-competent tubulin from diverse eukaryotic sources. Mol. Biol. Cell. 2012;23:4393–4401. doi: 10.1091/mbc.E12-06-0444. [DOI] [PMC free article] [PubMed] [Google Scholar]