

SUMMARY
Homologous recombination (HR) helps maintain genome integrity, and HR defects give rise to disease, especially cancer. During HR, damaged DNA must be aligned with an undamaged template through a process referred to as the homology search. Despite decades of study, key aspects of this search remain undefined. Here, we use single-molecule imaging to demonstrate that Rad54, a conserved Snf2-like protein found in all eukaryotes, switches the search from the diffusion-based pathways characteristic of the basal HR machinery to an active process in which DNA sequences are aligned via an ATP-dependent molecular motor-driven mechanism. We further demonstrate that Rad54 disrupts the donor template strands, enabling the search to take place within a migrating DNA bubble-like structure that is bound by replication protein A (RPA). Our results reveal that Rad54, working together with RPA, fundamentally alters how DNA sequences are aligned during HR.
Graphical Abstract
In Brief
Rad54 promotes an active mechanism for homology searching during homologous recombination.
INTRODUCTION
Homologous recombination (HR) is a major pathway for repairing DNA double-strand breaks (DSBs), single-stranded DNA (ssDNA) gaps, and stalled or collapsed replication forks (Kowalczykowski, 2015; Mehta and Haber, 2014; San Filippo et al., 2008). HR also provides an alternative pathway for telomere maintenance and ensures proper chromosome segregation during meiosis (Heyer et al., 2010; San Filippo et al., 2008). Aberrant HR underlies chromosomal rearrangements often associated with cancer, cancer-prone syndromes, and other genetic diseases (Heyer et al., 2010; San Filippo et al., 2008).
DSB repair in S. cerevisiae is an important paradigm for studying HR (Kowalczykowski, 2015; Mehta and Haber, 2014; Symington et al., 2014). During HR repair, the DNA ends at the break are processed by 5′→3′ strand resection, yielding 3′ ssDNA overhangs that become coated by the heterotrimeric ssDNA-binding protein replication protein A (RPA) (Chen and Wold, 2014; Kowalczykowski, 2015). RPA is replaced by Rad51 to form the presynaptic complex (PSC) (Heyer et al., 2010; Kowalczykowski, 2015; Mehta and Haber, 2014; San Filippo et al., 2008; Symington et al., 2014). Rad51 is a member of the Rad51/RecA family of DNA recombinases, which form extended helical filaments on ssDNA (Kowalczykowski, 2015). These filaments catalyze strand invasion, in which presynaptic ssDNA is paired with a homologous double-stranded DNA (dsDNA) donor template to form a D-loop intermediate that can be shunted through a number of pathways, including DSB repair (DSBR), synthesis-dependent strand annealing (SDSA), and break-induced replication (BIR) (Kowalczykowski, 2015; Mehta and Haber, 2014; Symington et al., 2014).
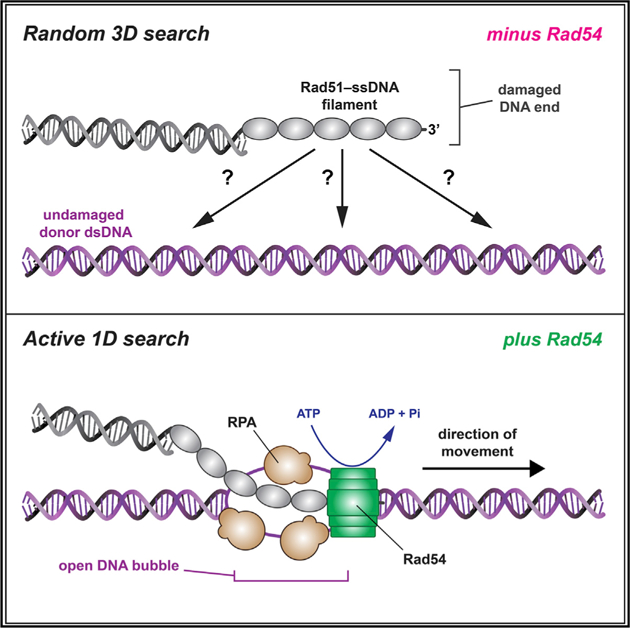
A central feature of HR is that the damaged DNA must be aligned with a homologous donor dsDNA that can be used as a template to guide repair (Haber, 2018). This alignment process is referred to as the “homology search” and is conceptually similar to target searches conducted by all other site-specific DNA-binding proteins (Hager et al., 2009; von Hippel and Berg, 1989). The importance of the homology search can be intuitively understood by recognizing that DNA sequence misalignment will cause loss of genetic information and can give rise to gross chromosomal rearrangements. A major challenge to establishing a comprehensive understanding of the homology search is the transient and highly dynamic nature of the relevant search intermediates (Barzel and Kupiec, 2008; Greene, 2016; Haber, 2018; Renkawitz et al., 2014). An emerging picture of the principles that underlie the homology search includes a combination of 3D intersegmental transfer, short-distance 1D diffusion, and length-dependent homology recognition (Adzuma, 1998; Forget and Kowalczykowski, 2012; Qi et al., 2015; Ragunathan et al., 2012). However, these mechanisms are only informative of the basal properties of Rad51/RecA recombinases and do not account for contributions of accessory factors. So, despite recent advances, we still do not have a firm grasp of the molecular mechanisms that contribute to DNA sequence alignment during HR (Barzel and Kupiec, 2008; Greene, 2016; Haber, 2018; Renkawitz et al., 2014).
Rad54, a member of Swi2/Snf2 family of DNA motor proteins, is a crucial HR factor that promotes strand invasion, catalyzes nucleosome remodeling, and removes Rad51 from dsDNA (Alexeev et al., 2003; Amitani et al., 2006; Ceballos and Heyer, 2011; Jaskelioff et al., 2003; Petukhova et al., 1998; Ristic et al., 2001; Wright and Heyer, 2014; Zhang et al., 2007). The importance of Rad54 is underscored by the sensitivity of S. cerevisiae rad54Δ strains and mouse and chicken DT40 RAD54−/− cells to DNA damage and by the association of human RAD54 mutations with breast cancer, colon cancer, lymphoma, and other cancers (cBioPortal for Cancer Genomics; Bezzubova et al., 1997; Cerami et al., 2012; Essers et al., 1997; Gao et al., 2013). Several lines of evidence implicate Rad54 as a participant in the homology search. First, the dsDNA-dependent ATP hydrolysis activity of Rad54 is stimulated by Rad51 ssDNA, and second, Rad54 renders dsDNA sensitive to the ssDNA-specific nuclease P1, and this activity is also stimulated by Rad51 ssDNA (Mazin et al., 2000a; Van Komen et al., 2000); both effects are independent of DNA sequence homology, implying that Rad54 acts prior to homology recognition. Third, the early stages of HR coincide with a Rad54-dependent increase in chromosome mobility, which may reflect long-range DNA movements during the homology search (Lisby and Rothstein, 2015). Fourth, Rad54 promotes co-association of a heterologous template with the PSC (Tavares et al., 2019). Finally, deletion of RAD54 disrupts the homology search in vivo, as evidenced by a reduced ability of the Rad51 PSC to scan chromosomes for homology in budding yeast strains that lack a homologous donor (Renkawitz et al., 2013). However, there is as yet no definitive description of the potential mechanistic role(s) of Rad54 in the homology search.
To address the role of Rad54 in the homology search, we developed a DNA curtain assay for visualizing search intermediates at the single-molecule level. Our results show that Rad54 reduces the dimensionality of the search by acting as a molecular motor to drive rapid ATP-dependent translocation of the PSC along the donor dsDNA. We show that the motor activity of Rad54 is coupled to transient perturbation of the donor dsDNA, consistent with formation of a bubble or underwound structure, which enables the ssDNA-binding protein RPA to track with the PSC during the homology search. Furthermore, we show that RPA dramatically affects homology recognition efficiency and enables simultaneous surveillance of both strands of the donor dsDNA. Altogether, our findings reveal that Rad54 fundamentally alters the homology search mechanism from a passive diffusion-based mechanism characteristic of the basal HR machinery to an active ATP-dependent molecular motor-based mechanism in which the PSC actively scans duplex DNA for potential sequence homology.
RESULTS
Rad54 Drives PSC Translocation along Donor DNA
We developed a DNA curtain assay for visualizing the homology search using total internal reflection fluorescence (TIRF) microscopy (Figure 1A). We tested unlabeled S. cerevisiae Rad54 or GFP-tagged Rad54, which retains ATPase activity, binds tightly to the PSC (Crickard et al., 2018), is targeted to DNA repair foci in vivo, and complements a rad54Δ strain (Lisby et al., 2004). Rad54 greatly stimulates formation of D-loop products by Rad51 (Mazin et al., 2000a; Petukhova et al., 1998, 1999). Biochemical D-loop assays using an Atto647N-labeled tailed duplex DNA fragment containing a 21-nt ssDNA 3′ overhang and 56 bp of dsDNA for assembly of the PSC (Figure 1B) showed that GFP-Rad54 strongly stimulated the D-loop reaction (Figure S1A). Biochemical assays also confirmed that the ATPase activity of GFP-Rad54 is stimulated by Rad51 ssDNA (Figures S1B–S1D). These results provide assurance that GFP-Rad54 possesses the biochemical functions of the untagged protein.
Figure 1. Rad54 Acts as a Molecular Motor during the Homology Search.
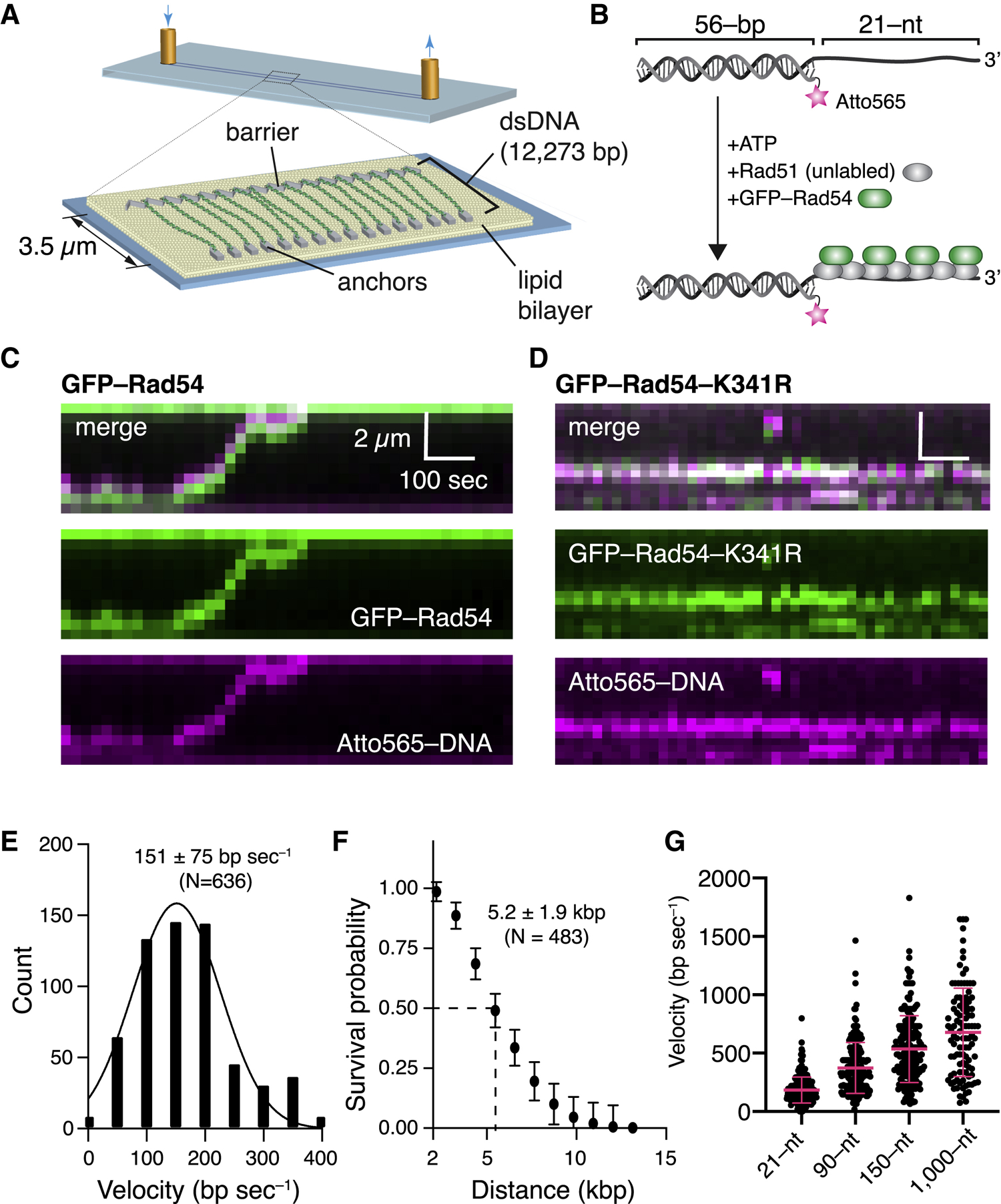
(A) DNA curtain assay.
(B) PSC preparation.
(C) Kymograph showing a PSC containing Rad51,GFP-Rad54 (green), and Atto565-DNA (magenta) translocating on the donor dsDNA.
(D) Kymograph illustrating the behavior of a GFP-Rad54-K341R PSC.
(E) Distribution of PSC translocation velocities(represents combined datasets for Rad54 and GFP-Rad54); the solid line is a Gaussian fit to the data.
(F) Linear translocation distance graphed as survival probability; error bars (SD) were generated by bootstrapping (represents combined datasets for Rad54 and GFP-Rad54).
(G) Distribution of PSC translocation velocities forthe tailed duplex DNA (21-nt ssDNA) and 90-, 150-, and 1,000-nt ssDNAs. Red lines represent mean and SD. In each case, the ssDNA is fully homologous to a specific target site on the donor dsDNA (Figure 2).
To visualize the homology search, we generated a CURMID (curtains plasmid; 12,273 kb, ~4.2 μm) substrate comprised of a pUC19 plasmid with an added 9.5-kb fragment of λ-phage DNA (Figure 1A). For DNA curtain assays, the linearized CURMID was labeled at one end with biotin and at the other end with digoxigenin (DIG). The labeled DNA was then aligned and anchored on the sample chamber surface (Greene et al., 2010). PSCs prepared with an Atto565-labeled DNA (Figure 1B) were monitored as they interacted with donor dsDNA (Figure 1C); this Atto565-labeled DNA is identical to that used in Figure S1B and is homologous to a specific site in the anchored CURMID DNA (see below). Unless stated otherwise, all subsequent assays with the 21-nt tailed duplex correspond to this substrate. These assays revealed extensive Rad51-dependent co-localization of GFP-Rad54 and the Atto565-labeled DNA on the donor dsDNA (93% co-localization, N = 196; Figure S1E). Control reactions lacking Rad51 or GFP-Rad54 exhibited little or no binding of the PSCs to the donor dsDNA (Figure S1F), consistent with the stringent requirement for Rad54 in bulk biochemical assays for D-loop formation with S. cerevisiae Rad51 (Ceballos and Heyer, 2011; Mazin et al., 2000a; Petukhova et al., 1998, 1999).
Remarkably, the PSCs were not stationary but instead underwent extensive 1D translocation along the donor dsDNA (Figure 1C). No evidence of translocation was observed for the ATP hydrolysis-deficient Rad54-K341R mutant (Figure 1D). Analysis of the translocation data yielded an average PSC velocity of 151 ± 75 bp s−1 (N = 636; Figure 1E) and an average processivity of 5.2 ± 1.9 kb (N = 483; Figure 1F); comparable results were obtained with either GFP-Rad54 or unlabeled Rad54 (Figure S2A). Translocation velocity increased to 302 ± 172 (N = 187), 477 ± 266 (N = 202), and 592 ± 421 (N = 105) for PSCs prepared with longer 90-, 150-, and 1,000-nt ssDNA substrates, respectively (Figures 1G and S2B–S2D). The increased velocity observed for substrates with longer lengths did not coincide with an increased rate of ATP hydrolysis by Rad54 in bulk biochemical assays (Figure S2E), suggesting that the longer PSCs may somehow allow Rad54 to more efficiently couple ATP hydrolysis to translocation along the donor dsDNA. Importantly, Rad51 alone does not allow 1D movement of the presynaptic ssDNA relative to the donor dsDNA (Lee et al., 2015; Qi et al., 2015), suggesting that the observed motion was driven by Rad54. This conclusion is consistent with previous studies demonstrating that Rad54 and its paralog Rdh54 are ATP-dependent dsDNA motor proteins (Amitani et al., 2006; Nimonkar et al., 2007; Petukhova et al., 2000; Prasad et al., 2007). Our findings show that Rad54 co-localizes with the PSC and promotes rapid ATP-dependent translocation of the PSC along the donor dsDNA.
Homology Recognition by the PSC
If the 1D movement represented behavior of bona fide search intermediates, then the PSCs should become arrested upon encountering homology in the donor dsDNA molecule. We tested this prediction using substrates that were homologous to one of two different regions on the donor dsDNA (Figure 2A). TIRF microscopy images and binding distribution histograms revealed PSC accumulation at the target sites on the donor dsDNA (Figures 2B, 2C, and S3A–S3D) whereas PSCs prepared with Rad54-K341R did not (Figure 2D). These results suggested that PSC translocation leads to homology recognition.
Figure 2. Target Recognition during the Homology Search.
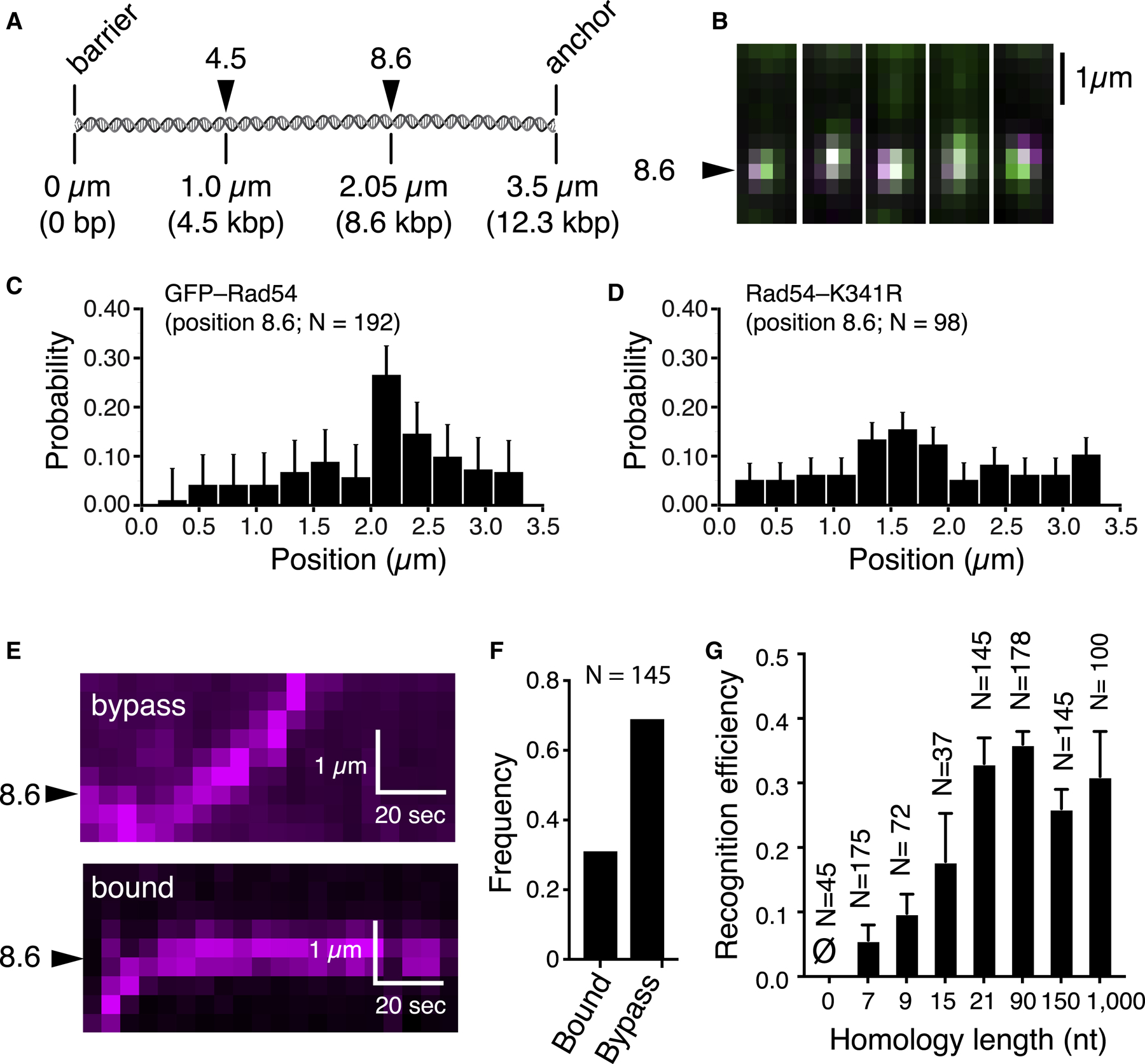
(A) Donor dsDNA schematic highlighting the locations of two different homologous targets.
(B) Images of PSCs bound at target 8.6.
(C) Binding distribution for PSCs containing Atto565-DNA homologous to target 8.6; error bars for all binding distributions (SD) were generated by bootstrapping.
(D) Distribution of PSCs containing GFP-Rad54-K341R and Atto565-DNA.
(E) Kymographs showing target recognition and target bypass.
(F) Homology recognition and bypass for PSCs with 21 nt of homology.
(G) Homology recognition efficiency for different lengths of homology; error bars represent SD of three independent experiments. Homology lengths of 0, 7, 9, 15, and 21 all correspond to the tailed duplex substrate with a 21-nt ssDNA overhang. The 90-, 150-, and 1,000-nt substrates were ssDNA molecules fully homologous to the 8.6 target site on the donor dsDNA.
An important question concerns the efficiency by which the PSC can recognize homology. To address this issue, we determined how frequently the translocating PSC halts its movement during the first encounter with the homologous target (Figures 2E and 2F). These experiments revealed that first-passage recognition efficiency scaled in proportion to the length of homology (Figures 2G and S3E). For PSCs bearing a fully complementary 21-nt ssDNA overhang, 33% ± 4.0% of the initial encounters with the homologous target resulted in recognition (Figure 2E–2G). PSCs prepared with longer 90-, 150-, or 1,000-nt ssDNA substrates yielded first-passage recognition efficiencies of 36% ± 2%, 26% ± 3%, and 31% ± 7%, respectively (Figure 2G). Recognition efficiency decreased for shorter lengths of homology, yielding values of 17.8% ± 7.5% and 9.8% ± 3.0% for 21-nt overhangs bearing either 15 or 9 nt of homology, respectively (Figure 2G). Recognition efficiency dropped to 5.6% ± 2.4% when the homology length was reduced to 7 nt (Figure 2G), and analysis of the corresponding binding distribution following 10-min incubation revealed overall poor target enrichment for this substrate (Figure S3E). PSCs prepared using a non-homologous 21-nt 3′ ssDNA overhang did not recognize the target site in the DNA curtain assay (Figure 2G) and failed to form D-loop products in the biochemical assay (Figure S4A and S4B). Even though we did not detect D-loop formation for substrates with 9–15 nt of homology in biochemical assays (Figures S4A and S4B), we could visualize target-bound PSCs in the DNA curtain analysis with these substrates (Figure S3E). We attribute this difference to the fact that the bulk reaction products must be deproteinized prior to gel electrophoresis, which likely disrupts less stable intermediates. In contrast, the DNA curtain assay detects intermediates in their native protein-bound states.
Sudden Reversals during the Homology Search
Rad54 and other Snf2 family proteins are thought to track in the 3′→5′ direction relative to the orientation of the DNA strand bound within their motor domain (Cairns, 2007). Rad54, its paralog Rdh54, and the chromatin remodeling complexes RSC and SWI/SNF can also undergo sudden changes in direction of movement (Amitani et al., 2006; Lia et al., 2006; Nimonkar et al., 2007; Prasad et al., 2007; Zhang et al., 2006). DNA translocation reversal may involve a switch from one strand of the donor dsDNA to the other so that the Snf2 motor-defined 3′→5′ directionality is maintained (Cairns, 2007). This mechanism may be comparable with the strand switching that takes place during DNA unwinding and re-zipping by the bacterial helicase UvrD (Comstock et al., 2015). Alternatively, reversal events may reflect the alternating actions of Rad54 complexes bound within the same PSC but organized in opposing orientations. We find that PSCs translocation typically occurs without reversal, accounting for 65% of all observed events (Figures 3A and 3B). However, the remaining PSCs exhibit one or more reversals (Figures 3A and 3B). One implication of this observation is that if the PSC fails to recognize homology during its first passage, then it might reverse direction and recognize homology during a repeated survey of the donor dsDNA.
Figure 3. Homology Recognition and Translocation Direction.
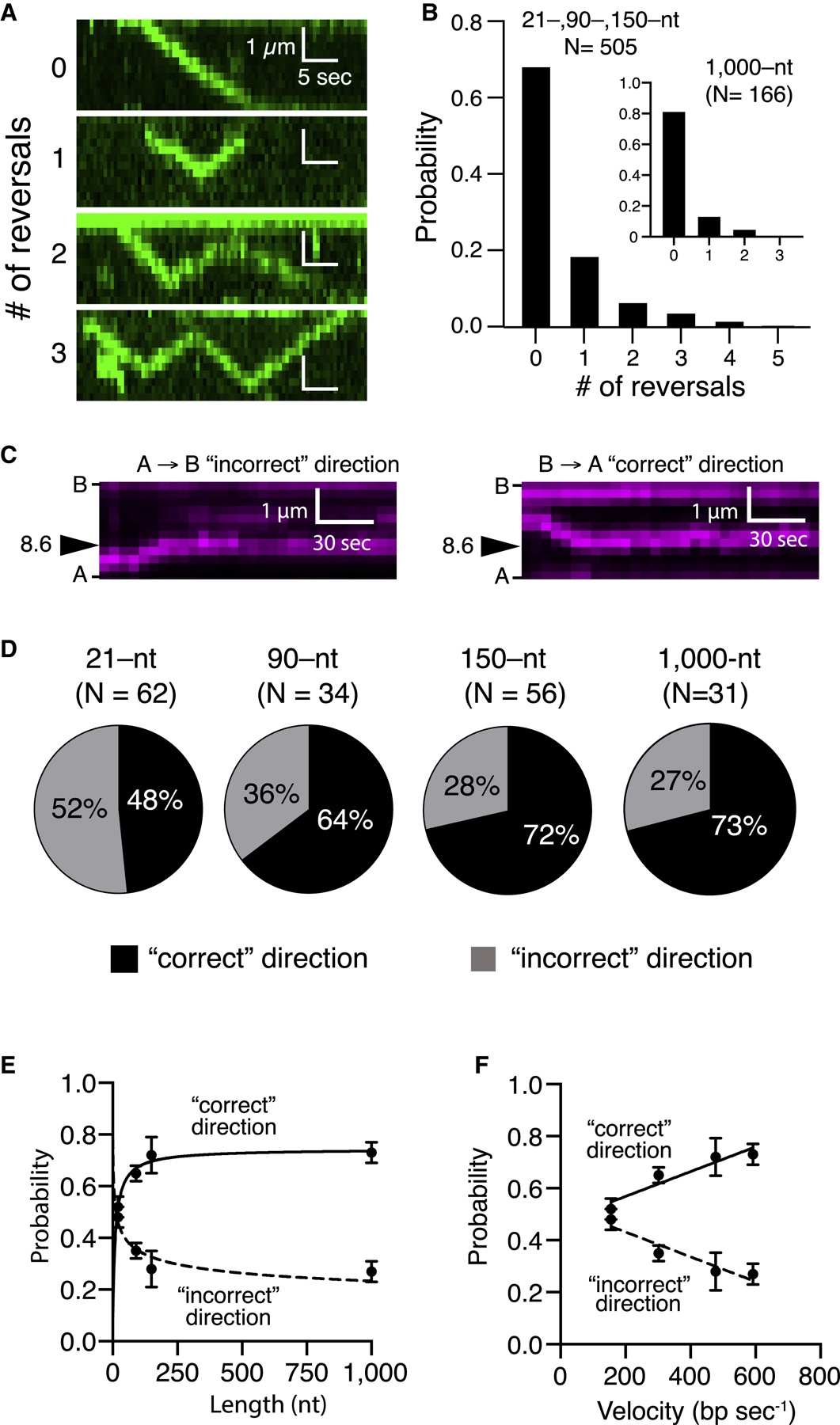
(A) Kymographs illustrating examples of varying numbers of reversal events by the PSC (unlabeled Rad51, GFP-Rad54 [green], and unlabeled 150-nt ssDNA).
(B) Frequency of observed translocation reversalevents; the main panel corresponds to pooled datasets for the tailed duplex (21-nt ssDNA), 90 nt and 150 nt, which were all similar in reversal characteristics, and the inset corresponds to the 1,000-nt ssDNA substrate.
(C) Kymographs illustrating examples of homologoustarget recognition for translocation events occurring in either direction. B, barrier; A, anchor (Figure 1A; unlabeled Rad51, unlabeled Rad54, and Atto565-DNA [magenta]).
(D) Relative fraction of first-passage recognition events occurring for PSC translocation in either direction for different-length PSC substrates.
(E and F) Relative fraction of first-passage recognition events for either direction as a function of (E) PSC ssDNA length or (F) translocation velocity. The error bars represent the SD for three independent experiments. (E) and (F) represent different presentations and analyses of the same experimental dataset.
Homology Recognition and Strand Polarity
The presynaptic ssDNA is homologous to only one of the two strands in the donor duplex. This asymmetry raises the question of whether the PSC displays a directional bias with respect to homology recognition. If the PSC can scan just one of the two donor dsDNA strands, then there should be a strong directional bias. In assays with the tailed duplex DNA, the homologous strand within the tethered donor dsDNA corresponds to the 5′ DIG-labeled strand being anchored to the pedestal. Assuming a directional bias model, one would predict that PSCs scanning in the direction from the barrier (B) to the anchors (As) should recognize the homologous target (for brevity, we designate this the B→A or “correct” direction; Figures 3C and 3D) whereas PSCs scanning in the opposite direction (the A→B or “incorrect” direction) should not. In contrast, if both donor DNA strands are sampled during the homology search regardless of strand polarity, then there should not be a directional preference. Remarkably, our data revealed that PSCs prepared with the tailed duplex could efficiently identify the homologous target regardless of direction of translocation (Figures 3C and 3D). PSCs prepared with the 90-, 150-, and 1,000-nt ssDNA substrates also recognized homology when approaching from either direction but showed reduced efficiency when approaching from the incorrect direction (Figure 3D). Interestingly, inspection of all three datasets revealed an inverse relationship between velocity and the probability of recognizing homology when approaching in the incorrect orientation (Figures 3E and 3F). These results suggest that the PSC can simultaneously scan both donor dsDNA strands while searching for homology, although the ability to recognize homology when approaching from the incorrect direction may be compromised at higher velocities.
ATP Hydrolysis Requirements for the Homology Search
PSCs prepared with Rad54-K341R exhibited no evidence of translocation activity (Figure 1D). Consistent with this result, these PSCs failed to accumulate at the homologous target (Figure 2D) and did not promote D-loop formation (Figures S4C and S4D). Similarly, no D-loop product was detected in control reactions with the non-hydrolyzable ATP analog AMP-PNP (Figures S4C and S4D). In contrast, D-loop products were readily observed with Rad54 and the Rad51-K191R mutant (Sung and Stratton, 1996), which binds ATP but is strongly attenuated for ATP hydrolytic activity (Figures S4C and S4D). PCSs comprised of Rad54 plus Rad51-K191R were also proficient for translocation and homology recognition (data not shown), consistent with the finding that Rad54 overexpression can suppress the ionizing radiation (IR) sensitivity of cells expressing Rad51-K191R (Fung et al., 2006). No translocation or homology recognition was observed in assays with Rad54 and Rad51 when ATP was replaced with AMP-PNP (see below). We conclude that the ATPase activity of Rad54 is essential for the homology search, whereas ATP binding by Rad51 alone is sufficient to sustain a robust search process. The inability of Rad54-K341R to support the homology search may explain why cells expressing this mutant display HR-related defects in yeast and mammals (Agarwal et al., 2011; Petukhova et al., 1999).
DNA and Protein Composition of the PSC
We conducted photobleaching measurements to count the number of Atto565-DNA molecules and GFP-Rad54 molecules within the PSCs. Atto565-DNA displayed well-defined single-step photobleaching events (Figures S5A and S5B), revealing an average of 1.9 ± 1.4 molecules of Atto565-DNA per PSC (N = 30; Figure S5C). The finding that PSCs contained supernumerary Atto565-DNA is reminiscent of in vivo DNA repair centers that harbor multiple DSB ends (Lisby and Rothstein, 2015) and is also consistent with the hypothesis that the two DSB ends may travel together during the homology search (Haber, 2018).
We did not detect single-step bleaching events for GFP-Rad54; instead, the photobleaching traces yielded a gradual decline in fluorescence (Figure S5A) commensurate with limitations of these measurements for counting molecules in large complexes. To overcome this challenge, we examined PSCs prepared with a 1 part in 6 mixture of GFP-Rad54 to unlabeled Rad54 and assumed that each photobleaching step reflects ~6 molecules of Rad54 (Figure S5D). Given this assumption, photobleaching assays with the tailed duplex revealed 2.2 ± 0.94 photobleaching steps corresponding to 13.2 ± 5.64 molecules of GFP-Rad54 (Figure S5E). Surprisingly, the 90- and 150-nt ssDNA substrates contained comparable amounts of Rad54 even though these longer PSCs traveled at much faster velocities (cf. Figures 1G and S5E). Cumulative analysis of all substrates (up to 150 nt in length) yielded an average of 2.2 ± 0.8 photobleaching steps corresponding to 13.2 ± 4.8 molecules of GFP-Rad54 (Figure S5E). These findings suggest that the number of Rad54 molecules bound to the PSC does not scale linearly with ssDNA length but may instead be related to the number of ssDNA and/or Rad51 filament ends. Interestingly, the 1,000-nt substrate had more bound GFP-Rad54, yielding 3.4 ± 1.47 photobleaching steps corresponding to 20.4 ± 8.82 molecules of GFP-Rad54 (Figure S5E); 23% of the 1,000-nt PSCs were bound by too much GFP-Rad54 to reliably discern individual photobleaching steps, which is consistent with the notion that these longer PSCs may be able to accommodate multiple higher-order Rad54 complexes.
If a single Rad54 monomer was responsible for driving translocation, then a mixture of Rad54 and Rad54-K341R would change the ratio of active to inactive complexes but should not affect translocation velocity. However, if translocation required the coordinated action of multiple Rad54 monomers, then the velocity of the PSCs should decrease in proportion with the concentration of Rad54-K341R. To distinguish between these possibilities, we conducted experiments with varying ratios of Rad54 and Rad54-K341R; the GFP-tagged variant of Rad54-K341R was used to visually confirm that the mutant protein was present in the PSCs. Remarkably, the presence of substoichiometric amounts of Rad54-K341R reduced the fraction of translocating PSCs (Figure S5F) and also caused a substantial reduction in translocation velocity (Figure S5G). Indeed, a 4:1 or 1:1 ratio of Rad54 to Rad54-K341R caused a 54% or 93% reduction in translocation velocity, respectively (Figure S5G). These results suggest that multiple Rad54 monomers within a higher-order complex contribute to PSC translocation.
RPA Tracks with the PSC during the Homology Search
All of the experiments described above contained RPA; moreover, we found that its presence is crucial for efficient product formation in the D-loop reaction (Figures S6A and S6B). To assess the behavior of RPA in these reactions, we performed homology search experiments while monitoring the fate of GFP-tagged RPA. Remarkably, RPA-GFP co-localized with the PSCs as they scanned the donor dsDNA (Figure 4A). Indeed, 82% of all PSCs (translocating and stationary; N = 157 of 191; Figure 4B) and ~64% of translocating PSCs co-localized with RPA-GFP (N = 59 of 92; Figure 4C). RPA co-localization was reduced to just 15% in reactions with Rad54-K341R (Figure 4B). In addition, the non-hydrolyzable ATP analog AMP-PNP reduced RPA co-localization with the PSC by ~60-fold (Figure 4B) and abolished PSC translocation (Figure 4C) and D-loop formation (Figures S4C and S4D). The requirement for ATP hydrolysis by Rad54 suggests that RPA co-localization with the PSC is coupled to Rad54 motor activity.
Figure 4. RPA Co-localizes with the PSC during the Homology Search.
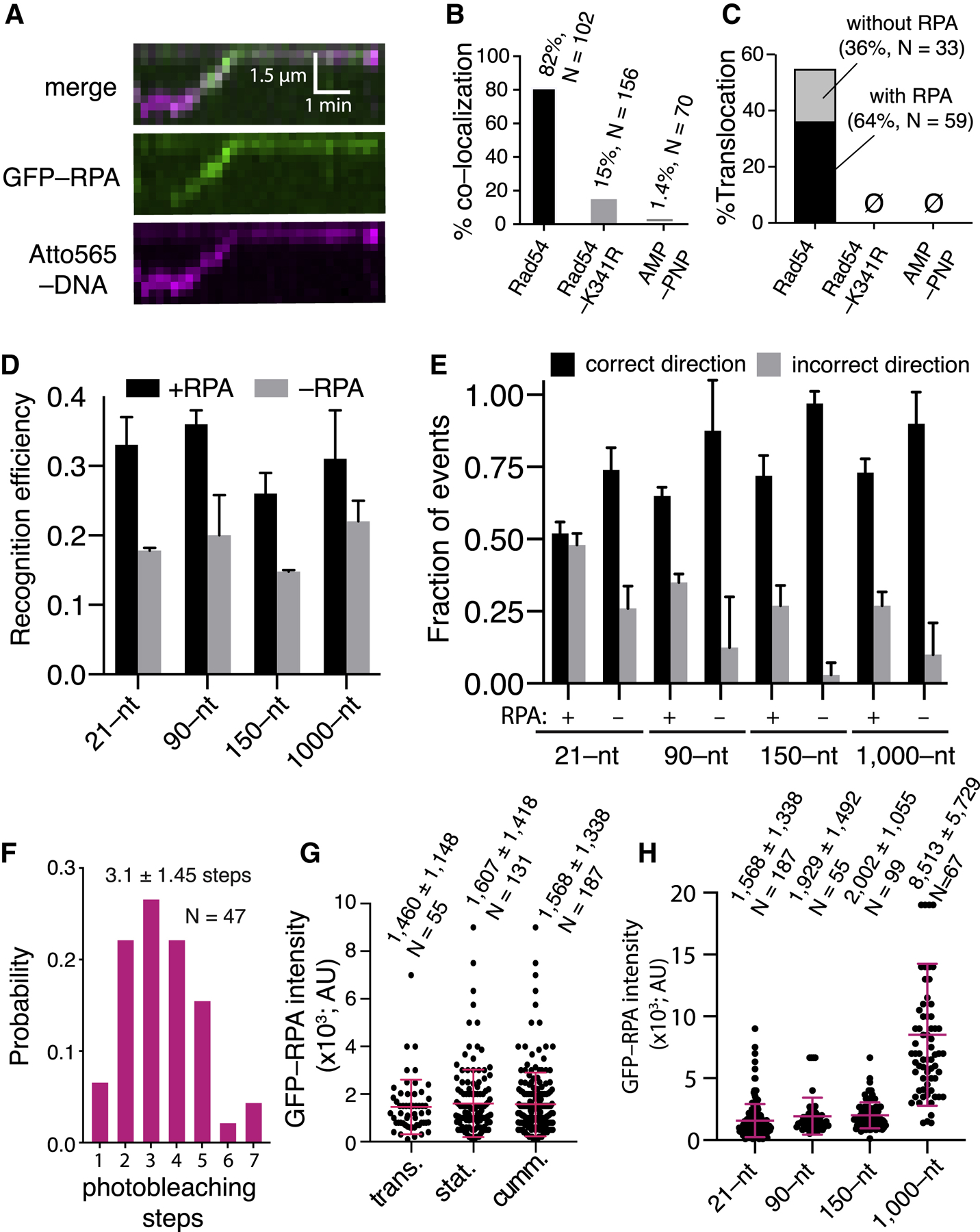
(A) Co-localization of RPA-GFP with the PSC.
(B) Fraction of PSCs (translocating and stationary) that co-localize with RPA.
(C) Fraction of PSCs that undergo translocationwith RPA co-localization.
(D) First-passage recognition efficiency for different-length PSCs in the presence and absence of RPA; this dataset is not segregated for direction of approach. The error bars represent the SD of three independent experiments.
(E) Fraction of first-passage recognition events forPSCs approaching the target site from the correct and incorrect directions for different-length PSCs in the presence and absence of RPA. The error bars represent the SD of three independent experiments.
(F) Photobleaching analysis to count the number ofRPA molecules associated with the PSC.
(G) RPA-GFP signal intensity for translocating andstationary PSCs and cumulative datasets. The red lines represent the mean and SD. (H) RPA-GFP signal intensity of different-length PSCs. The red lines represent the mean and SD.
RPA Promotes Bidirectional Homology Recognition
Interestingly, PSC translocation was not altered when RPA was omitted from the reactions (Figures S6C and S6D). However, there was an ~47% decrease in overall first-passage recognition efficiency in reactions with the 21-nt tailed duplex upon omitting RPA (Figure 4D), commensurate with the reduced efficiency of D-loop formation in biochemical assays without RPA (Figures S6A and S6B). This effect was also observed with the 90-, 150-, and 1,000-nt ssDNA substrates, yielding reductions in first-passage recognition efficiency of ~44%, ~43%, and 31%, respectively, when RPA was omitted (Figure 4D). Remarkably, analysis of the data to consider strand polarity revealed that omission of RPA caused an even more drastic reduction in first-passage recognition when the PSCs approached from the incorrect direction (Figure 4E). With the 21-nt tailed duplex, in reactions without RPA, the fraction of incorrect direction recognition decreased by ~65% compared with recognition from the correct direction (Figure 4E). This effect was exacerbated with the 90-, 150-, and 1,000-nt ssDNA substrates where the fraction of incorrect direction recognition was reduced by ~86%, ~97%, and 90% in the absence of RPA, respectively (Figure 4E). These findings suggest that RPA is responsible for allowing the PSC to concurrently sample both strands of the donor dsDNA for homology regardless of strand polarity.
Quantitation of RPA Interactions with the PSC
Photobleaching measurements revealed that PSCs prepared with the tailed duplex substrate co-localized with 3.2 ± 1.4 molecules of RPA-GFP (Figures 4F and S7A–S7C). Comparison of the RPA-GFP signal indicated that a comparable amount of RPA was present in translocating and stationary PSCs (Figure 4G). Even though most (85%) of the PSCs prepared with Rad54-K341R did not co-localize with RPA (Figure 4B), the remaining 15% that did contained similar levels of RPA-GFP as observed for PSCs with Rad54 (Figure S7D). Interestingly, for PSCs containing Rad54, there was a lag time of 0.96 ± 1.5 min (N = 184) prior to the appearance of the RPA-GFP, but this lag increased ~3-fold in reactions with Rad54-K341R (Figure S7E), suggesting the possibility that these association mechanisms occurred through kinetically distinct processes. We speculate that, with Rad54-K341R, RPA co-localization may have occurred as Rad51 attempted to pair with a heterologous target, resulting in partial displacement of the non-complementary strand. The binding behavior of RPA was dynamic and exhibited a lifetime of 68 ± 7 s while tracking with the PSC (N = 105; Figures S7F and S7G); this dissociation behavior was slow relative to the rate of translocation, indicating that RPA was indeed tracking with the PSCs. Remarkably, a similar amount of RPA-GFP was present for PSCs prepared with each of the different-length PSCs up to 150 nt, suggesting that the RPA content is perhaps related to the amount of Rad54 present in the complexes (Figure 4H). There was, however, more RPA-GFP associated with the longer 1,000-nt substrate (Figure 4H), which we attribute to the greater amount of Rad54 bound to these PSCs.
Rad54 Alone Is Responsible for RPA Co-localization
To rule out the possibility that RPA might simply bind to Rad51 or the Atto565-labeled substrate, we took advantage of the fact that Rad54 alone is able to translocate on dsDNA (Amitani et al., 2006; Ristic et al., 2001). We observed translocation of GFP-Rad54 on dsDNA (Figure 5A), as reported previously (Amitani et al., 2006), revealing a velocity of 96 ± 61 bp s−1 (Figure 5B) and a processivity of 5.0 ± 3.2 kb (Figure 5C); the concentration of GFP-Rad54 had to be increased 5-fold to detect dsDNA binding in the absence of the PSC. The velocity of Rad54 on its own was slower than when it was in complex with the tailed duplex PSC (~100 bp s−1 versus ~150 bp s−1; p = 0.009, Student’s t test) and substantially slower than PSCs prepared with the longer ssDNAs (cf. Figures 1G and 5B). Nevertheless, the translocation parameters for GFP-Rad54 alone were not significantly different in reactions with or without RPA (Figures 5B and 5C). We could detect RPA-mCherry association with the translocating GFP-Rad54, but assays with GFP-Rad54-K341R revealed no such co-localization (Figures 5D and 5E). We conclude that RPA can track with Rad54 alone through a mechanism that requires the ATPase activity of Rad54.
Figure 5. Rad54 Alone Can Open dsDNA Strands during Translocation.
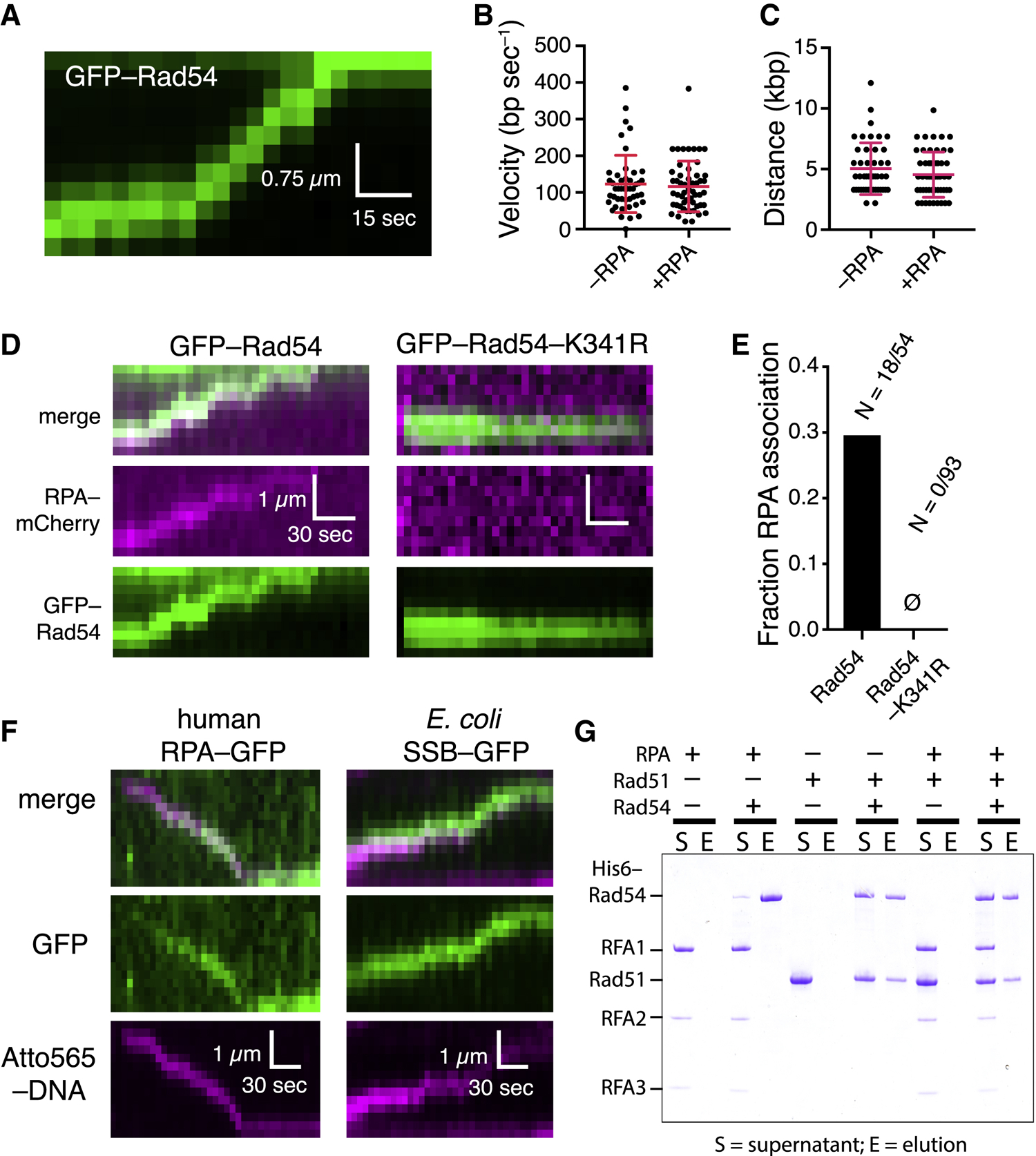
(A) Kymograph showing GFP-Rad54 (green) translocation.
(B) Comparison of translocation velocities of GFP-Rad54 only in the presence and absence of RPA; red lines represent mean and SD.
(C) GFP-Rad54 processivity in the presence andabsence of RPA; red lines represent mean and SD.
(D) Kymographs showing GFP-Rad54 or GFP-Rad54-K341R in the presence of RPA-mCherry.
(E) Fraction of GFP-Rad54 and GFP-Rad54-K341R that co-localize with RPA-mCherry.
(F) Kymographs showing human RPA and E. coli SSB tracking with the translocating S. cerevisiae PSC.
(G) Pull-down assay showing that Rad54 and RPAdo not interact in solution.
Remarkably, human RPA and E. coli SSB could also track with the S. cerevisiae PSC, arguing against the existence of a species-specific protein-protein interaction to allow for RPA co-localization (Figure 5F). Furthermore, we saw no evidence of any protein-protein interaction between RPA and Rad54 in an affinity pull-down assay (Figure 5G). These findings are consistent with published reports showing that RPA and Rad54 do not interact with one another in the yeast 2-hybrid assay (Jiang et al., 1996) and do not co-localize at DSBs in vivo in the absence of Rad51 (Lisby et al., 2004), nor do they interact with one another in vitro (Crickard et al., 2018). These data provide evidence that Rad54 is responsible for RPA co-localization with the PSC and further indicate that co-localization does not involve specific protein contacts.
Nucleosome Remodeling by the PSC
Rad54 belongs to the Swi2/Snf2 family of enzymes capable of remodeling nucleosomes (Alexeev et al., 2003; Alexiadis and Kadonaga, 2002; Ceballos and Heyer, 2011; Zhang et al., 2007). Therefore, we sought to examine the behavior of the PSC upon encountering single nucleosomes on the donor DNA (Figure 6A). In assays using the tailed duplex (21–nt ssDNA) and nucleosomes labeled with Atto565–H2A, the PSCs either stall upon encountering the nucleosomes (6%, N = 7/116, remodel the nucleosomes (51%, N = 59/116) or bypass the nucleosome (43%, N = 50/116; Figures 6B and 6C). Similar results were obtained with nucleosomes labeled with Alexa488–H4 (Figures 6B and 6C). Remodeling events could be subdivided into outcomes that involved nucleosomes sliding (83%, N = 49/59 for reactions with Atto565–H2A; 74%, N = 37/50 for reactions with Alexa488–H4) or events that resulted in complete eviction of the nucleosomes from the donor DNA (17%, N = 10/59 for reactions with Atto565–H2A; 26% for reactions with Alexa488–H4; Figure 6D). As indicated above, a sizable fraction of the nucleosome encounters resulted in the PSC bypassing the nucleosomes (Figure 6B) but with no evident effect on the nucleosomes themselves (Figure 6E), although in some instances the PSC would pause for relatively long periods (49.6 ± sec, N = 14) before bypass occurred (Figures 6E and 6F).
Figure 6. Nucleosome Remodeling and Bypass during the Homology Search.
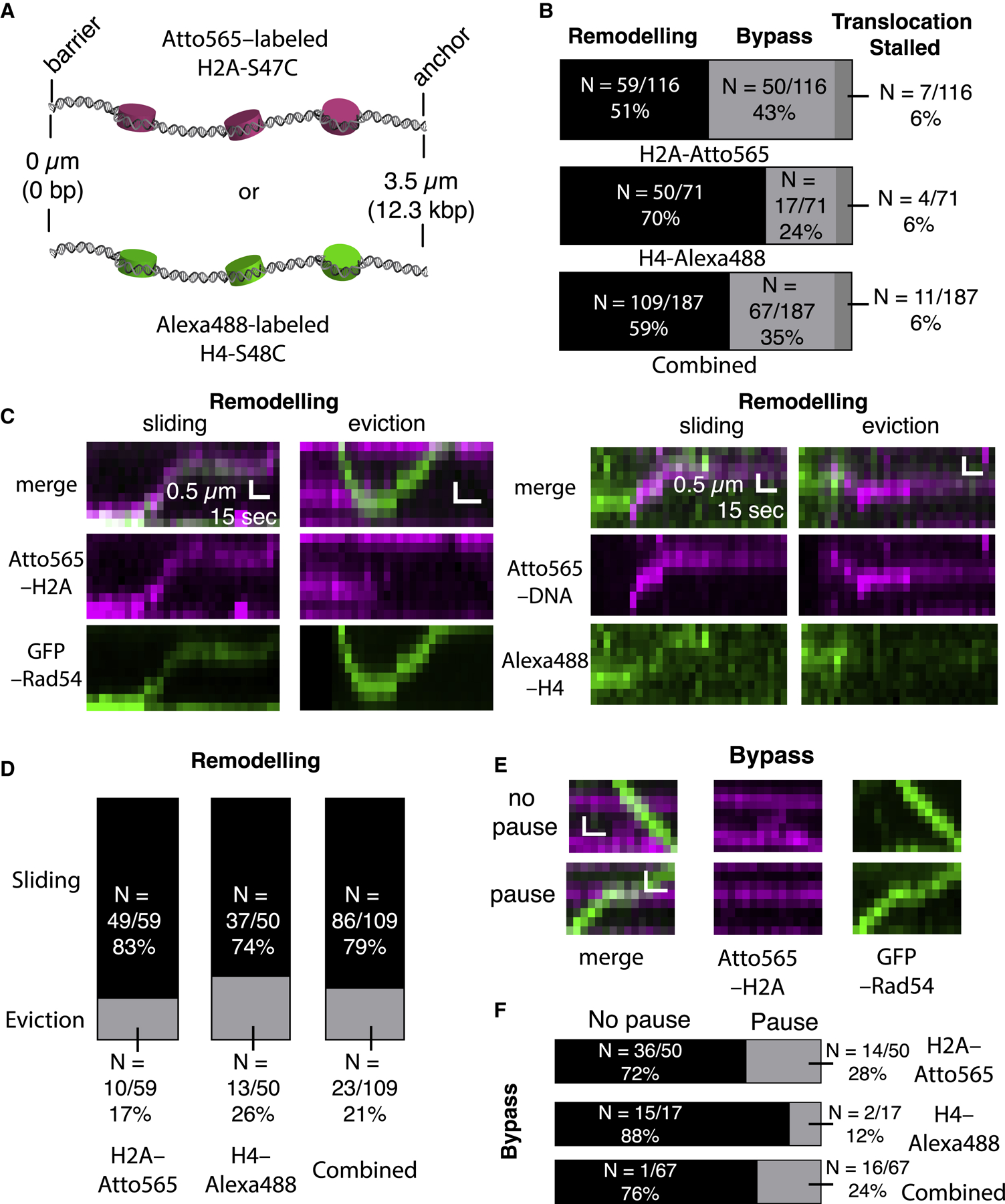
(A) Schematic of nucleosome-bound donor dsDNA substrates labeled with Atto565-H2A or Alexa 488-H4.
(B) Summary of different outcomes during PSC encounters with single nucleosomes for labeled H2A or labeled H4 with PSCs prepared with the tailed duplex (21-nt ssDNA) substrate. The bottom graph shows the combined labeled H2A and labeled H4 datasets.
(C) Kymographs showing examples Atto565-H2A nucleosomes (magenta) being remodeled by PSCs labeled with GFP-Rad54 (green, left panels) and examples of Alexa 488-H4 nucleosomes (green) being remodeled by PSCs labeled with Atto565-labeled tailed duplex DNA (magenta, right panels).
(D) Distributions of nucleosome remodeling events, sliding or eviction, for H2A-Atto565-labeled, H4-Alexa 488-labeled, and combined datasets.
(E) Kymographs depicting nucleosomes (magenta) being bypassed by translocating PSCs (green) with or without evident PSC pausing at the nucleosome.
(F) Fraction of bypass events where the PSCs pause during nucleosome bypass for nucleosomes labeled with H2A-Atto565, H4-Alexa 488, and combined datasets.
DISCUSSION
Our work establishes that Rad54 changes the homology search from the diffusion-based mechanism characteristic of the basal HR machinery to a motor-guided mechanism. Importantly, Rad54 catalyzes formation of a migrating DNA structure in which the strands are unwound to enhance homology recognition and DNA joint formation. Our findings also reveal new and unexpected roles of RPA in homology recognition; specifically, we show that Rad54 and RPA work together to enable bidirectional homology sampling by the PSC while translocating on the donor DNA. These findings have crucial implications for understanding how DNA sequences are aligned during genetic recombination.
Molecular Recognition during HR
Initial interactions between the PSC and a potential donor dsDNA are governed by contact probability, which reflects the probability that two DNA segments will come into close physical proximity through random diffusion (Haber, 2018; Lee et al., 2016; Zhang and Dudko, 2016). We anticipate that Rad54 would reinforce the effects of contact probability by ensuring that the first dsDNA encountered remains tightly captured and can be thoroughly scanned for homology (Figure 7A). Newly replicated sister chromatids are linked by cohesin during the S and G2 phases of the cell cycle, and homologous sequences may also have a higher probability of being in physical contact, even in other cell cycle stages (AlHaj Abed et al., 2019; Barzel and Kupiec, 2008; Gladyshev and Kleckner, 2014, 2017; Kim et al., 2017; Weiner and Kleckner, 1994). Therefore, under normal circumstances, the homology search may be spatially confined so that the first dsDNA encountered by the PSC will have a greater probability of being the correct donor template.
Figure 7. Model Describing the Influence of Rad54 on the Homology Search Mechanism.
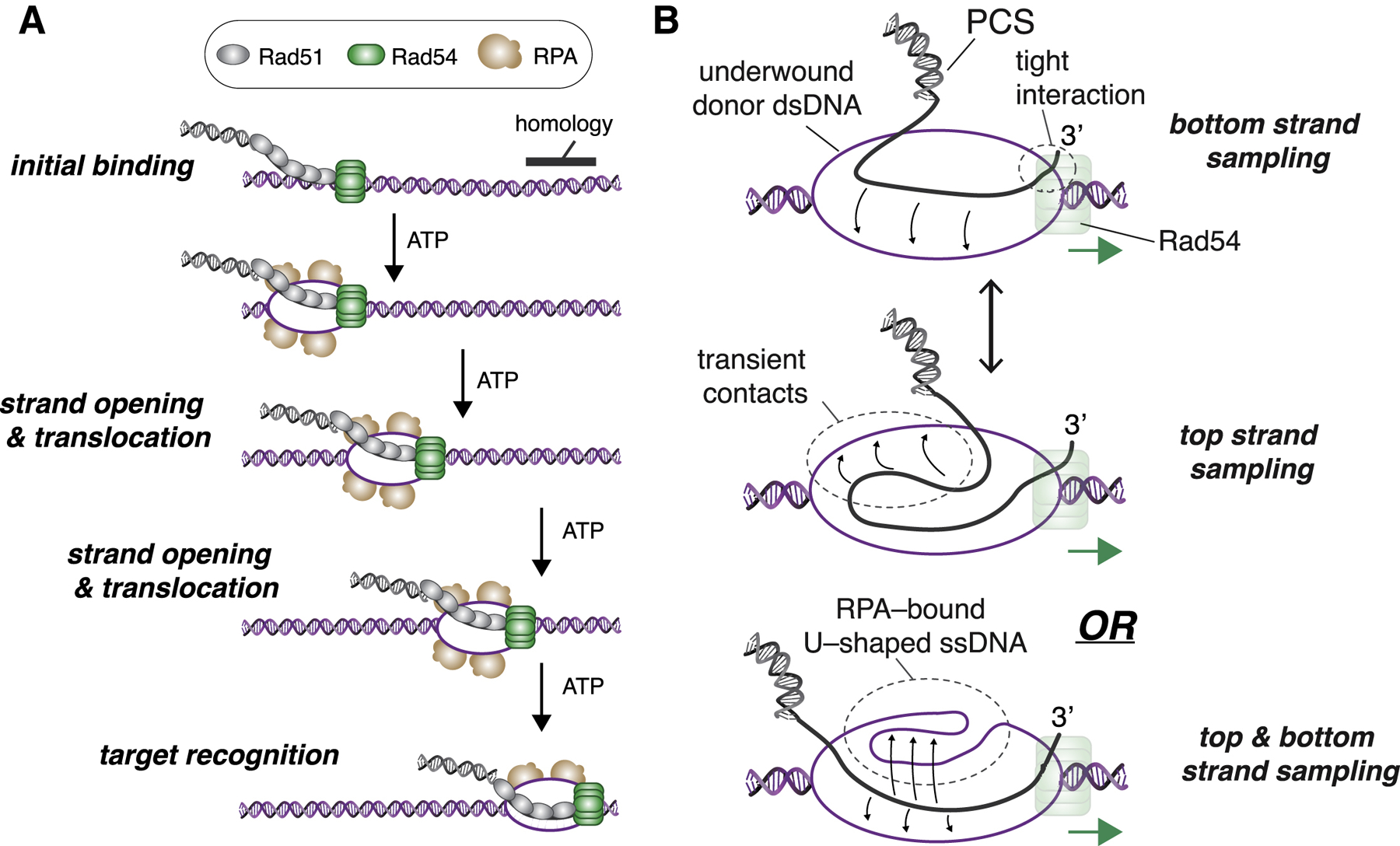
(A) Schematic depiction of the PSC linked to a donor dsDNA template via the binding activity of Rad54, where ATP-dependent forward progression of the complex is coupled to deformation of the DNA duplex, enabling RPA association, which, in turn, promotes homology recognition.
(B) Model depicting rapid sampling of donor DNA by Rad51 ssDNA within the translocating PSC. In the cartoon schematic, Rad51 is not depicted for clarity, and Rad54 is shown bound at or near the 3′ end of the PSC ssDNA; similar principles may apply regardless of where Rad54 is located within the PCS. Additional details are in the main text.
Superimposed on the issue of contact probability is the question of how many physical encounters between two interacting entities are necessary to yield a productive interaction. This molecular recognition problem is shared among all bimolecular interactions but remains difficult to assess because there are limited means of measuring transient nonproductive encounters. Our assays now allow direct measurement of first-passage recognition efficiency during the homology search. Remarkably, the data suggest an approximately linear relationship between first-passage recognition efficiency and the predicted stability of the resulting heteroduplex DNA joint, with a peak first-passage recognition efficiency of ~35% for substrates with melting temperatures on the order of approximately ≥ 60°C (Figures 2G and S3F). Notably, the average first-passage recognition efficiency (in the presence of RPA) is comparable for substrates ranging from 21 nt to 1,000 nt of homology (Figure 2G). This outcome differs from the in vivo requirements for efficient HR, which requires on the order of 35–70 nt of homology (Haber, 2018; Ira and Haber, 2002; Mehta et al., 2017). Indeed, substrates 33 nt in length can undergo a Rad51-independent BIR process that is enhanced by RAD51 deletion (Ira and Haber, 2002). A key difference between our in vitro work and the in vivo studies is that we are not assaying for completion of HR; we are instead capturing and characterizing transient intermediates prior to and during homology recognition under well-defined conditions. Our findings imply that there is no inherent deficiency in the ability of short substrates to undergo the homology search and homology recognition; instead, the defects observed with shorter homology lengths in the in vivo settings may reflect competition between protein binding (e.g., for the Rad51-dependent versus Rad51-independent reactions) or defects in processing the resulting D-loop intermediates (e.g., polymerase recruitment and 3′ end extension).
Length-Dependent PSC Translocation Velocity
Our data reveal that the PSC undergoes faster Rad54-dependent translocation with longer ssDNA with a velocity that peaks at ~150 nt; however, there is no change in the amount of bound Rad54 (for substrates ≤ 150 nt in length), and there is no corresponding change in ATP hydrolysis rates for the different-length PSCs. From these results, we infer that coupling between ATP hydrolysis and motor activity is affected by PSC length and that shorter substrates prevent effective coupling. Future work will be necessary to establish complete molecular understanding of how ATP hydrolysis is coupled to PSC translocation velocity during the homology search.
The ssDNA ends at native processed DSBs may be longer (~2–4 knt) than the substrates used in our model system (Chung et al., 2010). Nevertheless, our finding that the translocation velocity and first-passage recognition efficiencies plateau for substrates ranging from 150–1,000 nt in length suggests that these model PSCs provide a reasonable representation of a possibly longer physiological search entity. Moreover, subdivision of a longer native PSCs into shorter, physically linked but semi-independent search entities would enable enhanced homology recognition through multipartite binding interactions and would also allow simultaneous interrogation of multiple potential donor DNA sites (Forget and Kowalczykowski, 2012; Oh et al., 2007; Piazza et al., 2017). Given our findings, an added benefit of this subdivision is that the unit length of the semi-independent search entities might be tuned to balance translocation velocity and recognition efficiency.
Transient Strand Opening during the Homology Search
RPA co-localizes with the PSC, and this co-localization is required for efficient homology recognition. Notably, Rad54 renders dsDNA sensitive to cleavage by the ssDNA-specific P1 nuclease, indicating that changes in supercoiling through the action of Rad54 are coupled to transient separation of dsDNA strands (Mazin et al., 2000a; Van Komen et al., 2000). These results are incorporated into our working model, in which Rad54 translocation is coupled to local structural perturbation of the donor dsDNA (Figure 7A). Local strand opening would enable Rad51 to probe for homology within the context of a dsDNA structure that is already unwound or partially unwound (Figures 7A and 7B). One molecule of RPA should have a binding site size of ~18–30 nt (Chen and Wold, 2014; Kumaran et al., 2006), so three RPA molecules would correspond to ~54–90 nt of ssDNA exposed by Rad54 during the homology search.
Rad54 does not possess any DNA helicase activity (Ceballos and Heyer, 2011), which raises the question of how it might perturb donor DNA structure. One possibility is that the donor dsDNA may be opened or partially opened simply because of the binding of Rad54. However, if Rad54 binding alters dsDNA structure, then this structural perturbation must be coupled to ATP hydrolysis. An alternative mechanism that would enable transient, local strand opening is changes in DNA topology. In this scenario, the DNA ahead of Rad54 will be positively supercoiled, whereas the DNA behind will be negatively supercoiled. The underwound DNA immediately behind Rad54 could provide an access point, enabling RPA to melt the donor DNA strands (Figure 7A). This model is consistent with the classical theory for how motor proteins affect DNA topology (Liu and Wang, 1987) and is also consistent with the ability of Rad54 to induce ATP-dependent changes in DNA supercoiling (Mazin et al., 2000a; Ristic et al., 2001; Van Komen et al., 2000). In the DNA curtain assays, supercoils would dissipate through twist diffusion because the linear substrates are not torsionally constrained. The underlying mechanistic processes involved in coupling protein translocation to local changes in DNA topology do not require topologically closed substrates. Indeed, changes in local supercoiling have been reported for other motor proteins acting on unconstrained linear dsDNA, including the Snf2-like chromatin remodeling complexes SWI/SNF, ISWI, Mi-2, and BRG1 (Havas et al., 2000); the bacterial motor protein FtsK (Aussel et al., 2002); and even RNA polymerase (Kouzine et al., 2013; Levens et al., 2016). Interestingly, the positive supercoils generated in front of RNA polymerase have been implicated in nucleosome destabilization (Teves and Henikoff, 2014), which may bear some resemblance to the ability of Rad54 to remodel nucleosomes.
A New Role of RPA in HR
RPA stabilizes the ssDNA that is generated during DSB end resection (Chen and Wold, 2014; Kowalczykowski, 2015) and also plays a postsynaptic role by stabilizing the displaced donor DNA strand after strand invasion (Eggler et al., 2002). Our work now reveals a new role of RPA in HR where the homology search and subsequent D-loop formation do not occur in the context of normal B-form dsDNA but, rather, in the context of mobile DNA structure that is perturbed through the combined action of Rad54 and RPA (Figure 7A). Moreover, the PCS can identify homology irrespective of orientation, indicating that both donor dsDNA strands are sampled for homology, which could reduce the time necessary for the homology search by half. Given that each PSCs contained an average of ~2 presynaptic ssDNA molecules, it is possible that one ssDNA is dedicated to scanning with the donor DNA top strand while the other scans the bottom strand. Such a model would be attractive, given that the two ends of the same DSB might remain associated with one another throughout the homology search (Haber, 2018). An alternative mechanism that could allow bidirectional homology recognition would be rapid, diffusion-driven spatial fluctuations of the Rad51 ssDNA (or perhaps fluctuations of the underwound donor template) to enable rapid sampling of the top and bottom strands (Figure 7B, top). We do not discount either of the aforementioned models; however, neither accounts for the striking difference in strand bias observed in the absence of RPA. A third and perhaps more likely possibility stems from the crystal structure of RPA, which reveals the bound ssDNA fragment bent into a U-shaped configuration where the polarity of one half of the ssDNA is reversed relative to the other half (Fan and Pavletich, 2012). If RPA bound to the underwound donor DNA were to adopt such a configuration, then the Rad51 ssDNA within the PSC would have access to underwound segments of the top and bottom donor DNA strands oriented with the correct polarity relative to the presynaptic ssDNA irrespective of translocation direction (Figure 7B, bottom).
Rad54 and Mating-Type Switching
Consistent with our findings, numerous studies have suggested pre- and postsynaptic roles for Rad54 in HR (Ceballos and Heyer, 2011; Kowalczykowski, 2015). Interestingly, a chromatin immunoprecipitation (ChIP)-based study looking at Rad51 association with a donor DNA locus during mating-type switching in budding yeast suggested that Rad54 plays only a postsynaptic role in recombination (Sugawara et al., 2003). On the other hand, our data suggest that the presynaptic effect of Rad54 would lead to enhancement of homology search kinetics. This kinetic effect might not have been evident because of low temporal resolution of ChIP-based sampling of mating-type switching (Ceballos and Heyer, 2011; Sugawara et al., 2003). Moreover, the initiating MAT locus and the donor sequence at HML or HMR are located on chromosome III, and the recombination event between these partners can also be facilitated by a resident recombination enhancer module (Lee and Haber, 2015; Thon et al., 2019). Therefore, it is possible that the intrachromosomal location of the recombining partners in mating-type switching innately favors pairing interactions even when Rad54 is absent. However, a similar in vivo assay reported evidence of extensive Rad54-dependent scanning by the PSC of neighboring chromosomal sites while undergoing a frustrated search for a non-existent donor locus (Renkawitz et al., 2013). One important effect of this frustrated donor-less search may have been to enhance experimental detection of Rad54-dependent homology search intermediates by forcing the search to take place outside of the specialized context of the MAT locus and its donor sequence, which could explain the discrepancy between these two studies. Notably, Sugawara et al. (2003) used a single probe and would not have been able to assess the broader region-wide association observed by Renkawitz et al. (2013).
The Search for Homology in Physiological Settings
In eukaryotes, HR must take place within chromatin. Rad54 can remodel nucleosomes in biochemical assays (Alexeev et al., 2003; Jaskelioff et al., 2003; Zhang et al., 2007) and greatly stimulates strand invasion in the context of chromatin (Alexiadis and Kadonaga, 2002). In agreement with these studies, we find that the PSC can displace and remodel isolated nucleosomes and even bypass nucleosomes without evicting them from the DNA. However, a fraction of the encounters with nucleosomes cause the PSC to stall, so other chromatin remodeling factors may come into play to help ensure that potential donor DNA sequences remain accessible to the HR machinery. Indeed, ATP-dependent chromatin remodeling complexes, such as RSC, SWI/SNF, Ino80, Fun30, and SWR, have all been implicated in the early stages of HR in yeast, whereas INO80, hSNF2, CHD4, and other enzymes may fulfill similar roles in human cells (Papamichos-Chronakis and Peterson, 2013; Peterson and Almouzni, 2013; Sinha and Peterson, 2009).
Prokaryotic versus Eukaryotic Homology Search Mechanisms
Rad54 is found in all eukaryotes but is notably absent from bacteria (Ceballos and Heyer, 2011; Kowalczykowski, 2015). There is no known prokaryotic motor protein that might contribute to the homology search in bacteria, and E. coli RecA seems to conduct its homology search through purely diffusion-based mechanisms (Adzuma, 1998; Forget and Kowalczykowski, 2012; Ragunathan et al., 2012). These considerations suggest that prokaryotes and eukaryotes use distinct mechanisms to align DNA sequences during HR. The emergence of an ATP-dependent motor-coupled homology search mechanism may have been necessary to accommodate the unique challenges (i.e., larger chromosomes bound by nucleosomes) associated with supporting efficient DNA repair of the eukaryotic genome. Finally, our work reveals that Rad54 actively pries open the donor dsDNA strands during the homology search. Given its more robust ATPase levels, it is possible that RecA alone may be able to pry open dsDNA strands more readily than Rad51 alone and that the strand-opening activity of Rad54 may have coevolved as an important component of the search process during HR in eukaryotic organisms.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Eric C. Greene (ecg2108@cumc.columbia.edu).
Materials Availability
All reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
All kymographs used as the original source for data analysis throughout the paper are available on Mendeley [https://doi.org/10.17632/x2cb2mkhty.1].
EXPERIMENTAL MODEL AND SUBJECT DETAILS
For single molecule and bulk biochemical assays S. cerevisiae GST–Rad54 and GFP–GST–Rad54 were overexpressed in S. cerevisiae (MAT alpha leu2 trp1 ura3–52 prb1–1122 his3::pGAL1–GAL) grown in Yeast Nitrogen Base (−URA) media plus 3% glycerol and 2% lactic acid. S. cerevisiae Rad51, RPA, RPA–mCherry, RPA–GFP, Nap1 and histones (H2A, H2B, H3 and H4) and were all overexpressed in E. coli BL21 (DE3) Rosetta2 cells (EMD Millipore Cat# 714003; F− ompT hsdSB(rB− mB−) gal dcm (DE3) pRARE2 (CamR)) grown in Luria Broth.
For affinity pulldown assays S. cerevisiae Rad51 was expressed in the yeast strain LYS0411 (MAT alpha leu2–3,112 trp1–1 can1–100 ura3–1 ade2–1 his3–11,15 rad51::URA3, rad5–535) transformed with pPM230–RAD51 (2 μ, PGK, leu–2d) and grown in synthetic medium lacking leucine. RPA was overexpressed in yeast strain RDKY2275 (MAT a, ura3–52 trp1 leu2Δ his3Δ200 pep4::HIS3 prb1D1.6R can1, GAL) grown in synthetic media lacking leucine, tryptophan and uracil. Rad54 protein containing N–terminal thioredoxin and His6 tags was expressed in E. coli Rosetta cells (EMD Millipore Cat# 714003; F− ompT hsdSB(rB− mB−) gal dcm (DE3) pRARE2 (CamR)) grown in 2x Luria Broth.
Growth temperature, expression and purification information for each different protein are provided below in the METHOD DETAILS.
Plasmids for CURMIDS and plasmids for general cloning were grown in E. coli DH5alpha (NEB Cat# C2987H; fhuA2 a(argF-lacZ) U169 phoA glnV44 a80a(lacZ)M15 gyrA96 recA1 relA1 endA1 thi-1 hsdR17) or E. coli NEB Turbo (NEB Cat# C2984H; F proA+B+ lacIqΔ lacZ M15/ fhuA2 Δ(lac-proAB) glnV gal R(zgb-210::Tn10)TetSendA1 thi-1 Δ(hsdS-mcrB)5).
METHOD DETAILS
Rad51 and RPA purification for single–molecule and bulk biochemical assays
Rad51, RPA, RPA–GFP and RPA–mCherry, (Crickard et al., 2018; Gibb et al., 2014; Sung and Stratton, 1996). Rad54 and GFP–Rad54 were also purified as previously described (Crickard et al., 2018; Solinger et al., 2001). In brief, a protease deficient yeast strain was transformed with GST–Rad54 or GST–GFP–Rad54 on 2–micron plasmids under the control of the Gal1 promoter. Cells were grown in Yeast Nitrogen base (−URA) plus 3% Glycerol and 2% lactic acid. When the cells reached and OD of 1.5, expression was induced by the addition of 2% galactose for 6 hours. Cells were harvested and stored in the −80°C. Cell pellets were re–suspended in Rad54 re–suspension buffer (30 mM Tris–HCl [pH 7.5], 1 M NaCl, 1 mM EDTA, 10% glycerol, 10 mM BME (β–mercaptoethanol), protease inhibitor cocktail (Roche Cat. No. 05892953001) and 2 mM PMSF. Cells were disrupted by manual bead beating, and the lysate was clarified by ultracentrifugation at 100,000×g for 1 hour. The lysate was fractionated by ammonium sulfate (AS) precipitation. AS was gradually added with mixing to a final concentration of 20% followed by centrifugation at 10,000 × g for 10 minutes. The supernatant was discarded, and the AS concentration was raised to 50% followed by centrifugation at 10,000 × g for 10 min. The protein pellet was re–suspended in PBS (phosphate buffered saline) plus 1M NaCl and 10 mM BME. The resulting re–suspended protein was then bound to pre–equilibrated GST resin in batch for 1 hour at 4°C. The GST resin was washed 2x with PBS plus 1000 mM NaCl, and 2x with PBS plus 500 mM NaCl. The protein was eluted in 20 mM glutathione in PBS plus 500 mM NaCl. The peak fractions were pooled and then applied to a Sephacryl S–300 High Resolution gel filtration column (GE Healthcare, Cat. No. 17–0599–10) pre–equilibrated with Rad54 SEC buffer (30 mM Tris–HCl [pH 7.5], 500 mM NaCl, 1 mM EDTA, 10% glycerol, and 10 mM BME. The protein eluted in two peaks, one peak occurred outside of the exclusion volume of the column and was discarded. The second peak eluted close to the expected MW of a Rad54 monomer. The monomeric Rad54 peak was pooled and dialyzed against Rad54 SEC buffer plus 50% glycerol and stored in at −80°C in single use aliquots.
6xHis–SUMO–Rad51 was overexpressed in E. coli BL21 (DE3) Rosetta2 cells at 37°C to an OD600 of 0.4–0.6. Expression was induced by addition of 0.5 mM IPTG for 3 hours at 37°C. overexpression cells were harvested and stored at −80°C. Cell were lysed by freeze–thaw in Cell Lysis Buffer (30 mM Tris–HCl [pH 8.0], 1 M NaCl, 10% Glycerol, 10 mM imidazole, 5 mM BME and protease inhibitor cocktail (Roche Cat. No. 05892953001)). Crude lysates were sonicated for 6 pulses of 30 s on and 2 minutes off, and then clarified by ultracentrifugation at 100,000 × g for 45 minutes. The extract was then bound to 1 mL of pre–equilibrated Ni–NTA resin for 1 hour with rotation. The resin was then washed 3X with CLB and eluted in CLB+200 mM imidazole. The protein was mixed with 400 units of the SUMO protease Ulp1 and dialyzed overnight at 4°C into Rad51 buffer (30 mM Tris–HCl [pH 8.0], 150 mM NaCl, 1 mM EDTA, 10% Glycerol, 10 mM imidazole). The 6xHis–SUMO tag and SUMO protease were removed by passing the dialyzed proteins over a second 1 mL Ni–NTA column. The purified Rad51 was then stored at −80°C in single use aliquots.
RPA, RPA–mCherry and RPA–GFP were overexpressed in E. coli BL21 (DE3) Rosetta2 grown at 37°C to an OD600 of 0.4–0.6. The temperature of the cultures was adjusted to 18°C and expression was induced with addition of 0.2 mM IPTG for 8–12 hours. After overexpression cells were harvested and stored at −80°C. Cells were lysed by freeze–thaw in Lysis buffer (40 mM NaHPO4 [pH 7.5], 600 mM KCl, 5% Glycerol, 5 mM imidazole, 0.1 mM tris– (2–carboxyethyl)phosphine (TCEP), 0.05% Tween 20, and protease inhibitor cocktail [Roche Cat. No. 05892953001). Crude lysates were sonicated on ice 6 3 30 s on 2 min off. The lysate was clarified by ultracentrifugation at 100,000 × g for 45 minutes. The lysate was bound to pre–equilibrated Talon (Clontech) resin for 30 min at 4°C. The bound protein was washed with nickel A buffer (40 mM NaHPO4 [pH 7.5], 300 mM KCl, 5% Glycerol, 5 mM imidazole, 0.02% Tween–20). The protein was then eluted in nickel A buffer + 200 mM imidazole. Peak fractions were then further fractionated on a Superdex 200 size exclusion column equilibrated with SEC buffer (40 mM NaHPO4 [pH 7.5], 200 mM KCl, 10% Glycerol, 0.02% Tween–20). Peak fractions, containing all three subunits of RPA, were then pooled and stored in single use aliquots at −80°C. For unlabeled RPA an additional purification step was included in between the Talon column and the size inclusion column. In this case Peak fractions from the Talon column were bound to Chitin resin (NEB) and then washed in nickel buffer A. The bound protein was then incubated overnight in nickel A buffer plus 100 mM DTT this removed the chitin tag and eluted the RPA protein. This step was followed by the size exclusion step listed above.
Histone purification and nucleosome reconstitution
Recombinant nucleosomes were prepared as previously described (Xue et al., 2019). Budding yeast histones were individually overexpressed in E. coli Rosetta2 cells. Bacterial cultures (6L) were grown at 37°C to an OD600 between 0.4 – 0.6 and expressed by addition of 0.45 mM IPTG for 3 hours. Cells were re–suspended in 80 mL of Tris–sucrose buffer (50 mM Tris–HCl [pH 8.0], 10% sucrose, 1 mM benzamidine), and then flash frozen in liquid nitrogen. Cells were lysed by freeze / thaw and then sonicated for 3 min at 60% power output, in sequences of 10 s on and 50 s off. Inclusion bodies were harvested by centrifugation at 20,000xg for 20 min. The inclusion bodies were then washed three times with 10 mL of inclusion body buffer (IBB); 5 mM BME, 1 mM benzamidine, 1% Triton X–100); and washed twice with IBB–Triton–x–100. The inclusion bodies were solubilized in 30 mL of unfolding buffer (20 mM Tris–HCl [pH 8.0], 7 M guanidium–HCl, 10 mM DTT) by rotation at 25°C for 1 hour. The resulting suspensions were then centrifuged at 20,000xg for 20 min to remove remaining insoluble material. The unfolded protein was then dialyzed (3,500 MWCO) against de–ionized Urea buffer (10 mM Tris–HCl [pH 8.0], 7 M Urea, 1 mM EDTA [pH 8.0], 5 mM BME and 100 mM NaCl) at 4°C with three buffer changes at ~2 hours each. Urea buffer de–ionization was achieved by incubating with AG501–X8 resin (BioRad, Cat. No. 1426424) for four hours at 25°C. The dialyzed histones were then loaded onto a tandem Q–Sepharose column (80 mL column volume, CV) followed by an SP–Sepharose column (80 mL CV) pre–equilibrated with Urea buffer at a flow rate of 1.0 mL min−1. The column was washed with 2 CV of Urea buffer and the Q–Sepharose column was then removed. The SP–Sepharose column was then developed with a linear gradient from 200 mM to 1000 mM NaCl over 2 CV’s. Peak fractions were pooled and then dialyzed against 10 mM Tris–HCl [pH 8.0] and 5 mM BME. Dialysis proceeded with 3x buffer changes at two–hour intervals. Purified histones were lyophilized and stored at −20°C
Octamer re–folding was performed by solubilizing individual histones in unfolding buffer and mixing histones in equimolar amounts. The reaction was diluted to a final concentration of 1 mg ml−1 and dialyzed into TEB2000 (10 mM Tris–HCl [pH 8.0], 1 mM EDTA [pH 8.0], 5 mM BME) for 48 hours with 4 buffer changes. Insoluble material was then removed by centrifugation at 20,000xg and the soluble material was concentrated to 1 mL and loaded onto a Superdex–200 16/60 size exclusion column (GE healthcare) pre–equilibrated with TEB2000. The histone octamer was resolved at a flow rate of 0.2 mL min−1. Peak fractions were pooled and concentrated using a Vivaspin 6 10,000 MWCO (GE Healthcare, Cat. No. 28–9322–96) to a final concentration of 20 μM. The concentrated octamer was mixed with an equal volume of 100% glycerol and stored at −20°C.
Histone H2A–S47C or H4–S48C labeling was achieved by incubating re–folded histone octamer (~1 nmol) with 10 mM TCEP (Tris (2–carboxyethyl) phosphine hydrochloride (Sigma–Aldrich, Cat. No. C4706) in TEB2000 for 10 min at 25°C. The solution was then added to an Amicon–Ultra 0.5 mL 10K MWWCO spin concentration device (Millipore, Cat. No. UFC501024) pre–equilibrated with TEB2000, and the TCEP removed. A ~10–fold molar excess of Atto565–maleimide (ATTO–TEC, Cat. No. AD565–41) or in the case of H2A–S47C or Alexa488–maleimide (Thermo Scientific, Cat. No. A10254) was added to the spin concentrator and incubated with the histone octamer for 1 hour at 25°C in the dark. Unreacted dye was removed by at least 5 sequential rounds of concentration and dilution. The labeling efficiency (~90%) was calculated by measuring the amount of dye relative to protein using a NanoDrop Spectrophotometer (ThermoFisher Scientific) and extinction coefficients of ϵ280 nm = 40,920 M−1cm−1 for the histone octamer, ϵ564 nm = 1.2×105 M−1cm−1 for Atto565, and ϵ495 nm = 73,000 M−1cm−1 for Alexa488.
Yeast Nap1 was purified from E. coli Rosetta2 cells. Cells (4 L) were grown to an OD600 of 0.6 and protein expression was induced by addition of 1 mM IPTG for 3 hours. Cells were harvested and re–suspended in Lysis buffer (20 mM Tris–HCl [pH 7.5], 100 mM KCl, 5 mM imidazole, 10% glycerol, 0.5% Triton X–100, 1 mM BME, 1 mM Benzamidine, PMSF), and flash frozen. Cells were lysed by freeze / thaw and then sonicated for 3 min with 60% power output with 30 s on and 1 min off. Insoluble material was removed by centrifugation at 90,000 g. The soluble material was then bound in batch to Talon resin (Clontech, Cat. No. 635503) 0.5 mL CV/L of culture for one hour with rotation at 4°C. The resin was then washed 2x in wash buffer A (20 mM Tris–HCl [pH 7.5], 100 mM KCl, 1 mM 2–mercaptoethanol, 10 mM imidazole, 10% Glycerol) followed by two washes with wash buffer B (20 mM Tris–HCl [pH 7.5], 500 mM KCl, 1 mM BME, 10 mM imidazole, 10% glycerol) followed by 1x wash with wash buffer A. The resin was then poured into a disposable column (VWR, Cat. No. 20170–333) and the protein was eluted with wash buffer A plus 150 mM imidazole. Peak fractions were pooled and loaded on to 1 mL CV Q–Sepharose FF column and washed with 10 CV Q–buffer A (20 mM Tris–HCl [pH 7.5], 100 mM KCl, 1 mM BME, 0.2 mM EDTA 10% glycerol). The column was then developed with a linear gradient from 100–1000 mM KCl over 10 CV’s. Peak fractions were then pooled and dialyzed against Q buffer A. The yield of Nap1 was measured using the A280. The protein was aliquoted and stored at −80°C.
Nucleosome reconstitutions were performed in HR buffer (30 mM Tris–OAc [pH 7.5], 20 mM MgOAc2, 50 mM KCl, 1 mM DTT, 0.1 mg ml−1 BSA) by mixing sufficient Atto565–labeled octamer to yield ~1–4 nucleosomes per donor dsDNA molecule, 750 nM Nap1 and incubating on ice for 20 min. CURMID DNA was then added to a final concentration of 30 ng μl−1 in 30 μl for four hours at 30°C. Insoluble material was removed by centrifugation at 10,000xg for 10 min at 4°C. Nucleosome–bound DNA curtains were then prepared as described above for naked dsDNA curtains, with the exception that nucleosomes curtains were washed for 10 min at 1 mL min−1 with buffer containing 0.5 mg ml−1 Salmon Sperm DNA (Sigma, Cat. No. D1626) to remove free histones and free NAP1. Nucleosomes prepared using these procedures were validated by confirming the characteristic 150–bp footprint using micrococcal nuclease digests in bulk biochemical assays and also by single–step photobleaching DNA curtain assays.
Bulk D–loop assays
Bulk strand invasion experiments were carried out essentially as described (Mazin et al., 2000b), using an Atto565– or Atto647N–labeled oligonucleotide substrate (15 nM) that contained a 21–nt 3′ ssDNA overhang homologous to pUC19 (2,686 bp) and a 56–bp region of dsDNA (Figure 1B); note, the Atto647N dye was used primarily for bulk biochemical experiments because of its better signal to noise ratio and the Atto565 dye was used for TIRFM experiments because of its brighter signal. All oligonucleotides were purchased from IDT and oligonucleotide sequences are listed in the Key Resources Table. Oligonucleotides were annealed by warming to 95°C and then gradually reducing the temperature to 25°C in a thermocycler. Annealed oligonucleotides were purified on a 12% Native PAGE in 0.5x TBE and eluted by a crush and soak method. Strand exchange reactions contained supercoiled pUC19 (0.3 nM plasmid) and were performed in HR buffer (30 mM Tris–OAc [pH 7.5], 20 mM MgOAc2, 50 mM KCl, 1 mM DTT, 0.1 mg ml−1 BSA and 5 mM ATP). The reactions also included 300 nM Rad51, 500 nM RPA, and Rad54 (varying concentrations, as indicated). Reactions were allowed to proceed at 30°C for 10 min and then quenched with stop solution (20 mM EDTA and 1% SDS). The reactions were then treated with Proteinase K (1 units) for 30 min at 37°C. Reaction products were resolved on a 0.9% TAE–agarose gel, and directly visualized by scanning the gel with a Typhoon FLA 9000 (GE Healthcare).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Digoxigenin Fab Fragment (from Sheep) | Roche | Cat# 11214667 |
| Bacterial and Virus Strains | ||
| E. coli Bl21 Rosetta2 (DE3) F−ompThsdSB(rB− mB−) gal dcm (DE3) pRARE2 (CamR) | EMD Millipore | Cat# 714003 |
| E. coli NEB Turbo F proA+B+ lacIqΔ lacZ M15/ fhuA2 Δ(lac-proAB) glnV gal R(zgb-210::Tn10)TetSendA1 thi-1 Δ(hsdS-mcrB)5 | NEB | Cat# C2984H |
| E. coli DH5alpha fhuA2 a(argF-lacZ)U169 phoA glnV44 a80a(lacZ)M15 gyrA96 recA1 relA1 endA1 thi-1 hsdR17 | NEB | Cat# C2987H |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Sc GST-Rad54 | This Study | N/A |
| Sc GST-GFP-Rad54 | This Study | N/A |
| Sc Rad51 | This Study | N/A |
| Sc RPA-6xHiS | This Study | N/A |
| Sc RPA-GFP-6xHis | This Study | N/A |
| Hs RPA-GFP-6xHis | This Study | N/A |
| Ec SSB-GFP-6xHis | This Study | N/A |
| Sc Nap1 | This Study | N/A |
| Sc Histone H2AS47C | This Study | N/A |
| Sc Histone H2B | This Study | N/A |
| Sc Histone H3 | This Study | N/A |
| Sc Histone H4 | This Study | N/A |
| Streptavidin from Streptomyces | Millipore | Cat# S4762 |
| Albumin from bovine serum | Sigma-Aldrich | Cat# A7030 |
| Sc Ulp1-SUMO Protease | Sigma-Aldrich | Cat# SAE0067 |
| Talon Resin | Clontech | Cat# 635503 |
| HisTrap prepacked column | GE Healthcare | Cat# 17-5247-01 |
| GST-Resin | GE Healthcare | Cat# 17-0756-01 |
| Hi-load 16/60 Superdex-200 | GE Healthcare | Cat# 17-1043-01 |
| Sephacryl-S-300 | GE Healthcase | Cat# 17-0599-10 |
| Q-Sepharose | GE Healthcare | Cat# 17-0510-01 |
| Dynabeads M-280 Streptavidin | Invitrogen | Cat# 11205D |
| 1,2-dioleoyl-sn-glycero-3-phosphocholine | Avanti Polar Lipids | Cat# 850375C |
| 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(cap biotinyl) (sodium salt) | Avanti Polar Lipids | Cat# 870273P |
| 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(methoxy(polyethylene glycol)-2000](ammonium salt) | Avanti Polar Lipids | Cat# 880130P |
| Critical Commercial Assays | ||
| In-Fusion HD Cloning plus Kit | Clontech | Cat# 638909 |
| Deposited data | ||
| Kymographs used for all data analysis | Mendeley | https://doi.org/10.17632/x2cb2mkhty.1 |
| Experimental Models: Organisms/Strains | ||
| S. cerevisiae (MAT alpha leu2 trp1 ura3–52 prb1–1122 pep4–3 his3::pGAL1-GAL4) | Gift from Symington Lab (Columbia University Medical Center) | LSY0929 |
| S. cerevisiae rad51Δ (MATalpha leu2–3,112 trp1–1 can1–100ura3–1 ade2-1 his3–11,15 rad51:: URA3, rad5–535) | Gift from Symington Lab (Columbia University Medical Center) | LSY0411 |
| S. cerevisiae (MAT a, ura3–52 trp1 leu2Δ his3Δ200 pep4::HIS3 prb1D1.6R can, GAL) | Gift from Symington Lab (Columbia University Medical Center) | RDKY2275 |
| Oligonucleotides | ||
| Position 8.6 (21 -nt homology) 5′-AAT TCT CAT TTT ACT TAC CGG ACG CTA TTA GCA GTG GGT GAG CAA AAA CAG GAA GGC-3′ | IDT, This Study | N/A |
| Position 8.6 (15-nt homology) 5′-AAT TCT CAT TTT ACT TAC CGG ACG CTA TTA GCA GTG ATG ATC CAA AAA CAG GAA GGC-3′ | IDT, This Study | N/A |
| Position 8.6 (9-nt homology) 5′-AAT TCT CAT TTT ACT TAC CGG ACG CTA TTA GCA GTG ATG ATC CAT ACT CAG GAA GGC-3′ | IDT, This Study | N/A |
| Position 8.6 (0-nt homology) 5′-AAT TCT CAT TTT ACT TAC CGG ACG CTA TTA GCA GTG CAT AGT ATC CCT AGT CCG TAA-3′ | IDT, This Study | N/A |
| Position 8.6 90 nt homology 5-GGT GCA CGA GTG GGT TAC ATC GAA CTG GAT CTC AAC AGC GGT AAG ATC CTT GAG AGT TTT CGC CCC GAA GAA CGT TTT CC A ATG ATG AGC | IDT, This Study | N/A |
| Position 8.6 150 nt homology GTA AAA GAT GCT GAA GAT CAG TTG GGT GCA CGA GTG GGT TAC ATC GAA CTG GAT CTC AAC AGC GGT AAG ATC CTT GAG AGT TTT CGC CCC GAA GAA CGT TTT CCA ATG ATG AGC ACT TTT AAA GTT CTG CTA TGT GGC GCG GTA TTA TCC | IDT, This Study | N/A |
| Position 4.5 (21-nt homology) 5′-AAT TCT CAT TTT ACT TAC CGG ACG CTA TTA GCA GTG GCG CGC CTG TGC ACT CTG TGG-3′ | IDT, This Study | N/A |
| Labeled strand 5′-Atto565-CAC TGC TAA TAG CGT CCG GTA AGT AAA ATG AGA ATT-3′ | IDT, This Study | N/A |
| Labeled strand 5′-Atto647N-CAC TGC TAA TAG CGT CCG GTA AGT AAA ATG AGA ATT-3′ | IDT, This Study | N/A |
| For additional oligonucleotide sequences see Key Resource Table S1 | This Paper | N/A |
| Other | ||
| Fisherfinest Premium Cover Glass | Fisher Scientific | 12-548-5P |
| Quartz Microscope Slides (1” × 3”) | Finkenbeiner | N/A |
| TIRFM Microscope | This Study | N/A |
| Recombinant DNA | ||
| pUC19 plasmid | New England Biolabs | Cat# N3041S |
| CURMIDDNA | This Study | N/A |
| pYes2-GST-Rad54 | WD Heyer Lab (University of California, Davis) | N/A |
| pYes2-GFP-GST-Rad54 | This Study | N/A |
| pET28D-Nap1 | K. Luger Lab (University of Colorado, Boulder) | N/A |
| pET11C-H2A, H2B, H3, H4 | K. Luger Lab (University of Colorado, Boulder) | N/A |
| pET11C-SUMO-Rad51 | This study | N/A |
| p11D-sctRPA, sctRPA-GFP-70, sctRPA, hstRPA-GFP-70 | M. Wold Lab (University of Iowa) | N/A |
| Software and Algorithms | ||
| Graphpad Prism 8.2 | Graphpad | https://www.graphpad.com/scientific-software/prism/ |
| Nikon Elements Ars 5.11 | Nikon | (https://www.nikon.com/products/microscope-solutions/lineup/img_soft/nis-elements/table_of_features.htm |
ATP hydrolysis assays
ATP hydrolysis assays were performed in HR buffer (30 mM Tris–OAc [pH 7.5], 20 mM MgOAc2, 50 mM KCl, 1 mM DTT, 0.1 mg ml−1 BSA in the presence of pUC19 plasmid (10 ng/μl) and 2 mM ATP with trace amounts of γ32P–ATP (3000 Ci/mmol). Reactions contained 30 nM Rad54 or GFP–Rad54 (as indicated), 300 nM Rad51 and 500 nM RPA. All reactions were performed at 30°C. Aliquots were removed at specified time points and quenched by mixing with an equal volume of 25 mM EDTA and 1% SDS. The quenched reactions were spotted on TLC plates (Millipore, Cat. No. HX71732079) and resolved in 0.5 M LiCl plus 1 M Formic acid. Dried TLC plates were exposed to phosphor–imaging screen and scanned with a Typhoon FLA 9000 (GE Healthcare).
DNA curtain preparation
All experiments were conducted with a custom–built prism–type total internal reflection fluorescence (TIRF) microscope (Nikon) equipped with a 488–nm laser (Coherent Sapphire, 200 mW), a 561–nm laser (Coherent Sapphire, 200 mW), and two Andor iXon EMCCD cameras (De Tullio et al., 2018). Metallic patterns (chromium, Cr) were deposited onto the surface of fused silica microscope slides by electron beam (e–beam) lithography (Greene et al., 2010). Briefly, holes were drilled into a quartz microscope slide with a diamond coated drill bit. The slides were then cleaned for 20 minutes in piranha solution (3 parts H2SO4 1 part H2O2). Followed by washing 3x with H2O. The slide surfaces were then coated first with Poly–methyl methacrylate (PMMA) 25 K, then PMMA 495K, and finally aquaSave (Mitsubishi). Patterns were written into the PMMA using e–beam lithography. Slides were developed in an Isopropanol: Methyl–Isobutyl Ketone solution (3:1) with sonication for 1 min at 0°C. Chromium was then deposited on the microscope surface using electron beam evaporation. The PMMA and excess chromium were then removed by lift–off in Acetone. The quality of the chromium features was checked by light microscopy. Flow cells were assembled and dsDNA curtains were prepared as previously described (Greene et al., 2010). First, using double–sided tape a glass coverslip was attached to the quartz microscope slide. The tape was melted in an oven to create a sealed reaction chamber. Inlet outlet nanoports were then glued over the holes that had been drilled in the microscope slide and the flow cell was completed. dsDNA curtains were prepared as previously described (Greene et al., 2010), with the exception that the DNA substrates were prepared with a CURMID (Curtains Plasmid; 12,273 kbp, ~4.2 mm) comprised of pUC19 plasmid containing a 9.5 kb fragment of λ–phage DNA; the additional length provided by the λ–phage DNA fragments facilitated preparation and visualization of the double–tethered DNA curtains. This pUC19–based substrate allowed use of the same Atto565–labeled DNA for both the bulk biochemical assays and DNA curtain assays, which greatly facilitated optimization of the reaction conditions. The distance between the barriers and the pedestals was 3.2 μm resulting in a mean extended length (〈X〉) of ~0.7 for the double–tethered CURMID substrates. To generate these substrates, the CURMID was grown in E. coli purified and then digested with the enzymes Nsi1 and PspOMI, generating an overhang for DNA handle ligation. Digested CURMIDs were then ligated to DNA handles that were either biotinylated or modified with digoxygenin, as indicated. Lipid bilayers were prepared with 91.5% DOPC (1,2–dioleoyl–sn–glycero–3–phosphocholine), 0.5% biotinylated–PE (1–oleoyl–2–(12–biotinyl(aminododecanoyl))–sn–glycero–3–phosphoethanolamine), and 8% mPEG 2000–DOPE (1,2–distearoyl–sn–glycero–3–phosphoethanolamine–N–[methoxy(polyethylene glycol)–2000] (ammonium salt)); all lipids were purchased from Avanti Polar Lipids. The flow cell was then attached to a microfluidic system and sample delivery was controlled using a syringe pump (KD Scientific). The donor dsDNA substrates were tethered through a biotin–streptavidin linkage to the lipid bilayer, then aligned at chromium (Cr) barriers through the application of buffer flow and then anchored to Cr pedestals via a digoxigenin–antibody linkage, as described (Greene et al., 2010).
Single–molecule DNA curtain assays
Single molecule experiments were performed essentially as described above for the bulk biochemical D–loop assays with a few changes. Experiments were performed in HR buffer at 30°C using DNA curtains comprised of the linear CURMID DNA as the donor dsDNA. PSCs were prepared by mixing 5 nM Atto565–labeled tailed duplex oligonucleotide DNA pre–incubated for 15 min at 30°C with 50 nM Rad51. 60 nM RPA and 5 nM Rad54 or GFP–Rad54 (as indicated) were added just prior to injection into the sample chamber. The presynaptic complexes were quickly injected into the sample chamber at a flow rate of 1 mL min−1 and then buffer flow was terminated. Data collection was initiated prior to sample injection and continued for a total of 15 to 20 min. Assays using the longer 90–,150–, and 1,000–nt ssDNA substrates were performed using the same reagent concentrations and under identical reaction conditions as those listed above for the tailed duplex DNAs.
Generation of long PSC ssDNA substrates
All of the longer ssDNA substrates used in this study were all fully homologous to the 8.6 locus depicted in Figure 2A. The 90– and 150–nt ssDNA were purchased from IDT. 1,000–nt substrate was generated by first creating 1,000–bp dsDNA by PCR with oligonucleotide primers labeled on the 5′ end with Atto565 for the (+) strand oligonucleotide or biotin on (−) strand oligonucleotide. The PCR product was then purified from an agarose gel to remove excess unreacted oligonucleotide primers. The gel purified PCR product was then immobilized for 10 min on 30 μl of M–280 Dynabeads (Invitrogen, Cat. No. 11205D) that had been washed 2x with H2O and 2x with 100 mM Tris–Cl (pH 8.0) with 1 M NaCl. After immobilization the beads were washed 2x with 100 mM Tris–Cl (pH 8.0) with 1 M NaCl. The immobilized DNA was then treated with 20 μl of 1 M NaOH for 10 minutes to separate the DNA strands and eluate the Atto565–labeled ssDNA strand. After 10 minutes the supernatant was neutralized with 80 μl of 1 M Tris–Cl (pH 8.0). The efficiency of the ssDNA elution was monitored by gel electrophoresis. The ssDNA product was quantified by UV spectrophotometry, and used in single molecule assays based on the measured concentration. Prior to use the ssDNA was heated to 65°C for 5 minutes to remove any potential secondary structure.
Protein purification for affinity pulldown assays
Rad51, His–Rad54, and RPA were purified as described with minor modifications (Raschle et al., 2004; Van Komen et al., 2006). S. cerevisiae Rad51 protein was expressed in the rad51Δ yeast strain LSY411 transformed with pPM230–RAD51 (2 μ, PGK, leu–2d) in synthetic medium lacking leucine at 30°C for 15 hr (Sherman, 1991). Cells from a 20 L culture were harvested by centrifugation and stored at −80°C. All the subsequent steps were carried out at 0–4°C. The frozen cell pellet (100 g) was resuspended in 200 mL of cell breakage buffer (50 mM Tris–HCl [pH 7.5], 10% sucrose, 10 mM EDTA, 0.6 M KCl, 1 mM DTT, 0.01% Igepal CA–630 (Sigma)) containing 1 mM phenylmethylsulfonyl fluoride (PMSF), 1 mM benzamidine HCl, and 1.25 mg/ml of the following protease inhibitors: aprotinin, chymostatin, leupeptin, and pepstatin A. Cell lysate was prepared in a bead beater using 0.5 mm glass beads. and clarified by ultracentrifugation (100,000 × g, 90 min). The supernatant was treated with ammonium sulfate at 40% saturation (0.24 g/ml) and the protein precipitate was collected by centrifugation at 15,000 × g, for 30 min. The ammonium sulfate precipitate was dissolved in 200 mL of T buffer (25 mM Tris–HCl [pH 7.5], 10% glycerol, 1 mM EDTA, 1 mM DTT, 0.01% Igepal CA–630) and applied onto a 20–ml Q–Sepharose column (GE Healthcare), which was developed with a 200–ml gradient of 0–650 mM KCl in T buffer. Fractions containing Rad51 were identified by SDS–PAGE and Coomassie Blue staining, pooled, and fractionated in a 10–ml Macroprep hydroxyapatite (Macro–HAP) column (Biorad) using a 120–ml gradient of 0–200 mM KH2PO4 in T buffer with 50 mM KCl. Rad51 fractions were pooled and further fractionated in a 1–ml Mono Q column (GE Healthcare), using a 60–ml gradient of 150–400 mM KCl in T buffer. The peak fractions of Rad51 were pooled and concentrated with an Amicon Ultra 4 concentrator (Millipore) before being divided into 5 μl aliquots and stored at −80°C.
S. cerevisiae Rad54 protein containing N–terminal thioredoxin and His6 tags was expressed in E. coli Rosetta cells (Novagen) transformed with pET32a–Rad54. Cells were grown in 2x Luria Broth at 37°C to A600 of 0.8. Protein expression was induced with 0.1 mM IPTG for 16 h at 16°C. Cells from a 24–L culture were harvested by centrifugation and stored at −80°C. All the subsequent steps were carried out at 0–4°C. The frozen cell pellet (50 g) was resuspended in 300 mL of K buffer (20 mM KH2PO4 [pH 7.5], 10% glycerol, 0.5 mM EDTA, 0.01% Igepal CA–360, 1 mM DTT, containing 150 mM KCl, 1 mM PMSF, 1 mM benzamidine HCl, and 1.25 μg/ml of the following protease inhibitors: aprotinin, chymostatin, leupeptin, and pepstatin A. Cell lysate was prepared by sonication and clarified by ultracentrifugation (100,000 × g, 90 min). The supernatant was applied sequentially onto a 20–ml Q–Sepharose column and a 20–ml SP–Sepharose column (GE Healthcare), which was developed with a 200–ml gradient of 150–600 mM KCl in K buffer. Fractions containing Rad54 were identified by SDS–PAGE and Coomassie Blue staining, pooled, and incubated with 3–ml Ni2+–NTA–agarose (QIAGEN) for 2 h. The resin was washed with 20 mL each of K buffer containing 350 mM KCl, 10 mM imidazole and 20 mM imidazole, followed by eluting bound proteins with a total of 30 mL of Buffer K containing 200 mM imidazole. The Rad54 pool was further fractionated in a 6–ml Macro–HAP column (Biorad) using a 60–ml gradient of 0–300 mM KH2PO4 [pH 7.5] in K buffer with 100 mM KCl. Fractions containing Rad54 were pooled and concentrated with an Amicon Ultra 4 concentrator (Millipore) before being divided into 2 ml aliquots and stored at −80°C.
S. cerevisiae RPA was overexpressed in yeast strain RDKY2275 transformed with three plasmids expressing RPA subunits under the GAL10 promoter. Cells were grown in synthetic medium lacking leucine, tryptophan, and uracil, and RPA expression was induced in YPG medium (1% yeast extract, 2% peptone, and 2% galactose) containing 3% glycerol and 3% lactic acid at 30°C for 15 hr (Sherman, 1991). Cells from a 40–L culture were harvested by centrifugation and stored at −80°C. All the subsequent steps were carried out at 0–4°C. The frozen pellet (150 g) was resuspended in T buffer containing 100 mM KCl, 1 mM PMSF, 1 mM benzamidine HCl, and 1.25 μg/ml of the following protease inhibitors: aprotinin, chymostatin, leupeptin, and pepstatin A. Cell lysate was prepared using a French press and clarified by ultracentrifugation (100,000 × g, 90 min). The supernatant was applied onto a 20–ml Affi–Gel Blue agarose (Bio–Rad) column, which was developed with a 120–ml gradient of 0.5–2.5 M NaSCN in T buffer. Fractions containing RPA were identified by SDS–PAGE and Coomassie Blue staining, pooled, and dialyzed against T buffer overnight and then applied onto an 8–ml Macro–HAP column and developed with a 100–ml gradient of 0–200 mM KH2PO4 in T buffer with 50 mM KCl. Fractions containing RPA were pooled and further fractionated in a 1–ml Mono Q column with a 100–ml gradient from 50 to 335 mM KCl in buffer T. RPA peak fractions were pooled and concentrated with an Amicon Ultra 4 concentrator (Millipore) before being divided into 5 μl aliquots and stored at −80°C.
Affinity pulldown assays
To test for protein–protein interactions, the indicated yeast RPA or yeast Rad51 were incubated with His6–tagged Rad54 (4 μg each) in 20 μL of T100–I20 buffer (25 mM Tris–HCl [pH 7.5], 0.5 mM EDTA, 10% glycerol, 0.01% Igepal CA–630, 100 mM KCl, 20 mM imidazole) supplemented with 125 U Benzonase (Sigma, Cat. No. E1014) on ice for 30 min. Then, the binding mixtures were incubated with 10 μL of Ni–NTA Superflow (QIAGEN) for 1 hr at 4°C. The beads were then separated by centrifugation and the supernatants were removed. After washing the beads three times with 100 mL of T100–I20, bound proteins were eluted with 25 μL of SDS–loading buffer (50 mM Tris–HCl [pH 6.8], 50 mM DTT, 2% SDS, 10% glycerol). Fifty percent of total supernatant (S) and elution (E) fractions were analyzed by SDS–PAGE and Coomassie Blue staining.
QUANTIFICATION AND STATISTICAL ANALYSIS
Single–molecule data collection and analysis
For all two–color images, we used a custom–built shuttering system to avoid signal bleed–through during image acquisition (De Tullio et al., 2018). With this system, images from the green (GFP) and the red (mCherry) channels are recorded independently, these recordings are offset by 100 ms such that when one camera records the red channel image, the green laser is shuttered off, and vice versa. This system prevents any possible signal bleed–though between the two channels. Images were captured at an acquisition rate of 1 frame per 10 s or 1 frame per 5 s with a 100–millisecond integration time, and the laser was shuttered between each acquired image to minimize photo–bleaching. For the longer ssDNA substrates, images were captured at an acquisition rate of 1 frame per 0.5 s with a 100–millisecond integration time. For Raw TIFF images were imported as image stacks into ImageJ, and kymographs were generated from the image stacks by defining a 1–pixel wide region of interest (ROI) along the long–axis of the individual dsDNA molecules. All data analysis was performed using the resulting kymographs. From these kymographs, the location of the PSC was defined based upon peak pixel signal intensity, the start of translocation was defined as when the peak signal intensive moved unidirectionally for ≥ 2 pixels, pauses were defined as when the peak pixel intensity stopped moving for 2 consecutive frames (5–10 s, depending upon acquisition rate), and the termination of translocation was defined as when the PSC completely stopped moving. Note, we report velocities as the pause–free values, so different segments of the same trajectory separated by a pause (≥2 consecutive frames) are analyzed separately. Velocity values were determined using the following formula: [(|Yf − Yi|) × 1, 111 bp]/[(|Xf − Xi|) × frame rate; where Yi and Yf correspond to the initial and final pixel position for the PSC along the long axis of the dsDNA and Xi and Xf correspond to the start and stop time (in seconds) for each observed translocation event. Graphs of individual velocities and processivities were created using Graphpad Prism 8. These values were fit to a Gaussian distribution and the mean was determined from these fits. Note that the reported means for the PSC velocities represent pause free velocities – in some cases molecules would pause for extended periods of time and then resume translocation – in these cases, translocation events were counted as separate velocity trajectories and the pause event was excluded from the calculation. Where indicated, all statistical tests for comparison of different datasets were conducted using a two–tailed Student’s t–test in Graphpad prism 8. Statistical tests for were conducted using the mean of the raw data and not the mean as determined by the Gaussian fitting.
Analysis of PSC binding distributions
The binding distribution of PSCs on the DNA was performed by determining the central pixel position for each binding event (using NIH ImageJ) after a ten–minute reaction. Bulk reactions are complete within ten minutes; therefore, ten minutes is a valid time frame to monitor binding position and effective target binding. The pixels were then converted to a physical distance with the conversion factor of 0.26 μm/pixel. Binding events were then placed in binding distribution histograms to observe binding site enrichment.
Analysis of PSC reversal frequency
To estimate the reversal frequency of Rad54–Rad51 PSC translocation events, kymographs were monitored for changes in translocation direction that extended over at least two pixels. If the event fit this criterion, then it was scored as a reversal. The number of reversal events was then placed into a frequency distribution to determine the prevalence of reversal during translocation.
First pass target recognition efficiency
Target binding efficiency was quantified as follows, for each translocation event it was determined if the Rad54–Rad51–ssDNA PSC passed over the spatial position of the expected donor DNA target. If the translocating PSC stopped at the target during its initial encounter then it was assigned a score of 0, indicating that the molecule did not miss the target. However, if the PSC bypassed the target it was given a score of 1, indicating a missed targeting event. The percentage of each event was then determined, and a target utilization efficiency was calculated as the number of targeting events. A similar analysis was used to determine the polarity of target recognition. In this case the data were segregated into PSCs that translocated from above the target and PSCs that translocated from below the target.
PSC processivity measurements
We defined processivity as the linear distance traveled by the PSCs in one direction. If a PSC reversed direction, then this was scored as a separate translocation event with respect to processivity. Using this criterion, the processivity was measured from kymographs by determining the starting and ending positions of the PSCs during each translocation event. The distance traveled (processivity) was then defined using the following: (|Yf − Yi|) × 1, 111 bp; where Yi and Yf correspond to the initial and final pixel position for the PSC along the long axis of the dsDNA. Note, that the PSCs stopped irreversibly upon encountering the chromium barriers or pedestals used to make the DNA curtains, so the reported values likely represent a lower bound on the linear processivity.
RPA–GFP binding analysis
For experiments measuring the association of RPA–GFP with the PSC, GFP signal intensities were measured directly from the kymographs. For calculating of percentage of PSCs associated with RPA–GFP association, the total population of bound Atto565–labeled PSCs was monitored for RPA–GFP co–localization over the course of the experiments. If RPA–GFP associated with the Atto565–DNA at any point in the experiment for longer than 2 frames it was scored as a positive binding event, and at the end of the experiment the RPA–GFP binding events were divided by the total Atto565–DNA binding events to convert the values to percentages. For measuring delays in RPA–GFP association, kymographs were visually inspected for the initial binding of the PSCs. Then intensity traces were generated from the binding event. This allowed a quantitative measure of the number of frames and by extension time before RPA–GFP associated with the bound PSC. This data was then compiled into a distribution and the mean time to RPA–GFP binding was compared for Rad54 and Rad54-K341R. For RPA–GFP binding lifetimes, intensity traces generated from kymographs were analyzed for the duration of each RPA binding event. Based on the lifetimes of these survival events a survival probability plot was generated from these results, and the data were fit by a single exponential decay curve, and the lifetime of RPA–GFP.
GFP–Rad54 and RPA–GFP photobleaching analysis
Rad54–GFP photobleaching experiments were conducted as described above with the exception that they were performed without shuttering. For analysis of photobleaching events, intensity traces were generated from kymographs. A quantitative step size for individual photobleaching events was determined by collecting the intensity information of single GFP photobleaching events. The mean of this data was then determining from a Gaussian distribution indicating that the step size of an individual GFP photobleaching event was 147 ± 20 a.u. (Crickard et al., 2018). The number of photobleaching events was quantified by measuring the number of steps that were greater than equal to this value; note that we scored steps sizes greater than 147 ± 20 a.u. as single bleaching events although they could be due to concurrent bleaching two fluorophores, so the reported values may represent a lower bound on the number of bound proteins. The number of photobleaching steps were counted, and frequency distributions were generated from the resulting data. When possible, these distributions were fit by a Gaussian and the mean number of steps was determined. To estimate the number of GFP–Rad54 within translocating PSCs, we measured the photobleaching steps in experiments containing a 5:1 mixture of unlabeled Rad54 to GFP–Rad54; this dilution was necessary to detect individual GFP photobleaching steps. The total number of Rad54 molecules present within the translocating PSCs was then estimated based upon the assumption that unlabeled Rad54 and GFP–Rad54 were present at a 5:1 molar ratio.
Analysis of PSC encounters with nucleosomes
For analysis of nucleosome outcomes experiments were performed as for the naked CURMID DNA substrate, with the exception that velocity traces were only included in the data analysis if a nucleosome was visibly being pushed by a Rad54–containing PSC. For analysis of collision outcomes, kymographs were scored for event type, nucleosome remodeling, bypass, or stop, based upon visual inspection. Nucleosome remodeling was further divided into two categories, nucleosome sliding and nucleosome eviction. These categories were defined as follows. If the nucleosomes being contacted by the PSC moved greater than 2–pixels (~2,200 bp) it was scored as nucleosome sliding. Likewise, if contact between the PSC and the nucleosome resulted in immediate loss of nucleosome signal, then it was scored as a nucleosome eviction event. Nucleosome bypass outcomes were divided into two categories, bypass with and without pausing within the nucleosome. Pausing within the nucleosome was defined by translocation events that would momentarily pause within the nucleosome for ≥ 2 frames, or in these experiments 20 s, and then resumed translocation. Categorized outcomes were then placed in distributions to visualize the percentage of population.
Supplementary Material
Highlights.
Rad54 acts as a molecular motor that guides Rad51 ssDNA along a donor dsDNA
Rad54 opens the donor DNA, allowing the search take place within an underwound bubble
Homology recognition is dependent on RPA and also on strand polarity
ACKNOWLEDGMENTS
We thank Rodney Rothstein and Lorraine Symington for helpful discussions and comments on the manuscript. This research was funded by NIH grants R35GM118026 (to E.C.G.), R01CA236606 (to E.C.G.), RO1ES007061 and R35CA241801 (to P.S.), P30CA054174 (to the Mays Cancer Center, University of Texas Health Science Center at San Antonio), and P01CA92584 (to E.C.G. and P.S.); and by Cancer Prevention & Research Institute of Texas (CPRIT) Recruitment of Established Investigators (REI) Award RR180029 (to P.S.). C.J.M. was supported by a Boehringer Ingelheim Fonds Ph.D. fellowship. J.B.C. is the Mark Foundation for Cancer Research Fellow for the Damon Runyon Cancer Research Foundation (DRG 2310-17). P.S. is the holder of the Robert A. Welch Distinguished Chair in Chemistry (AQ-0012).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.cell.2020.04.056.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Adzuma K (1998). No sliding during homology search by RecA protein. J. Biol. Chem 273, 31565–31573. [DOI] [PubMed] [Google Scholar]
- Agarwal S, van Cappellen WA, Guénolé A, Eppink B, Linsen SE, Meijering E, Houtsmuller A, Kanaar R, and Essers J (2011). ATP-dependent and independent functions of Rad54 in genome maintenance. J. Cell Biol 192, 735–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexeev A, Mazin A, and Kowalczykowski SC (2003). Rad54 protein possesses chromatin-remodeling activity stimulated by the Rad51-ssDNA nucleoprotein filament. Nat. Struct. Biol 10, 182–186. [DOI] [PubMed] [Google Scholar]
- Alexiadis V, and Kadonaga JT (2002). Strand pairing by Rad54 and Rad51 is enhanced by chromatin. Genes Dev. 16, 2767–2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AlHaj Abed J, Erceg J, Goloborodko A, Nguyen SC, McCole RB, Saylor W, Fudenberg G, Lajoie BR, Dekker J, Mirny LA, and Wu CT (2019). Highly structured homolog pairing reflects functional organization of the Drosophila genome. Nat. Commun 10, 4485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amitani I, Baskin RJ, and Kowalczykowski SC (2006). Visualization of Rad54, a chromatin remodeling protein, translocating on single DNA molecules. Mol. Cell 23, 143–148. [DOI] [PubMed] [Google Scholar]
- Aussel L, Barre FX, Aroyo M, Stasiak A, Stasiak AZ, and Sherratt D (2002). FtsK Is a DNA motor protein that activates chromosome dimer resolution by switching the catalytic state of the XerC and XerD recombinases. Cell 108, 195–205. [DOI] [PubMed] [Google Scholar]
- Barzel A, and Kupiec M (2008). Finding a match: how do homologous sequences get together for recombination? Nat. Rev. Genet 9, 27–37. [DOI] [PubMed] [Google Scholar]
- Bezzubova O, Silbergleit A, Yamaguchi-Iwai Y, Takeda S, and Buerstedde JM (1997). Reduced X-ray resistance and homologous recombination frequencies in a RAD54−/− mutant of the chicken DT40 cell line. Cell 89, 185–193. [DOI] [PubMed] [Google Scholar]
- Cairns BR (2007). Chromatin remodeling: insights and intrigue from single-molecule studies. Nat. Struct. Mol. Biol 14, 989–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceballos SJ, and Heyer WD (2011). Functions of the Snf2/Swi2 family Rad54 motor protein in homologous recombination. Biochim. Biophys. Acta 1809, 509–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. (2012). The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, and Wold MS (2014). Replication protein A: single-stranded DNA’s first responder: dynamic DNA-interactions allow replication protein A to direct single-strand DNA intermediates into different pathways for synthesis or repair. BioEssays 36, 1156–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung WH, Zhu Z, Papusha A, Malkova A, and Ira G (2010). Defective resection at DNA double-strand breaks leads to de novo telomere formation and enhances gene targeting. PLoS Genet. 6, e1000948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comstock MJ, Whitley KD, Jia H, Sokoloski J, Lohman TM, Ha T, and Chemla YR (2015). Protein structure. Direct observation of structure-function relationship in a nucleic acid-processing enzyme. Science 348, 352–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crickard JB, Kaniecki K, Kwon Y, Sung P, Lisby M, and Greene EC (2018). Regulation of Hed1 and Rad54 binding during maturation of the meiosis-specific presynaptic complex. EMBO J. 37, e98728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Tullio L, Kaniecki K, and Greene EC (2018). Single-Stranded DNA Curtains for Studying the Srs2 Helicase Using Total Internal Reflection Fluorescence Microscopy. Methods Enzymol. 600, 407–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggler AL, Inman RB, and Cox MM (2002). The Rad51-dependent pairing of long DNA substrates is stabilized by replication protein A. J. Biol. Chem 277, 39280–39288. [DOI] [PubMed] [Google Scholar]
- Essers J, Hendriks RW, Swagemakers SM, Troelstra C, de Wit J, Bootsma D, Hoeijmakers JH, and Kanaar R (1997). Disruption of mouse RAD54 reduces ionizing radiation resistance and homologous recombination. Cell 89, 195–204. [DOI] [PubMed] [Google Scholar]
- Fan J, and Pavletich NP (2012). Structure and conformational change of a replication protein A heterotrimer bound to ssDNA. Genes Dev. 26, 2337–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forget AL, and Kowalczykowski SC (2012). Single-molecule imaging of DNA pairing by RecA reveals a three-dimensional homology search. Nature 482, 423–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung CW, Fortin GS, Peterson SE, and Symington LS (2006). The rad51-K191R ATPase-defective mutant is impaired for presynaptic filament formation. Mol. Cell. Biol 26, 9544–9554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. (2013). Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 6, pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibb B, Ye LF, Gergoudis SC, Kwon Y, Niu H, Sung P, and Greene EC (2014). Concentration-dependent exchange of replication protein A on single-stranded DNA revealed by single-molecule imaging. PLoS ONE 9, e87922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladyshev E, and Kleckner N (2014). Direct recognition of homology between double helices of DNA in Neurospora crassa. Nat. Commun 5, 3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladyshev E, and Kleckner N (2017). DNA sequence homology induces cytosine-to-thymine mutation by a heterochromatin-related pathway in Neurospora. Nat. Genet 49, 887–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene EC (2016). DNA Sequence Alignment during Homologous Recombination. J. Biol. Chem 291, 11572–11580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene EC, Wind S, Fazio T, Gorman J, and Visnapuu ML (2010). DNA curtains for high-throughput single-molecule optical imaging. Methods Enzymol. 472, 293–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber JE (2018). DNA Repair: The Search for Homology. BioEssays 40, e1700229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hager GL, McNally JG, and Misteli T (2009). Transcription dynamics. Mol. Cell 35, 741–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havas K, Flaus A, Phelan M, Kingston R, Wade PA, Lilley DM, and Owen-Hughes T (2000). Generation of superhelical torsion by ATP-dependent chromatin remodeling activities. Cell 103, 1133–1142. [DOI] [PubMed] [Google Scholar]
- Heyer WD, Ehmsen KT, and Liu J (2010). Regulation of homologous recombination in eukaryotes. Annu. Rev. Genet 44, 113–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ira G, and Haber JE (2002). Characterization of RAD51-independent break-induced replication that acts preferentially with short homologous sequences. Mol. Cell. Biol 22, 6384–6392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaskelioff M, Van Komen S, Krebs JE, Sung P, and Peterson CL (2003). Rad54p is a chromatin remodeling enzyme required for heteroduplex DNA joint formation with chromatin. J. Biol. Chem 278, 9212–9218. [DOI] [PubMed] [Google Scholar]
- Jiang H, Xie Y, Houston P, Stemke-Hale K, Mortensen UH, Rothstein R, and Kodadek T (1996). Direct association between the yeast Rad51 and Rad54 recombination proteins. J. Biol. Chem 271, 33181–33186. [DOI] [PubMed] [Google Scholar]
- Kim S, Liachko I, Brickner DG, Cook K, Noble WS, Brickner JH, Shendure J, and Dunham MJ (2017). The dynamic three-dimensional organization of the diploid yeast genome. eLife 6, e23623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzine F, Gupta A, Baranello L, Wojtowicz D, Ben-Aissa K, Liu J, Przytycka TM, and Levens D (2013). Transcription-dependent dynamic supercoiling is a short-range genomic force. Nat. Struct. Mol. Biol 20, 396–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalczykowski SC (2015). An Overview of the Molecular Mechanisms of Recombinational DNA Repair. Cold Spring Harb. Perspect. Biol 7, a016410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumaran S, Kozlov AG, and Lohman TM (2006). Saccharomyces cerevisiae replication protein A binds to single-stranded DNA in multiple salt-dependent modes. Biochemistry 45, 11958–11973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CS, and Haber JE (2015). Mating-type Gene Switching in Saccharomyces cerevisiae. Microbiol. Spectr 3, Mdna3-0013-2014. [DOI] [PubMed] [Google Scholar]
- Lee JY, Terakawa T, Qi Z, Steinfeld JB, Redding S, Kwon Y, Gaines WA, Zhao W, Sung P, and Greene EC (2015). DNA RECOMBINATION. Base triplet stepping by the Rad51/RecA family of recombinases. Science 349, 977–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CS, Wang RW, Chang HH, Capurso D, Segal MR, and Haber JE (2016). Chromosome position determines the success of double-strand break repair. Proc. Natl. Acad. Sci. USA 113, E146–E154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levens D, Baranello L, and Kouzine F (2016). Controlling gene expression by DNA mechanics: emerging insights and challenges. Biophys. Rev 8, 259–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lia G, Praly E, Ferreira H, Stockdale C, Tse-Dinh YC, Dunlap D, Croquette V, Bensimon D, and Owen-Hughes T (2006). Direct observation of DNA distortion by the RSC complex. Mol. Cell 21, 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisby M, and Rothstein R (2015). Cell biology of mitotic recombination. Cold Spring Harb. Perspect. Biol 7, a016535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisby M, Barlow JH, Burgess RC, and Rothstein R (2004). Choreography of the DNA damage response: spatiotemporal relationships among checkpoint and repair proteins. Cell 118, 699–713. [DOI] [PubMed] [Google Scholar]
- Liu LF, and Wang JC (1987). Supercoiling of the DNA template during transcription. Proc. Natl. Acad. Sci. USA 84, 7024–7027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazin AV, Bornarth CJ, Solinger JA, Heyer WD, and Kowalczykowski SC (2000a). Rad54 protein is targeted to pairing loci by the Rad51 nucleoprotein filament. Mol. Cell 6, 583–592. [DOI] [PubMed] [Google Scholar]
- Mazin AV, Zaitseva E, Sung P, and Kowalczykowski SC (2000b). Tailed duplex DNA is the preferred substrate for Rad51 protein-mediated homologous pairing. EMBO J. 19, 1148–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta A, and Haber JE (2014). Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harb. Perspect. Biol 6, a016428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta A, Beach A, and Haber JE (2017). Homology Requirements and Competition between Gene Conversion and Break-Induced Replication during Double-Strand Break Repair. Mol. Cell 65, 515–526.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimonkar AV, Amitani I, Baskin RJ, and Kowalczykowski SC (2007). Single molecule imaging of Tid1/Rdh54, a Rad54 homolog that translocates on duplex DNA and can disrupt joint molecules. J. Biol. Chem 282, 30776–30784. [DOI] [PubMed] [Google Scholar]
- Oh SD, Lao JP, Hwang PY, Taylor AF, Smith GR, and Hunter N (2007). BLM ortholog, Sgs1, prevents aberrant crossing-over by suppressing formation of multichromatid joint molecules. Cell 130, 259–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papamichos-Chronakis M, and Peterson CL (2013). Chromatin and the genome integrity network. Nat. Rev. Genet 14, 62–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson CL, and Almouzni G (2013). Nucleosome dynamics as modular systems that integrate DNA damage and repair. Cold Spring Harb. Perspect. Biol 5, a012658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petukhova G, Stratton S, and Sung P (1998). Catalysis of homologous DNA pairing by yeast Rad51 and Rad54 proteins. Nature 393, 91–94. [DOI] [PubMed] [Google Scholar]
- Petukhova G, Van Komen S, Vergano S, Klein H, and Sung P (1999). Yeast Rad54 promotes Rad51-dependent homologous DNA pairing via ATP hydrolysis-driven change in DNA double helix conformation. J. Biol. Chem 274, 29453–29462. [DOI] [PubMed] [Google Scholar]
- Petukhova G, Sung P, and Klein H (2000). Promotion of Rad51-dependent D-loop formation by yeast recombination factor Rdh54/Tid1. Genes Dev. 14, 2206–2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piazza A, Wright WD, and Heyer WD (2017). Multi-invasions Are Recombination Byproducts that Induce Chromosomal Rearrangements. Cell 170, 760–773.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad TK, Robertson RB, Visnapuu ML, Chi P, Sung P, and Greene EC (2007). A DNA-translocating Snf2 molecular motor: Saccharomyces cerevisiae Rdh54 displays processive translocation and extrudes DNA loops. J. Mol. Biol 369, 940–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Z, Redding S, Lee JY, Gibb B, Kwon Y, Niu H, Gaines WA, Sung P, and Greene EC (2015). DNA sequence alignment by microhomology sampling during homologous recombination. Cell 160, 856–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragunathan K, Liu C, and Ha T (2012). RecA filament sliding on DNA facilitates homology search. eLife 1, e00067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raschle M, Van Komen S, Chi P, Ellenberger T, and Sung P (2004). Multiple interactions with the Rad51 recombinase govern the homologous recombination function of Rad54. J. Biol. Chem 279, 51973–51980. [DOI] [PubMed] [Google Scholar]
- Renkawitz J, Lademann CA, Kalocsay M, and Jentsch S (2013). Monitoring homology search during DNA double-strand break repair in vivo. Mol. Cell 50, 261–272. [DOI] [PubMed] [Google Scholar]
- Renkawitz J, Lademann CA, and Jentsch S (2014). Mechanisms and principles of homology search during recombination. Nat. Rev. Mol. Cell Biol 15, 369–383. [DOI] [PubMed] [Google Scholar]
- Ristic D, Wyman C, Paulusma C, and Kanaar R (2001). The architecture of the human Rad54-DNA complex provides evidence for protein translocation along DNA. Proc. Natl. Acad. Sci. USA 98, 8454–8460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- San Filippo J, Sung P, and Klein H (2008). Mechanism of eukaryotic homologous recombination. Annu. Rev. Biochem 77, 229–257. [DOI] [PubMed] [Google Scholar]
- Sherman F (1991). Getting started with yeast. Methods Enzymol. 194, 3–21. [DOI] [PubMed] [Google Scholar]
- Sinha M, and Peterson CL (2009). Chromatin dynamics during repair of chromosomal DNA double-strand breaks. Epigenomics 1, 371–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solinger JA, Lutz G, Sugiyama T, Kowalczykowski SC, and Heyer WD (2001). Rad54 protein stimulates heteroduplex DNA formation in the synaptic phase of DNA strand exchange via specific interactions with the presynaptic Rad51 nucleoprotein filament. J. Mol. Biol 307, 1207–1221. [DOI] [PubMed] [Google Scholar]
- Sugawara N, Wang X, and Haber JE (2003). In vivo roles of Rad52, Rad54, and Rad55 proteins in Rad51-mediated recombination. Mol. Cell 12, 209–219. [DOI] [PubMed] [Google Scholar]
- Sung P, and Stratton SA (1996). Yeast Rad51 recombinase mediates polar DNA strand exchange in the absence of ATP hydrolysis. J. Biol. Chem 271, 27983–27986. [DOI] [PubMed] [Google Scholar]
- Symington LS, Rothstein R, and Lisby M (2014). Mechanisms and regulation of mitotic recombination in Saccharomyces cerevisiae. Genetics 198, 795–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavares EM, Wright WD, Heyer WD, Le Cam E, and Dupaigne P (2019). In vitro role of Rad54 in Rad51-ssDNA filament-dependent homology search and synaptic complexes formation. Nat. Commun 10, 4058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teves SS, and Henikoff S (2014). Transcription-generated torsional stress destabilizes nucleosomes. Nat. Struct. Mol. Biol 21, 88–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thon G, Maki T, Haber JE, and Iwasaki H (2019). Mating-type switching by homology-directed recombinational repair: a matter of choice. Curr. Genet 65, 351–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Komen S, Petukhova G, Sigurdsson S, Stratton S, and Sung P (2000). Superhelicity-driven homologous DNA pairing by yeast recombination factors Rad51 and Rad54. Mol. Cell 6, 563–572. [DOI] [PubMed] [Google Scholar]
- Van Komen S, Macris M, Sehorn MG, and Sung P (2006). Purification and assays of Saccharomyces cerevisiae homologous recombination proteins. Methods Enzymol. 408, 445–463. [DOI] [PubMed] [Google Scholar]
- von Hippel PH, and Berg OG (1989). Facilitated target location in biological systems. J. Biol. Chem 264, 675–678. [PubMed] [Google Scholar]
- Weiner BM, and Kleckner N (1994). Chromosome pairing via multiple interstitial interactions before and during meiosis in yeast. Cell 77, 977–991. [DOI] [PubMed] [Google Scholar]
- Wright WD, and Heyer WD (2014). Rad54 functions as a heteroduplex DNA pump modulated by its DNA substrates and Rad51 during D loop formation. Mol. Cell 53, 420–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue C, Wang W, Crickard JB, Moevus CJ, Kwon Y, Sung P, and Greene EC (2019). Regulatory control of Sgs1 and Dna2 during eukaryotic DNA end resection. Proc. Natl. Acad. Sci. USA 116, 6091–6100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, and Dudko OK (2016). First-Passage Processes in the Genome. Annu. Rev. Biophys 45, 117–134. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Smith CL, Saha A, Grill SW, Mihardja S, Smith SB, Cairns BR, Peterson CL, and Bustamante C (2006). DNA translocation and loop formation mechanism of chromatin remodeling by SWI/SNF and RSC. Mol. Cell 24, 559–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Fan HY, Goldman JA, and Kingston RE (2007). Homologydriven chromatin remodeling by human RAD54. Nat. Struct. Mol. Biol 14, 397–405. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All kymographs used as the original source for data analysis throughout the paper are available on Mendeley [https://doi.org/10.17632/x2cb2mkhty.1].