

A single-cell atlas defines cell type composition and cell state transitions in zebrafish caudal fin regeneration.
Abstract
Zebrafish faithfully regenerate their caudal fin after amputation. During this process, both differentiated cells and resident progenitors migrate to the wound site and undergo lineage-restricted, programmed cellular state transitions to populate the new regenerate. Until now, systematic characterizations of cells comprising the new regenerate and molecular definitions of their state transitions have been lacking. We hereby characterize the dynamics of gene regulatory programs during fin regeneration by creating single-cell transcriptome maps of both preinjury and regenerating fin tissues at 1/2/4 days post-amputation. We consistently identified epithelial, mesenchymal, and hematopoietic populations across all stages. We found common and cell type–specific cell cycle programs associated with proliferation. In addition to defining the processes of epithelial replenishment and mesenchymal differentiation, we also identified molecular signatures that could better distinguish epithelial and mesenchymal subpopulations in fish. The insights for natural cell state transitions during regeneration point to new directions for studying this regeneration model.
INTRODUCTION
The ability to regenerate complex body parts varies considerably in the animal kingdom. While planarian and hydra are able to regenerate their entire bodies, many avian and mammalian species mostly stop at the wound healing stage without a reparative regeneration process (1). This disparity may result from complexity differences among organisms by nature, yet it leaves us the hope that we may learn from highly regenerative species to improve our own regenerative potential.
Zebrafish is known for its ability to regenerate multiple complex body structures (2). Among regenerable tissues, the caudal fin serves as a great model due to its faithful and rapid regeneration, ease of manipulation, and relatively low complexity. A key step in regeneration is the formation of the blastema, a layer of proliferative and undifferentiated cells that accumulates between the wound site and the wound epidermis following initial wound closure. This step occurs in response to appendage loss and is one of the key features that separates regenerative systems from nonregenerative systems. At later stages of regeneration, the blastema further proliferates and differentiates to regenerate the missing complex structures.
However, the molecular signatures of blastemal cell state transitions during regeneration in zebrafish remain elusive. The state of a cell can be represented by its collective gene expression profile, which has only been measured in bulk for all genes or in specific lineages of cells for a subset of genes during caudal fin regeneration. Prior work has shown that both proliferation of progenitors and dedifferentiation of adult lineage cells contribute to the blastema (3–8). Progenitors respond to injury cue and proliferate as in normal development. Cells derived from mature adult lineages, however, lose their lineage-specific markers while obtaining progenitor-like markers when they proliferate. Neither type of cell gains multipotency, but rather, they proliferate and regenerate with lineage restrictions. The limited resolution and throughput of these approaches have prevented a more systematic understanding of blastema cells. The advent of single-cell transcriptomic technologies promises to reveal signals masked at the bulk tissue level (9), granting us an opportunity to define and monitor cellular state transition in regenerating fin at an unprecedented resolution.
In this study, we generated single-cell transcriptomic maps of regenerating fin tissue. These maps allowed us to separate the contribution from different cell types and track the transcriptomic dynamics in cell state transitions during regeneration. By comparing with the profiles obtained from uninjured fin tissue, we identified cell types involved in regeneration. We demonstrated the activation of cell cycle–related programs shared across cell types as well as cell type–specific programs. Furthermore, we defined the heterogeneity in both epithelial and blastemal populations and their functional relations to the regeneration process.
RESULTS
Regenerating fins comprise the same cell types as uninjured fins
To better understand cell type involvement in fin regeneration, we characterized single-cell transcriptional landscapes for both preinjury and regenerating caudal fin tissues using the 10x Genomics platform (see Materials and Methods and table S1) (9). We sampled regenerating fins from 1, 2, and 4 days post-amputation (dpa) time points to interrogate the stages of blastema formation, outgrowth, and maintenance (Fig. 1A). Fin samples were collected from multiple fish to control for individual variation while at the same position along the proximal-distal axis to avoid positional effects. To establish the transcriptional ground states for each cell type in the fin tissue, we first focused on cells collected from the preinjury time point. Via an unsupervised clustering of 4134 cells, we identified epithelial cells (epcam and cdh1), hematopoietic cells (mpeg1.1 and cxcr3.2), and mesenchymal cells (msx1b and twist1a) (fig. S1, A and B) (10–14). Epithelial cells are from three transcriptionally distinct subgroups, representing the superficial (krt4), intermediate (tp63), and basal layers (tp63 and krtt1c19e) of the epithelium (fig. S1, A and B) (15, 16).
Fig. 1. Cell type identification in zebrafish caudal fins.
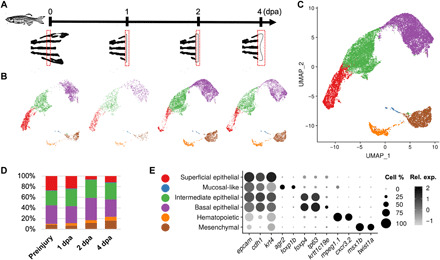
(A) General experimental design. Zebrafish caudal fin tissues at preinjury and 1/2/4 dpa stages were collected. (B) Clustering assignments for caudal fin cells collected from each stage. Uniform Manifold Approximation and Projection (UMAP) axes were calculated from the integrated cells dataset as in (C). (C) Clustering assignments for caudal fin cells collected from both preinjury and regenerating stages. Cells were plotted on UMAP axes. Color coding is the same as in (E). (D) Percentage distribution of the major cell types captured in caudal fin, grouped by their stage of collection. Color coding is the same as in (E). (E) Differential expressions of the key marker genes by the identified major cell types. Color gradient: normalized relative expression level. Dot size: percentage of cells in the cluster that express the specified gene.
To determine whether the same cell types existed in the regenerating stages, we performed analysis using two different approaches: (i) Cells from each stage were clustered independently, and (ii) cells from both uninjured fins and injured fins were integrated through the anchoring approach (see Materials and Methods; Fig. 1, B, C, and E; and table S2) (17). For both approaches, we regressed out cell cycle effects before principal components analysis (PCA). Agreement between cluster assignments was measured using Hubert and Arabie’s adjusted Rand index (ARI). An average ARI of 0.86 (preinjury, 0.86; 1 dpa, 0.85; 2 dpa, 0.90; and 4 dpa, 0.83) indicated that clustering results generated using the two approaches were highly consistent. Cell types identified in the preinjury cells presented consistently across all regenerating stages, suggesting that regenerating fins contain the same cell types as the preinjury fins.
Common and cell type–specific programs that regulate cell cycle reentry
New regenerates are built up by the proliferation and migration of cells located at a number of fin segments away from the amputation plane (2). In response to injury cues, these cells gained the ability to detach from local tissue, enter cell cycle, and migrate toward the wound site while undergoing transcriptional reprogramming. We computationally separated S phase, G2-M phase, and G1-phase cells based on the expression level of cell cycle–related genes and performed clustering analysis using only S phase cells (see Materials and Methods and fig. S2A). In this cycling cell population, we identified epithelial, mesenchymal, and hematopoietic cell groups as before (Fig. 2, A to C, and table S3). Our data support a model in which cells likely keep their original identities during proliferation.
Fig. 2. Cell type identification of cycling cells.
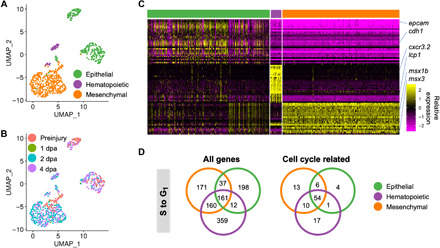
(A) Cell type clustering of S phase cells plotted onto UMAP axes calculated by S phase cell only. Cells are colored by the general cell types merged from major cells types in Fig. 1B. (B) Stage distribution of S phase cells. Cells were plotted on the same UMAP axes as in (A) and colored by stage when the cells were collected. (C) Relative expression levels of the top 30 differentially expressed genes from each cluster of only S phase cells. (D) Venn diagrams of numbers of genes shared between the cell cycle–activated genetic programs. Left, included all genes; right, included only cell cycle–related genes (see Materials and Methods).
Next, we asked whether different regenerating cell types exhibited similar or distinct cell cycle regulations. To this end, we identified genes up-regulated in S phase cells compared to G1 phase cells in each cell type, respectively (logFC, >0.25; minimum percentage, >10%). Of the 1098 differentially expressed genes, 161 were shared across all three groups of comparisons (Fig. 2D and table S4). Of these shared genes, at least 54 genes were related to cell cycle regulation, underscoring a shared program governing cell cycle reentry (criteria described in Materials and Methods). In contrast, hundreds of genes differentially highly expressed in S phase exhibited cell type–specific pattern, of which dozens were related to cell cycle (Fig. 2D). We next evaluated the degree of conservation of these enriched genes by asking what fraction did not have human orthologs that had been curated in the Metascape database (18). Twenty-five percent of genes in the epithelial cell–specific group had no human ortholog, whereas all shared groups had at most 15% genes without a human ortholog, suggesting that enriched genes shared by cell types were more evolutionarily conserved (fig. S2C).
Some cell type–specific S-G1 enriched genes were also expressed in a cell type–specific manner regardless of their cell cycle phases: For example, psmb8a and psmb9a shared similar epithelial-hematopoietic enrichments (fig. S2D). The human homologs of these genes (PSMB8 and PSMB9) encode β5i and β1i subunits of the immunoproteasome (19). Together with β2i and PA28 subunits of the proteasome, they turn the proteasome into immunoproteasome and take part in immune response (20). Immunoproteasome digests peptides more efficiently, promoting antigen presentation by a major histocompatibility complex (MHC) class I molecule. Although they did not pass the differential expression criteria in the S-G1 comparison, zebrafish psmb10, psme1, and psme2 shared a differential expression signature similar to that of psmb8a and psmb9a, suggesting that zebrafish might use the same group of subunits for the assembly of immunoproteasomes that might help increase immune responses during regeneration, especially in epithelial and hematopoietic cells (fig. S2, D and E). In addition, we found three genes that shared the same expression signature with the immunoproteasome subunits (psmb13a, psmb12, and psma6l) (fig. S2E) without known human or mouse homologs, suggesting that they might form zebrafish-specific proteasomes with functional relevance to regeneration (19).
Diverse epithelial populations are involved in regeneration
Consistent with current knowledge, we observed three transcriptionally distinct subgroups in the preinjury epcam+ epithelial cells, representing the superficial, intermediate, and basal layers of the adult zebrafish epithelium (Fig. 3A and fig. S1B) (15, 16). By integrating cells from all stages during regeneration, we found clusters of cells that corresponded to all three layers of the epithelium after injury (Fig. 1, B and C). In addition, we captured a rare agr2+ population (referred to as “mucosal-like epithelium” herein) that was too small to be clustered by itself in the preinjury stage (Fig. 1E). These cells shared general epithelial features with the other epithelial layers but exhibited higher expression of a unique set of 200 genes. We examined the expression distribution of the orthologs of these genes in human tissues (The Human Protein Atlas, http://proteinatlas.org/) (21). Among the top 30 genes with human orthologs, 11 showed enriched expressions in proximal digestive or gastrointestinal tract and another 11 in bone marrow of blood lineages, suggesting that this population is analogous to cells in the mucosa in mammalian systems (table S2). In zebrafish, agr2 is required for the differentiation of the mucosal-producing goblet cells in the intestinal epithelium (22). To confirm the cell type–specific expression pattern of this gene in the fin tissue, we performed in situ hybridization on agr2 in both uninjured and regenerating fin tissues (see Materials and Methods). agr2 transcripts are scattered within the epithelium regardless of the sample collection stage and reflect a round morphology of the cell expressing it (fig. S3, A, C, E, and G to I). A proportion of agr2+ cells overlap with positive dark blue staining of Alcian blue in serial sections, suggesting that these cells are mucous cells that are known to exist in the caudal fin epithelium (fig. S3, B, D, and F) (23).
Fig. 3. Epithelial cell diversity though regeneration.
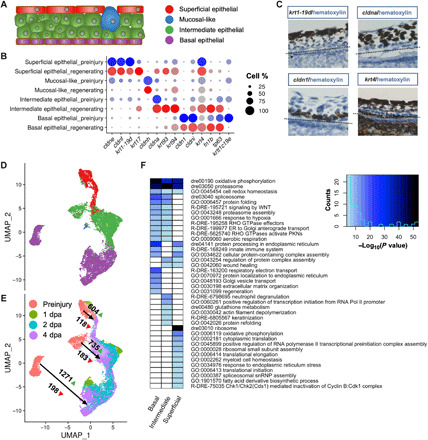
(A) Diagram of the stratified adult zebrafish epithelium. (B) Differential expressions of claudin family and keratin family genes in epithelial subgroups shown as a dot plot. Known epithelial markers krt4, fn1b, tp63, and krtt1c19e were included for comparison. Cells were first grouped by major cell types and then separated into preinjury and regenerating stages. Darkness of dot color: relative expression level. Dot size: percentage of cells in the cluster that express the specified gene. (C) In situ hybridization targeting krt1-19d, cldna, cldn1, and krt4 of 4-dpa fin tissues. Brown dots indicate positive RNA signals from target genes, while pale blue blocks represent hematoxylin-stained cell nuclei. Zoomed-in views are presented. Original images can be found in fig. S4. All epithelial layers are above the black dotted lines. (D) Clustering assignment of epithelial cells plotted on UMAP axes calculated with only epithelial cells. Cells are colored by their epithelial layer identity as in (A). (E) The same UMAP visualization as in (D), with cells colored by stage of collection. Arrows connect the groups of comparison, with a direction from preinjury stage to regenerating stages (1, 2, and 4 dpa). Numbers next to the green triangle: number of genes up-regulated in regenerating stage. Numbers next to the red triangle: number of genes down-regulated in regenerating stage. (F) Clustered GO enrichment for genes up-regulated in regenerating basal, intermediate, and superficial epithelial cells comparing to their preinjury counterparts. GTPase, guanosine triphosphatase; ER, endoplasmic reticulum; PKN, protein kinases N; snRNP, small nuclear ribonucleoprotein.
Although the same three-layer classification of epithelial cells could be defined when cells from regenerating stages were integrated with the preinjury cells, the expression of the commonly used layer-specific marker genes changed dramatically during regeneration: Superficial epithelial marker krt4 expanded into basal and intermediate layers of the epithelium, the intermediate layer marker fn1b was also highly expressed in the basal layer, and the basal epithelial marker krtt1c19e was barely detectable in the postinjury cell populations (Fig. 3B) (15, 16). To better understand the molecular features of the epithelial populations, we identified genes significantly more highly expressed in epithelial cells than in hematopoietic and mesenchymal cells and found that cell-cell junction genes ranked high in the list. Among these, genes from the claudin and keratin families were detected at a ratio 20-fold higher than that in randomly selected detectable genes (χ2 test, P value of <0.0001). We focused on expression patterns of all claudin and keratin genes in zebrafish and found that cldne, cldnf, krt1-19d, and krt17 labeled the superficial cluster; cldnh labeled the mucosal-like cluster; cldna, krt93, and krt94 labeled the intermediate cluster; and cldn1 and cldni labeled the basal cluster (Fig. 3B). Claudin genes are expressed in a tissue-specific manner in zebrafish and are generally considered to be the proteins responsible for regulating the paracellular permeability in the vertebrate epithelium (24). Their differential expression signature in both uninjured and regenerating tissues suggests that they might play important roles in maintaining the permeability in each epithelial population. On the other hand, the expression of keratin genes displayed less restriction across the three layers relative to claudin genes but stronger dependence on regenerating states (Fig. 3B). The differential expression signature suggests that they might perform epithelial subtype–related function in regeneration. To verify their expression pattern, we performed RNA in situ hybridization targeting the known marker krt4 and new candidates, including krt1-19d, cldna, and cldn1 (Fig. 3C) as well as cldne, krt94, and cldni (fig. S4, A to H). Comparing with the known marker krt4, these genes exhibited more restricted expression patterns in epithelial layers, better representing the molecular signatures of different epithelial populations in the fin tissue regardless of regeneration status (Fig. 3, B and C).
The three epithelial layers were present across the regeneration stages albeit with varying proportions (Fig. 1D). The proportion of basal epithelial cells peaked at 2 dpa, reaching up to 42%, whereas the superficial layer epithelial cells decreased from 27 to 6% at 2 dpa (the coefficient of variations of cell proportions between biological replicates is below 15%). The observed compositional change of the two epithelial populations is consistent with a previous finding that the initial migration of superficial layer cells to the new regenerates is followed by replenishment by basal epithelial cells (25). This basal replenishment was also reflected in the two-dimensional Uniform Manifold Approximation and Projection (UMAP) calculation from only epithelial cells, in which preinjury cells were separated by their respective layers, whereas regenerating cells were closer in the projection space (Fig. 3, D and E). Superficial layer cells from before and after injury stages were in juxtaposition to each other, consistent with our knowledge that this layer of epithelial cells directly migrates to and covers the wound site (25). On the other hand, basal layer cells from before and after injury stages were more distantly separated, suggesting more dramatic changes between resting and regenerating basal epithelial cells.
To understand the mechanisms of epithelium regeneration, we compared the transcriptome between preinjury and regenerating cells for the three epithelial layers. Basal layer cells up-regulated 1271 genes and down-regulated 198 genes during regeneration; both were the highest numbers across all comparisons (numbers of differentially expressed genes were from Wilcoxon rank sum test, adjusted P value of < 0.01; Fig. 3E). We performed gene ontology (GO) enrichment analysis on genes up-regulated in the regenerating stage by layer and found both common and layer-specific programs associated with regeneration (18). All three layers were enriched for oxidative phosphorylation (dre00190), proteasome (dre03050), and cell redox homeostasis (GO:0045454). While basal and intermediate layer cells could be regulated by Rho guanosine triphosphatase–mediated Wnt signaling for extracellular matrix organization and actin filament depolymerization, respectively (R-DRE-195258, R-DRE-5625740, R-DRE-195721, GO:0030198, and GO:0030042), superficial layer cells showed enrichment mainly for general transcriptional and translational regulations (Fig. 3F). When comparing the expression profiles between regenerating superficial epithelial and basal epithelial, we saw enrichment for antigen presentation and apoptosis features in the superficial layer (table S5). In addition, the superficial layer contained the lowest proportion of cells in S phase or G2-M phase, further supporting that superficial layer epithelium was most likely maintained by migration and proliferation from other layers (fig. S2B).
Subcluster identification within regenerating basal epithelial cells revealed two subgroups that represented different functionalities during regeneration, one labeled by distally distributed fgf24, while the other by proximally distributed lef1 (fig. S5, A to C) (26, 27). We compared expression profiles between group I (distal) and group II (proximal) cells and found that their suggested functionalities were consistent with their expected roles in regeneration: The distal subgroup (or distal wound epidermis) up-regulated genes associated with extracellular matrix degradation, and the proximal subgroup (or proximal wound epidermis) up-regulated genes associated with organization of extracellular matrix, skeletal system development, and negative regulation of locomotion (fig. S5, D and E). In addition, the increase of proximal cell proportion was accompanied by the decrease of distal cell proportion, suggesting that basal layer epithelium become gradually active in promoting blastema proliferation and differentiation during the initial regeneration process (fig. S5C). To confirm the distribution of these cells, we performed RNA in situ hybridization targeting two candidate genes, stmn1b and sema3b, one from each cluster. The expression of stmn1b was first observed at the basal layer of the wound epidermis at 1 dpa but diminished as regeneration proceeded (fig. S4, I to K). On the contrary, sema3b showed expression at later stages and was enriched in the relatively proximal portion of the basal layer epithelium (fig. S4, L to N). The expression dynamics of these two genes matched the predicted proportion changes of the two clusters (fig. S5C). While sema3b was more restricted to the basal layer, stmn1b showed low expression levels in the intermediate layer as well, potentially suggesting that this subpopulation could be labeling cells transitioning from the basal layer to the other layers of epithelium.
Hematopoietic cell activation during regeneration
We next performed subcluster analysis within the hematopoietic cluster and found four subpopulations (Fig. 4, A to C and table S6). The first three populations were enriched for the macrophage marker mpeg1.1, with the cluster H1 being M1-like (lgals2+ and lcp1+) and the cluster H3 M2-like (ctsc+ and lgmn+) (Fig. 4D) (12). We speculated that the cluster H2 represented a transition state between M1-like and M2-like or a state before the macrophages differentiate toward M1-like or M2-like. From 1 to 4 dpa, the proportion of M1-like macrophages remained at a constant level, while that of M2-like macrophages expanded (Fig. 4B), potentially suggesting a shift in the function of macrophages in the new regenerates from pro-inflammatory toward anti-inflammatory as regeneration proceeded. Macrophages are important for proper blastema proliferation (28). The change in the proportions of M1/M2-like macrophage in our data matched with that observed in the larvae fin, suggesting that the adults followed a similar rule for immune cell recruitment after injury.
Fig. 4. Subtypes in the hematopoietic cell cluster.
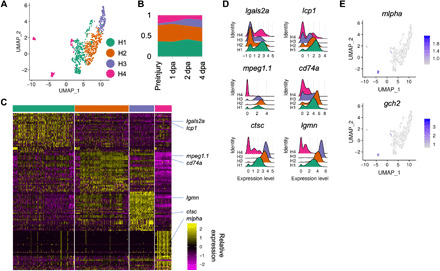
(A) Subcluster assignments of the hematopoietic cells. Cells were plotted on UMAP axes. The same color code is used for (B) to (D). (B) Proportion of subgroups of hematopoietic cells. (C) Expression enrichment of the top 30 differentially expressed genes in the four subclusters within hematopoietic cluster shown as a heatmap. (D) Expression distribution of genes associated with macrophage activation grouped by subclusters. Expression levels were log normalized by Seurat. y axis: cluster identity. z axis: cell density. (E) Expressions of pigment cell markers gch2 and mlpha in the hematopoietic population.
The cluster H4 enriched for genes including mlpha and gch2, both well-characterized markers for the chromatophore lineages in zebrafish (Fig. 4E) (29). Chromatophores are derived from neural crest lineage, yet here, they clustered with macrophages that were from hematopoietic lineage. One possibility is that this clustering result could be driven by features related to antigen presentation via MHC class II, a feature of pigment cells based on studies using human melanocytes (30). The proportion of this cluster decreased as regeneration started, agreeing with the known pattern of fin stripe recovery after amputation (Fig. 4B) (31).
Mesenchymal regenerations—Building and supporting the bone
To understand the component and function of the cells in the mesenchymal cell cluster before and during fin regeneration, we focused on genes enriched in this cluster and found previously identified blastema marker genes that are required for fin regeneration, including the muscle segment homeobox family members msx1b and msx3 and the insulin-like growth factor signaling ligand igf2b (logFC, >0.25; minimum percentage, >25%; and adjusted P value of <1 × 10−5, as listed in table S1) (2, 13). The mesenchymal cluster expressed these genes nearly exclusively, confirming their blastema identity in regenerating stages (fig. S6A). In addition, we found key genes involved in zebrafish bone development and regeneration: twist1a, the transcription factor that controls the skeletal development by regulating the expression of runx2 (14); cx43, the gap junction protein required for building the fin ray up to the right length; and hapln1a and serpinh1b, two genes downstream of cx43 (32, 33). By performing conserved marker analysis using Seurat, we found that msx1b and twist1a were also among the markers conserved across all stages, underscoring shared features that existed between regenerating and preinjury mesenchymal cells (maximum P values across stages: 4.72 × 10−10 and 2.84 × 10−9 for msx1b and twist1a, respectively). This theme of building and supporting bone tissues in mesenchymal cells was not only reflected by a handful of genes: GO analysis of all the detected up-regulated genes in this cluster revealed significant enrichment of genes associated with GO terms, including fin regeneration (GO:0031101) and skeletal system development (GO:0001501) (fig. S6B). When more stringent criteria for differential expression were used, genes were also significantly enriched for GO terms, including skeletal system morphogenesis (GO:0048705) and extracellular matrix organization (GO:0030198) (fig. S6C).
Previous work has shown that blastema comprises bone cells and non-bone cells but has not defined the cell types and the regeneration process of each type (23, 34, 35). To better understand the regeneration process by cell type, we performed clustering analysis within the mesenchymal cluster and identified nine distinct subgroups (Fig. 5A and fig. S6D). Of the two preinjury subgroups, M-2 represented the mature bone lineage, which was enriched for expressions of bglap, mgp, and sost (fig. S6E) (36, 37). Comparing to M-2, cluster M-1 presented low expression levels of bglap, mgp, and sost and high expression levels of a group of other genes, including fhl1a, fhl2a, and tagln (fig. S6E). Mammalian orthologs of these genes are required for chondrogenesis and osteogenesis, leading us to speculate that cluster M-1 could represent the supporting non-bone cell lineage in the preinjury state (38, 39).
Fig. 5. Putative bone and non-bone cell lineages in the blastema.
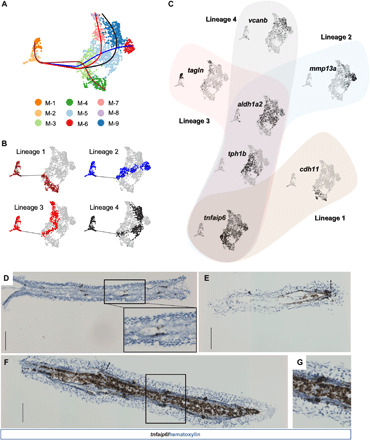
(A) Subclustering assignments of mesenchymal cells shown on UMAP axes. Cells are colored by their cluster assignments and connected by Slingshot-reconstructed trajectories. Lineage 1: 1-2-3-4; lineage 2: 1-2-3-5-6; lineage 3: 1-2-3-5-7-8; lineage 4: 1-2-3-5-9. (B) By-lineage highlighting of mesenchymal cells. Cells with colors other than gray represent the cells included in each corresponding lineage in (A). (C) Expression distribution of genes labeling cell lineages and cell states in mesenchymal cells. Gene feature plots were connected by estimated lineages using the same lineage color code as in (A). (D to G) In situ hybridization targeting the tnfaip6 gene in (D) preinjury, (E) 1-dpa, and [(F) and (G)] 4-dpa fin tissues. Brown dots indicate positive RNA signals from target genes, while pale blue blocks represent hematoxylin-stained cell nuclei. A zoomed-in view for the region inside the focused rectangle is provided within (D). (G) Zoomed-in view for the region highlighted by a rectangle in (F). Dotted lines indicate the amputation plane. All scale bars, 100 μm.
The remaining seven populations came from regenerates. Pseudotime analysis via Slingshot (40) suggested that these subgroups formed four trajectories, all initiated from the tnfaip6+ cluster (M-3), which was composed mainly of 1-dpa cells (Fig. 5, B and C, and fig. S6D). tnfaip6 was ranked top by an adjusted P value in the differentially expressed genes labeling the regeneration initiation cluster and was also expressed exclusively in the mesenchymal cluster (Fig. 5C and fig. S6A). The mammalian ortholog of this gene is required for proliferation and proper differentiation of mesenchymal stem cells (MSCs) and balances the mineralization via osteogenesis inhibitions (41). The expression of tnfaip6 in the postinjury zebrafish fin suggested that it could also be required in the early stages of regeneration for promoting mesenchymal proliferation. To confirm the expression pattern of tnfaip6, we performed RNA in situ hybridization for uninjured and regenerating fin tissues targeting this gene (Fig. 5, D and E). In the uninjured fin, tnfaip6 was expressed in a segmental pattern, presumably enriching at joints between bone segments. At 1 dpa, tnfaip6 was expressed not only near the bony rays but also in the cavity, showing a general activation in the mesenchymal population. As regeneration proceeded from 1 to 4 dpa, mesenchymal cells divided into cdh11+ (M-4) and tph1b+ (M-5) branches, with the latter further divided into mmp13a+ (M-6), tagln+ (M-7), and vcanb+ (M-9) branches (Fig. 5C and fig. S6D). The mmp13+ (M-6) cluster maintained a high-level tnfaip6 expression, whereas all other branches had a lower but detectable tnfaip6 expression. This was consistent with the observation we made from in situ hybridization at 4 dpa targeting tnfaip6: the broad expression in the mesenchymal population and segmental enrichments similar to that in the uninjured fin (Fig. 5, F and G).
The four trajectories initiated from the tnfaip6+ cluster revealed four putative lineages representing bone and non-bone cells in the blastema. cdh11+ lineage 1 specifically expressed runx2 and osterix/sp7, which are the key transcription factors regulating osteogenesis (fig. S6E) (42). Mammalian ortholog of cdh11 could induce Sp7-dependent bone and cartilage formation in vivo, suggesting that the cdh11+ branch in the blastema represented the regenerating osteoblasts (43). Genes highly expressed at the end of this lineage (M-4) compared to the initiation point (M-3) were associated with bone mineralization and skeletal system development, further supporting their bone cell identity (table S7).
Mesenchymal cells outside the osteoblast branch shared enrichment for tph1b and aldh1a2 expressions at 2 dpa, followed by and1 expression at 4 dpa (Fig. 5C and fig. S6F). These three genes had been suggested to label joint fibroblasts, fibroblast-derived blastema cells, and actinotrichia-forming cells in the blastema, respectively (34, 35, 44). However, their expression signatures implied that instead of labeling separate populations in the blastema, they might be labeling different states of the same non-osteoblastic cells at the early stage of fin regeneration.
Upon 4 dpa, these non-osteoblastic cells diverged into three groups (Fig. 5C and fig. S6D). To understand this separation, we performed differential expression analysis for each branch between cells at the end of the lineage tree (lineage 1, M-4; lineage 2, M-6; lineage 3, M-7 and M-8; and lineage 4, M-9) and cells in the initiation cluster (M-3). Genes highly expressed at the lineage end points were included for GO analysis for functional predictions (logFC, >0.25; minimum percentage of >25%; and adjusted P value of <0.01). These three lineages were also associated with skeletal system development or extracellular matrix organizations as were the bone cell lineage; however, the association was driven by a nearly completely different set of genes (table S7). Unlike the osteoblast lineage, none of these three non-bone cell lineages showed enrichment for bone mineralization, suggesting that these cells might indirectly contribute to bone formation. In lineage 2, top differentially expressed genes mmp13a and ogn both have mammalian orthologs that are associated with bone formation (Fig. 5C and fig. S6F) (45, 46). In addition, this lineage presented up-regulation of DLX family genes, especially dlx5a, suggesting the reactivation of fin outgrowth–related developmental programs during regeneration (fig. S6F and table S7) (47). Lineages 3 and 4 both enriched for estrogen response and expressed the retinoic acid (RA) synthesis gene aldh1a2. However, only lineage 3 displayed up-regulation of the RA-degrading enzyme cyp26b1 (fig. S6F and table S7). The cyp26b1high-aldh1a2low pattern helped to reduce RA levels in the blastema, promoting redifferentiation of the osteoblasts (44). The differentiation-promoting signature was also reflected in the enrichment of genes, including col6a1 and tagln, whose mammalian orthologs are essential for bone formation (fig. S6F and table S7) (39, 48). These genes were also enriched in the preinjury non-bone cell population, suggesting a connection between this subset of the non-bone cells and their preinjury counterparts (Fig. 5C and fig. S6F). Top up-regulated genes in lineage 4, on the other hand, were main contributors of the extracellular matrix, including and1/2, loxa, and vcanb (35, 49, 50). Enriched expression of these genes suggested that this lineage could be responsible for creating and organizing the fibrous environment. Together, the various non-osteoblastic cells could potentially work collaboratively with the osteoblasts in creating the environment for bone tissue regeneration.
Genes that had been suggested to label progenitors contributing to fin regeneration (mmp9 and cxcl12a) and several orthologs of known mammalian MSC markers (lrrc15, prrx1a/b, and pdgfra) (6, 7, 51, 52) were expressed almost exclusively in the mesenchymal cluster (fig. S6A). Consistent with the observations made in the lineage-tracing study, the mmp9 expression was associated with the regenerating bone cell lineage (lineage 1; Fig. 5B and fig. S6E) (7). However, mmp9 was detected only in a small portion of the mesenchymal cells and was highly expressed in the basal epithelium cells at similar proportions. On the other hand, we observed coenrichment of cxcl12a (previously known as sdf-1) and orthologs of the known mammalian MSC markers in the preinjury population (fig. S6E). cxcl12a-expressing cells in zebrafish were found to carry osteogenic, adipogenic, and chondrogenic characteristics in vitro like MSCs would do and contributed to the mesenchyme of the newly developing bony rays during fin regeneration (6, 53). The coenrichment pattern suggested that some of the preinjury cxcl12a-expressing cells could be MSCs in the fin tissue, which contribute to fin regeneration.
DISCUSSION
Zebrafish caudal fin is a unique regeneration system to model the injury response and regeneration of vertebrate appendages despite being a simple structure without muscular and adipose tissues. Major components of the regenerating caudal fin are epithelial cells covering the wound site and blastemal cells producing the connective tissue and bone matrices. Early studies established that actively proliferating blastema is the key to regeneration. Formed by cell migration and proliferation, this layer of cells continues in outgrowth and differentiation, rebuilding the complex body structure. Despite efforts in understanding its importance, basic questions regarding the formation of blastema remained: (i) Which type of cells contributes to the blastema and (ii) how do they shape the regeneration process?
Using single-cell transcriptomes, we defined cell types in both preinjury and postinjury fin tissues. Although regenerating cells were drastically different from their preinjury counterparts, both stage-specific and integrated clustering analysis revealed the same major cell type compositions in the fin tissues regardless of their time of collection. Common cell types detected include epithelial cells from all three layers, hematopoietic cells, and mesenchymal cells. Our data lay a foundation for lineage-targeted analysis to investigate the role of epithelial layers and subtypes in fin regeneration.
For each cell type to be a consistent component in the regenerated fin, cell cycle entry is required. We found that both common and unique cell cycle programs activated in the regenerating fin, with the shared ones appearing to be more evolutionarily conserved than the unique ones. Among the genes showing cell type–specific S phase enrichment, several immunoproteasome subunits also showed a clear cell type–specific expression. We speculated that the increasing level of immunoproteasome subunits in epithelial and hematopoietic cells specifically might accelerate antigen processing and presentation, which could be important for immune cell recruitment and tumor necrosis factor–α–induced blastemal proliferation (54).
Epithelial cells were the most abundant cell type in the profiled fins and could be clustered into four different subgroups, including the three layers in the adult fish epithelium and the mucosal-like cells within the intermediate layer. However, markers labeling these layers did not perform well in separating cell groups when only regenerating cells were considered. An unbiased differential expression test suggested that some members of the krt and cldn families were expressed in specific layers more consistently throughout regeneration. RNA in situ hybridization targeting cldne, krt1-19d, cldna, krt94, cldni, and cldn1 confirmed their exclusive layer-specific expression pattern, underscoring their potential to serve as markers for the distinct epithelial layers during regeneration. Our epithelium-specific analysis suggested that basal layer epithelial cells proliferate and could be the main source for replenishing the other two layers of the epithelium, similar to findings in a previous study based on genetic lineage tracing in zebrafish and echoing findings made using the axolotl limb regeneration model (25, 55). We observed higher apoptosis and lower proliferation features in the superficial epithelial layer compared to the other layers. At the same time, we observed transition patterns in gene expression, connecting the basal to the intermediate and the superficial layer during regeneration.
The behavior of mucosal-like cells during regeneration had been rarely reported for zebrafish in literature. We found in this study that this group of cells was an integral part of the regeneration process. Enrichment of foxp1b in this population and enrichment of foxp4 in basal and intermediate epithelial cells supported that zebrafish foxp homologs could be involved in regulating agr2 expression as does the Fox family in mice and, furthermore, the mucin production in the epithelium during regeneration (Fig. 1E) (56). The protein encoded by amphibian homologs of agr2, nAG (from newts) and aAG (from axolotl), are necessary and sufficient for salamander limb regeneration (57, 58). They are expressed in both dermal glands and the nerve sheath—the pattern of which has also been recovered from single-cell RNA sequencing (scRNA-seq) analysis (55). Regeneration deficiencies caused by denervation before amputation can be rescued by the ectopic expression of nAG. Although we do not have data supporting the nerve sheath expression pattern, as shown for the amphibian models, we hypothesize that agr2 could similarly mediate neuronal signals in zebrafish during regeneration.
Macrophages are critical players in the zebrafish caudal fin regeneration (28, 54). We observed subgroups of the mpeg1.1+ macrophage population in the regenerating fin tissue, resembling M1 and M2 macrophages in mammalian systems. However, we were not able to recover other immune cell population in the hematopoietic cells. This could potentially be due to the systematic bias against certain cell types during tissue dissociation and droplet incorporation in the microfluidic device. The same bias might also explain why we were not able to recover some other known players in the regenerating fin tissue, including neurons and endothelial cells (4). Increasing the number of cells sampled for scRNA-seq or performing scRNA-seq on sorted hematopoietic lineage cells would help to better understand the involvement of these populations in the regeneration process.
The expression profiles of mesenchymal cells captured from the postinjury stages resembled those of blastema in histology studies. We found four connected but distinct lineages representing both bone and non-bone cells in the blastema. All four lineages initiated from one cluster mostly consisted of 1-dpa cells and enriched for the tnfaip6 expression. A similar scenario has been observed in the axolotl limb regeneration model. By using scRNA-seq on a lineage-labeled axolotl model, Gerber et al. (58) found that connective tissue cells funnel into a progenitor state at initiation. Whether the cluster identified in our study represented a shared cell origin for the blastema or a shared state across mesenchymal cell types in the initial blastema-formation stage requires further investigation. High proportion of epithelial population in the fins could also hamper the discovery of relatively rare population with multipotency. Finer dissection before single-cell profiling might help in future study designs in capturing these populations.
While the bone cell lineage has been well studied in the regenerating fin, non-bone cells had been labeled by different markers and given different names and their intercorrelations left to be clarified. We found that tph1b, aldh1a2, and and1/and2 genes were shared among the non-bone cell lineages and could be labeling states instead of types of blastemal cells during regeneration. Meanwhile, differential analysis revealed similar enrichment for bone formation in all lineages yet distinct associations with reactivation of developmental programs, RA signaling, and collagen metabolism, underscoring their collaborative and complementary roles in the regeneration process.
Our scRNA-seq data also provided more details about the fish system we are working with. For all sample collections, we used the transgenic strain Tg(sp7:EGFP)b1212, which specifically labels osteoblast lineage in the fish (59). It was reported that green fluorescence signal could be detected in the fish skin after 72 hours post-fertilization. This ectopic expression, however, does not interfere with confocal imaging of skeletal structures of fish at any stage due to the fact that they lie in different planes of focus. What these cells are and why they expressed the transgene were unclear. In this study, we obtained a holistic view of the transgene expression pattern in the fin region regardless of whether that was associated with the cell type of interest, i.e., osteoblasts in this context. Unsupervised clustering on the expression profiles from single fin cells suggested that green fluorescent protein (GFP) is not only expressed in the mesenchymal but also highly enriched in the superficial layer epithelium (table S2). A closer examination of this classic reporter gene construct revealed that the regulatory region of sp7 used for the construction of the transgene did not exactly represent the endogenous sp7 regulatory region. Tg(sp7:EGFP)b1212 was generated from bacterial artificial chromosome transgenesis using CH73-243G6 as the backbone, which did not contain the first exon of sp7 according to the annotation of the current genome assembly (chr6:58630884-58720045 and GRCz10), leading to the usage of a regulatory sequence different from the endogenous version. Whether this usage difference contributed to the ectopic expression pattern of the transgene requires further study. This finding points to the potential of using single-cell–based approaches in reporter line validation and more thorough analysis of the transgene behavior.
MATERIALS AND METHODS
Fish husbandry and procedures
All zebrafish were used in accordance with protocol no. 20190041 approved by the Washington University Institutional Animal Care and Use Committee. Wild-type and Tg(sp7:EGFP) strains are maintained under standard husbandry in the Washington University Fish Facility, with the system water temperature at 28.5°C and a day-night cycle controlled as 14-hour light/10-hour dark. For fin amputation, we anesthetized 1-year-old fish with MS-222 (0.16 g/liter) in the system water and then removed the distal half of their caudal fin with sterilized razor blades. The fish were then sent back to circulating water system for recovery. We collected regenerating fin tissue from 39 fish by doing secondary fin amputation at the primary cutting plane with the same anesthesia and recovery procedures.
Cell preparation and scRNA-seq
Collected fin tissues were digested by Accumax (Innovative Cell Technologies), filtered through 40-μm cell strainers, and washed with 1× Dulbecco’s phosphate-buffered saline (DPBS)–0.04% bovine serum albumin to generate single-cell suspensions. Libraries were constructed from these cell suspensions following the instruction of the Chromium Single Cell Gene Expression Solution 3’ v2 (10x Genomics) and were subsequently sequenced on HiSeq2500 (Illumina) with read lengths of 26 + 75 (Read1 + Read2). Raw reads were processed by Cell Ranger (10x Genomics) with default parameters for read tagging, alignment to zebrafish reference genome (GRCz10), and feature counting based on Ensembl release 91 (cellranger count). EGFP sequence was added into the reference genome as a separate chromosome for mapping reads from the reporter gene.
Unsupervised clustering and cell type identification
We performed unsupervised clustering using Seurat v3.0 following the procedure of normalization (SCTransform), highly variable gene detection, dimensional reduction (principal components analysis), and cells clustering (Louvain clustering at resolutions from 0.1 to 0.6) (17). For integrating the four stages in finding conserved cell types, we used the anchoring approach provided by Seurat v3. Cell clustering was based on the top principal components that account for most of the cell-cell variances. The same set of principal components was used in UMAP calculation for visualization as well.
We found differentially expressed genes in each cluster by comparing the expression profiles of them with those of the rest of the cells using Wilcoxon rank sum–based approach with the criteria of log fold change more than 0.25 and a minimum cell percentage of 0.25. The same criteria were applied to all pairwise comparisons, unless stated otherwise. We made functional connections between the list of differentially expressed genes and the type of cell that they most likely represent by testing for GO term enrichment (18) and manual curation by searching The Zebrafish Information Network database and PubMed. Certain cell clusters were taken as independent samples for secondary clustering following the same unsupervised clustering procedures.
Cell cycle analysis
We calculated the by-cell average expression level of a set of S phase or G2-M phase markers suggested by Seurat that are detected in our zebrafish dataset and normalized by subtracting aggregated expression of control genes. Although G1 phase cells are also within cell cycle, they are hardly separable from G0 cells. To avoid false-positive labeling for active cycling cells, we set stringent thresholds and only included cells with |S.score − G2M.score| > 0.1 in the S or G2-M group, while cells with both S.score and G2M.score below zero as G1. Other cells were not included in this part of the analysis. Differentially expressed genes were also identified by Wilcoxon rank sum–based approach. These differentially expressed genes were considered to be cell cycle related if they were in the list of genes associated with R-DRE-1640170 Cell Cycle and/or cycling marker genes used for cell cycle phase score calculations.
RNA in situ hybridization and Alcian blue/PAS staining
We collected uninjured and regenerating fin tissues from casper (nacrew2/w2;roya9/a9) fish and fixed in 4% paraformaldehyde overnight (60). Fixed tissues were subsequently submerged in 10% sucrose in 1× PBS, 20% sucrose in 1× PBS, and 30% sucrose in 1× PBS for 4 hours each. After sucrose exchange, tissues were embedded in Optimal Cutting Temperature (O.C.T.) compound (Fisher Healthcare Tissue-Plus) and snap frozen on dry ice. The frozen tissue blocks were then processed into 15-μm sections on a Leica CM1950 cryostat. We performed RNA in situ hybridization targeting krt4, cldne, krt1-19d, cldna, krt94, cldni, cldn1, agr2, sema3b, stmn1b, and tnfaip6 for mRNA detection using an RNAscope kit (Advanced Cell Diagnostics, Hayward, CA, USA). Alcian blue/periodic acid–Schiff (PAS) staining was subsequently performed on the same section or separately on a consecutive serial section following the manufacturer’s protocol (Newcomer Supply). Microscopic images were taken by ZEISS Axio Observer.
Lineage reconstructions
Cell trajectories were constructed using Slingshot v1.3.1 (40). Through initial subclustering and cell type identifications, we found one subcluster with high epcam expression, potentially a doublet cell contamination from the major cell type classifications. We removed this group of cells from all downstream analysis within the mesenchymal cluster. We used UMAP embedding and subclustering assignments as input for the Slingshot calculation.
Statistical information
We performed nonparametric Wilcoxon rank sum test to identify differentially expressed genes across cell groups as implemented in Seurat. P values were adjusted by all features in the dataset using Bonferroni correction.
Supplementary Material
Acknowledgments
We thank Z. Kupchinsky and other staff members in the fish facility of the Washington University in St. Louis for basic fish husbandry. We thank C. Sawyer and other staff members at the Genome Technology Access Center (GTAC) of the Washington University in St. Louis for generating and sequencing scRNA-seq libraries. We dedicate this work in memory of S.L.J. Funding: This work is supported by the Children’s Discovery Institute of Washington University and the St. Louis Children’s Hospital through microgrants from the Zebrafish Models for Pediatric Research Services Cooperative (ZRSC). Y.H. is, in part, supported by the Philip and Sima Needleman Student Fellowship in Regenerative Medicine. Y.H., H.J.L., Y.C., and T.W. are also supported by the NIH grants R01HG007175, U24ES026699, U01HG009391, and U41HG010972 and by the American Cancer Society Research Scholar grant RSG-14-049-01-DMC. Author contributions: Y.H., T.W., G.Z., and M.H.M. conceived the idea and designed the experiments for this project. Y.H., J.G., F.O.I.O., and A.R.M. performed the experiments. Y.H., H.J.L., Y.C., G.Z., and T.W. analyzed and interpreted the sequencing data. Y.H., G.Z., and T.W. wrote the manuscript with feedback from all other authors. Competing interests: The authors declare that they have no competing interests or financial ties to disclose. Data and materials availability: All raw and processed sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://ncbi.nlm.nih.gov/geo/) under accession number GSE137971. Interactive cell clustering and gene expression analyses based on the sequencing data processed in the current study are available through the following URL: https://epigenome.wustl.edu/FinRegenerationSCRNA/.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/33/eaba2084/DC1
REFERENCES AND NOTES
- 1.Bely A. E., Nyberg K. G., Evolution of animal regeneration: Re-emergence of a field. Trends Ecol. Evol. 25, 161–170 (2010). [DOI] [PubMed] [Google Scholar]
- 2.Gemberling M., Bailey T. J., Hyde D. R., Poss K. D., The zebrafish as a model for complex tissue regeneration. Trends Genet. 29, 611–620 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sousa S., Afonso N., Bensimon-Brito A., Fonseca M., Simões M., Leon J., Roehl H., Cancela M. L., Jacinto A., Differentiated skeletal cells contribute to blastema formation during zebrafish fin regeneration. Development 138, 3897–3905 (2011). [DOI] [PubMed] [Google Scholar]
- 4.Tu S., Johnson S. L., Fate restriction in the growing and regenerating zebrafish fin. Dev. Cell 20, 725–732 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stewart S., Stankunas K., Limited dedifferentiation provides replacement tissue during zebrafish fin regeneration. Dev. Biol. 365, 339–349 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lund T. C., Patrinostro X., Kramer A. C., Stadem P., Higgins L. A., Markowski T. W., Wroblewski M. S., Lidke D. S., Tolar J., Blazar B. R., sdf1 expression reveals a source of perivascular-derived mesenchymal stem cells in zebrafish. Stem Cells 32, 2767–2779 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ando K., Shibata E., Hans S., Brand M., Kawakami A., Osteoblast production by reserved progenitor cells in zebrafish bone regeneration and maintenance. Dev. Cell 43, 643–650 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Knopf F., Hammond C., Chekuru A., Kurth T., Hans S., Weber C. W., Mahatma G., Fisher S., Brand M., Schulte-Merker S., Weidinger G., Bone regenerates via dedifferentiation of osteoblasts in the zebrafish fin. Dev. Cell 20, 713–724 (2011). [DOI] [PubMed] [Google Scholar]
- 9.Zheng G. X. Y., Terry J. M., Belgrader P., Ryvkin P., Bent Z. W., Wilson R., Ziraldo S. B., Wheeler T. D., Mc Dermott G. P., Zhu J., Gregory M. T., Shuga J., Montesclaros L., Underwood J. G., Masquelier D. A., Nishimura S. Y., Schnall-Levin M., Wyatt P. W., Hindson C. M., Bharadwaj R., Wong A., Ness K. D., Beppu L. W., Deeg H. J., Farland C. M., Loeb K. R., Valente W. J., Ericson N. G., Stevens E. A., Radich J. P., Mikkelsen T. S., Hindson B. J., Bielas J. H., Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 8, 14049 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Slanchev K., Carney T. J., Stemmler M. P., Koschorz B., Amsterdam A., Schwarz H., Hammerschmidt M., The epithelial cell adhesion molecule EpCAM is required for epithelial morphogenesis and integrity during zebrafish epiboly and skin development. PLOS Genet. 5, e1000563 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gumbiner B., Stevenson B., Grimaldi A., The role of the cell adhesion molecule uvomorulin in the formation and maintenance of the epithelial junctional complex. J. Cell Biol. 107, 1575–1587 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zakrzewska A., Cui C., Stockhammer O. W., Benard E. L., Spaink H. P., Meijer A. H., Macrophage-specific gene functions in Spi1-directed innate immunity. Blood 116, e1–e11 (2010). [DOI] [PubMed] [Google Scholar]
- 13.Akimenko M. A., Johnson S. L., Westerfield M., Ekker M., Differential induction of four msx homeobox genes during fin development and regeneration in zebrafish. Development 121, 347–357 (1995). [DOI] [PubMed] [Google Scholar]
- 14.Yang D.-C., Tsai C.-C., Liao Y.-F., Fu H.-C., Tsay H.-J., Huang T.-F., Chen Y.-H., Hung S.-C., Twist controls skeletal development and dorsoventral patterning by regulating runx2 in zebrafish. PLOS ONE 6, e27324 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shibata E., Ando K., Murase E., Kawakami A., Heterogeneous fates and dynamic rearrangement of regenerative epidermis-derived cells during zebrafish fin regeneration. Development 145, dev162016 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Lee R. T. H., Asharani P. V., Carney T. J., Basal keratinocytes contribute to all strata of the adult zebrafish epidermis. PLOS ONE 9, e84858 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W. M. III, Hao Y., Stoeckius M., Smibert P., Satija R., Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou Y., Zhou B., Pache L., Chang M., Khodabakhshi A. H., Tanaseichuk O., Benner C., Chanda S. K., Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 10, 1523 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McConnell S. C., Hernandez K. M., Wcisel D. J., Kettleborough R. N., Stemple D. L., Yoder J. A., Andrade J., de Jong J. L. O., Alternative haplotypes of antigen processing genes in zebrafish diverged early in vertebrate evolution. Proc. Natl. Acad. Sci. U.S.A. 113, E5014–E5023 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tanoka K., Kasahara M., The MHC class I ligand-generating system: Roles of immunoproteasomes and the interferon-4gMY-inducible proteasome activator PA28. Immunol. Rev. 163, 161–176 (1998). [DOI] [PubMed] [Google Scholar]
- 21.Uhlén M., Fagerberg L., Hallström B. M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson Å., Kampf C., Sjöstedt E., Asplund A., Olsson I. M., Edlund K., Lundberg E., Navani S., Szigyarto C. A.-K., Odeberg J., Djureinovic D., Takanen J. O., Hober S., Alm T., Edqvist P.-H., Berling H., Tegel H., Mulder J., Rockberg J., Nilsson P., Schwenk J. M., Hamsten M., von Feilitzen K., Forsberg M., Persson L., Johansson F., Zwahlen M., von Heijne G., Nielsen J., Pontén F., Tissue-based map of the human proteome. Science 347, 1260419 (2015). [DOI] [PubMed] [Google Scholar]
- 22.Chen Y.-C., Lu Y.-F., Li I.-C., Hwang S.-P. L., Zebrafish Agr2 is required for terminal differentiation of intestinal goblet cells. PLOS ONE 7, e34408 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marí-Beffa M., Santamaría J. A., Fernández-Llebrez P., Becerra J., Histochemically defined cell states during tail fin regeneration in teleost fishes. Differentiation 60, 139–149 (1996). [Google Scholar]
- 24.Kolosov D., Bui P., Chasiotis H., Kelly S. P., Claudins in teleost fishes. Tissue Barriers 1, e25391 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen C.-H., Puliafito A., Cox B. D., Primo L., Fang Y., Talia S. D., Poss K. D., Multicolor cell barcoding technology for long-term surveillance of epithelial regeneration in zebrafish. Dev. Cell 36, 668–680 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Poss K. D., Shen J., Keating M. T., Induction of lef1 during zebrafish fin regeneration. Dev. Dyn. 219, 282–286 (2000). [DOI] [PubMed] [Google Scholar]
- 27.Poss K. D., Shen J., Nechiporuk A., McMahon G., Thisse B., Thisse C., Keating M. T., Roles for Fgf signaling during zebrafish fin regeneration. Dev. Biol. 222, 347–358 (2000). [DOI] [PubMed] [Google Scholar]
- 28.Petrie T. A., Strand N. S., Tsung-Yang C., Rabinowitz J. S., Moon R. T., Macrophages modulate adult zebrafish tail fin regeneration. Development 141, 2581–2591 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Higdon C. W., Mitra R. D., Johnson S. L., Gene expression analysis of zebrafish melanocytes, iridophores, and retinal pigmented epithelium reveals indicators of biological function and developmental origin. PLOS ONE 8, e67801 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Le Poole I. C., Mutis T., van den Wijngaard R. M., Westerhof W., Ottenhoff T., de Vries R. R., Das P. K., A novel, antigen-presenting function of melanocytes and its possible relationship to hypopigmentary disorders. J. Immunol. 151, 7284–7292 (1993). [PubMed] [Google Scholar]
- 31.Rawls J. F., Johnson S. L., Zebrafish kit mutation reveals primary and secondary regulation of melanocyte development during fin stripe regeneration. Development 127, 3715–3724 (2000). [DOI] [PubMed] [Google Scholar]
- 32.Govindan J., Iovine M. K., Hapln1a is required for connexin43-dependent growth and patterning in the regenerating fin skeleton. PLOS ONE 9, e88574 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bhadra J., Iovine M. K., Hsp47 mediates Cx43-dependent skeletal growth and patterning in the regenerating fin. Mech. Dev. 138, 364–374 (2015). [DOI] [PubMed] [Google Scholar]
- 34.Tornini V. A., Thompson J. D., Allen R. L., Poss K. D., Live fate-mapping of joint-associated fibroblasts visualizes expansion of cell contributions during zebrafish fin regeneration. Development 144, 2889–2895 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.König D., Page L., Chassot B., Jaźwińska A., Dynamics of actinotrichia regeneration in the adult zebrafish fin. Dev. Biol. 433, 416–432 (2018). [DOI] [PubMed] [Google Scholar]
- 36.Gavaia P. J., Simes D. C., Ortiz-Delgado J. B., Viegas C. S. B., Pinto J. P., Kelsh R. N., Sarasquete M. C., Cancela M. L., Osteocalcin and matrix Gla protein in zebrafish (Danio rerio) and Senegal sole (Solea senegalensis): Comparative gene and protein expression during larval development through adulthood. Gene Expr. Patterns 6, 637–652 (2006). [DOI] [PubMed] [Google Scholar]
- 37.Ofer L., Dean M. N., Zaslansky P., Kult S., Shwartz Y., Zaretsky J., Griess-Fishheimer S., Monsonego-Ornan E., Zelzer E., Shahar R., A novel nonosteocytic regulatory mechanism of bone modeling. PLoS Biol. 17, e3000140 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee H.-H., Lee J.-Y., Shih L.-H., Proper Fhl1 expression as Wnt signaling is required for chondrogenesis of ATDC5 cells. Animal Cells Syst. 17, 413–420 (2013). [Google Scholar]
- 39.Elsafadi M., Manikandan M., Dawud R. A., Alajez N. M., Hamam R., Alfayez M., Kassem M., Aldahmash A., Mahmood A., Transgelin is a TGFβ-inducible gene that regulates osteoblastic and adipogenic differentiation of human skeletal stem cells through actin cytoskeleston organization. Cell Death Dis. 7, e2321 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Street K., Risso D., Fletcher R. B., Das D., Ngai J., Yosef N., Purdom E., Dudoit S., Slingshot: Cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genomics 19, 477 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tsukahara S., Ikeda R., Goto S., Yoshida K., Mitsumori R., Sakamoto Y., Tajima A., Yokoyama T., Toh S., Furukawa K.-I., Inoue I., Tumour necrosis factor alpha-stimulated gene-6 inhibits osteoblastic differentiation of human mesenchymal stem cells induced by osteogenic differentiation medium and BMP-2. Biochem. J. 398, 595–603 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li N., Felber K., Elks P., Croucher P., Roehl H. H., Tracking gene expression during zebrafish osteoblast differentiation. Dev. Dyn. 238, 459–466 (2009). [DOI] [PubMed] [Google Scholar]
- 43.Okazaki M., Takeshita S., Kawai S., Kikuno R., Tsujimura A., Kudo A., Amann E., Molecular cloning and characterization of OB-cadherin, a new member of cadherin family expressed in osteoblasts. J. Biol. Chem. 269, 12092–12098 (1994). [PubMed] [Google Scholar]
- 44.Blum N., Begemann G., Osteoblast de- and redifferentiation are controlled by a dynamic response to retinoic acid during zebrafish fin regeneration. Development 142, 2894–2903 (2015). [DOI] [PubMed] [Google Scholar]
- 45.Yamamoto K., Okano H., Miyagawa W., Visse R., Shitomi Y., Santamaria S., Dudhia J., Troeberg L., Strickland D. K., Hirohata S., Nagase H., MMP-13 is constitutively produced in human chondrocytes and co-endocytosed with ADAMTS-5 and TIMP-3 by the endocytic receptor LRP1. Matrix Biol. 56, 57–73 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ge G., Seo N.-S., Liang X., Hopkins D. R., Höök M., Greenspan D. S., Bone morphogenetic protein-1/tolloid-related metalloproteinases process osteoglycin and enhance its ability to regulate collagen fibrillogenesis. J. Biol. Chem. 279, 41626–41633 (2004). [DOI] [PubMed] [Google Scholar]
- 47.Heude É., Shaikho S., Ekker M., The dlx5a/dlx6a genes play essential roles in the early development of zebrafish median fin and pectoral structures. PLOS ONE 9, e98505 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Smeriglio P., Lee J., Bhutani N., Soluble collagen VI treatment enhances mesenchymal stem cells expansion for engineering cartilage. Bioeng. Transl. Med. 2, 278–284 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lucero H. A., Kagan H. M., Lysyl oxidase: An oxidative enzyme and effector of cell function. Cell. Mol. Life Sci. 63, 2304–2316 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zimmermann D. R., Ruoslahti E., Multiple domains of the large fibroblast proteoglycan, versican. EMBO J. 8, 2975–2981 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang Y., Liu Y., Zhang M., Lv L., Zhang X., Zhang P., Zhou Y., LRRC15 promotes osteogenic differentiation of mesenchymal stem cells by modulating p65 cytoplasmic/nuclear translocation. Stem Cell Res. Ther. 9, 313 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Miwa H., Era T., Tracing the destiny of mesenchymal stem cells from embryo to adult bone marrow and white adipose tissue via Pdgfrα expression. Development 145, dev155879 (2018). [DOI] [PubMed] [Google Scholar]
- 53.Dominici M., Le Blanc K., Mueller I., Slaper-Cortenbach I., Marini F. C., Krause D. S., Deans R. J., Keating A., Prockop D. J., Horwitz E. M., Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 8, 315–317 (2006). [DOI] [PubMed] [Google Scholar]
- 54.Nguyen-Chi M., Laplace-Builhé B., Travnickova J., Luz-Crawford P., Tejedor G., Lutfalla G., Kissa K., Jorgensen C., Djouad F., TNF signaling and macrophages govern fin regeneration in zebrafish larvae. Cell Death Dis. 8, e2979 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Leigh N. D., Dunlap G. S., Johnson K., Mariano R., Oshiro R., Wong A. Y., Bryant D. M., Miller B. M., Ratner A., Chen A., Ye W. W., Haas B. J., Whited J. L., Transcriptomic landscape of the blastema niche in regenerating adult axolotl limbs at single-cell resolution. Nat. Commun. 9, 5153 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li S., Wang Y., Zhang Y., Lu M. M., De Mayo F. J., Dekker J. D., Tucker P. W., Morrisey E. E., Foxp1/4 control epithelial cell fate during lung development and regeneration through regulation of anterior gradient 2. Development 139, 2500–2509 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kumar A., Nevill G., Brockes J. P., Forge A., A comparative study of gland cells implicated in the nerve dependence of salamander limb regeneration. J. Anat. 217, 16–25 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gerber T., Murawala P., Knapp D., Masselink W., Schuez M., Hermann S., Gac-Santel M., Nowoshilow S., Kageyama J., Khattak S., Currie J. D., Camp J. G., Tanaka E. M., Treutlein B., Single-cell analysis uncovers convergence of cell identities during axolotl limb regeneration. Science 362, eaaq0681 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.De Laurier A., Eames B. F., Blanco-Sánchez B., Peng G., He X., Swartz M. E., Ullmann B., Westerfield M., Kimmel C. B., Zebrafish sp7:EGFP: A transgenic for studying otic vesicle formation, skeletogenesis, and bone regeneration. Genesis 48, 505–511 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.White R. M., Sessa A., Burke C., Bowman T., Blanc J. L., Ceol C., Bourque C., Dovey M., Goessling W., Burns C. E., Zon L. I., Transparent adult zebrafish as a tool for in vivo transplantation analysis. Stem Cells 2, 183–189 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/33/eaba2084/DC1