

Abstract
Various ligands and receptors of the transforming growth factor-β superfamily have been found upregulated following traumatic brain injury; however, the role of this signalling system in brain injury pathophysiology is not fully characterized. To address this, we utilized an acute stab wound brain injury model to demonstrate that hallmarks of transforming growth factor-β superfamily system activation, such as levels of phosphorylated Smads, ligands and target genes for both transforming growth factor-β and bone morphogenetic protein pathways, were upregulated within injured tissues. Using a bone morphogenetic protein-responsive reporter mouse model, we showed that activation of the bone morphogenetic protein signalling pathway involves primarily astrocytes that demarcate the wound area. Insights regarding the potential role of transforming growth factor-β superfamily activation in glia cells within the injured tissues were obtained indirectly by treating purified reactive astrocytes and microglia with bone morphogenetic protein-4 or transforming growth factor-β1 and characterizing changes in their transcriptional profiles. Astrocytes responded to both ligands with considerably overlapping profiles, whereas, microglia responded selectively to transforming growth factor-β1. Novel pathways, crucial for repair of tissue-injury and blood–brain barrier, such as activation of cholesterol biosynthesis and transport, production of axonal guidance and extracellular matrix components were upregulated by transforming growth factor-β1 and/or bone morphogenetic protein-4 in astrocytes. Moreover, both ligands in astrocytes and transforming growth factor-β1 in microglia shifted the phenotype of reactive glia cells towards the anti-inflammatory and tissue reparatory ‘A2’-like and ‘M0/M2’-like phenotypes, respectively. Increased expression of selected key components of the in vitro modulated pathways and markers of ‘A2’-like astrocytes was confirmed within the wound area, suggesting that these processes could also be modulated in situ by the integrated action of transforming growth factor-β and/or bone morphogenetic protein-mediated signalling. Collectively, our study provides a comprehensive comparative analysis of transforming growth factor-β superfamily signalling in reactive astrocytes and microglia and points towards a crucial role of both transforming growth factor-β and bone morphogenetic protein pathways in modulating the inflammatory and brain injury reparatory functions of activated glia cells.
Keywords: astrocytes, BMP, microglia, TGFβ, traumatic brain injury
The current study provides a comprehensive comparative analysis of transforming growth factor-β and bone morphogenetic protein activation in the context of acute stab wound brain injury in glia cells and points towards a crucial role of both branches of the transforming growth factor-β superfamily signalling system in modulating the inflammatory and tissue reparatory activity of reactive astrocytes and microglia.
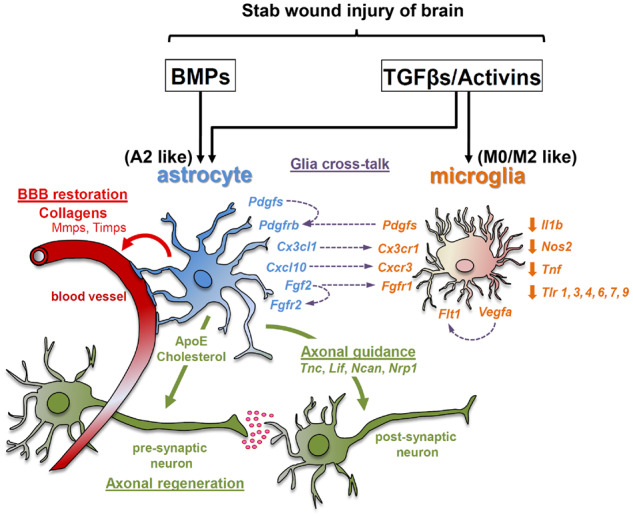
Graphical Abstract
Graphical Abstract.
Introduction
Traumatic brain injury (TBI), defined as alteration in brain function provoked by external mechanical force (Menon et al., 2010), is the main cause of injury-related death, disability and mental disorders, thus representing major public health issue (Majdan et al., 2016; Dewan et al., 2018). According to recent epidemiological studies, TBI affects annually more than 60 million individuals, justifiably characterized as ‘silent epidemic’ (Maas et al., 2017; Dewan et al., 2018).
TBI pathophysiology is characterized by acute necrotic or delayed apoptotic neuronal death, cytokine and chemokine production, infiltration of peripheral immune cells and activation of astrocytes and microglia (Helmy et al., 2011; Burda et al., 2016; Jassam et al., 2017). Astrocytes play important roles in the CNS providing structural support, maintaining blood–brain barrier integrity, regulating the neuronal microenvironment through clearance of excessive neurotransmitters (e.g. glutamate and GABA), promoting formation and function of synapses, pruning synapses by phagocytosis and secreting a variety of trophic and growth factors (Sofroniew and Vinters, 2010; Chung et al., 2013; Bylicky et al., 2018). Due to blood–brain barrier restriction in lipoprotein exchange, astrocytes are the main producers and exclusive providers of cholesterol in the adult brain (Pfrieger and Ungerer, 2011; Sidoryk-Wegrzynowicz et al., 2011).
In pathological situations astrocytes undergo dramatic transformation, become ‘reactive’ and depending on the conditions acquire neurotoxic or neuroprotective phenotypes (Myer et al., 2006; Burda et al., 2016; Bylicky et al., 2018). Following a terminology that parallels the ‘M1’ and ‘M2’ macrophage nomenclature (Biswas and Mantovani, 2010; Sica and Mantovani, 2012), neurotoxic/pro-inflammatory astrocytes, generated by inflammatory insult, are named ‘A1’, whereas, neuroprotective/anti-inflammatory astrocytes known as ‘A2’ are induced in the context of various types of brain injury such as ischaemic damage and produce neurotrophic and tissue-repair factors (Zamanian et al., 2012; Liddelow et al., 2017). The two types of astrocytes can be distinguished on the basis of functionality and characteristic gene expression profiles (Zamanian et al., 2012; Liddelow et al., 2017).
Microglia are heterogeneous resident immune cells within the CNS, surveying the brain parenchyma and reacting to any insult that disrupts homeostasis (Szepesi et al., 2018). Like astrocytes, microglia upon activation, and depending on the context, acquire pro-inflammatory/neurotoxic or anti-inflammatory/neuroprotective phenotypes. In line with the ‘M1’/‘M2’ macrophage nomenclature, the two microglial phenotypes are frequently referred to as ‘M1’- and ‘M2’-microglia, respectively (Fumagalli et al., 2011; Michell-Robinson et al., 2015; Loane and Kumar, 2016). In the context of neuropathology, cross-talk between microglia and astrocytes is critical for coordination of inflammation and tissue-injury repair (Karve et al., 2016; Donat et al., 2017). Pertinently, recent studies showed that reactive inflammatory microglia guide astrocytes towards the ‘A1’ phenotype (Liddelow et al., 2017) and, conversely, earlier studies demonstrated that activated astrocytes can exert inhibitory effects on microglial activation (Gao et al., 2013).
Among the molecular systems implicated in TBI pathophysiology is the transforming growth factor-β (TGFβ)-superfamily (Schachtrup et al., 2010; Chen et al., 2018), which encompasses structurally related polypeptides including, among others, TGFβs, activins and bone morphogenetic proteins (BMPs; Shi and Massague, 2003). TGFβ-superfamily members signal via hetero-tetrameric complexes of type-II and type-I receptors, which possess Ser/Thr kinase activity and activate ‘canonical’ and ‘non-canonical’ pathways. Canonical pathways involve phosphorylation-dependent activation of downstream effectors, the receptor-regulated Smads (R-Smads), which are organized into two branches, the TGFβ/Activin and BMP branches that involve Smads 2/3 or Smads 1/5/8, respectively (Shi and Massague, 2003). Phosphorylated R-Smads form complexes with the ‘common’ Smad 4, enter the nucleus and regulate gene expression (Moustakas et al., 2001; Budi et al., 2017). Activation of canonical pathways up-regulates Smads 6 and 7, which function as negative feedback regulators (Miyazawa and Miyazono, 2017). ‘Non-canonical’ pathways are activated in parallel and involve pathways such as mitogen-activated protein kinases (MAPKs), Rho-like GTPases and phosphatidylinositol-3-kinase (PI3K)/AKT (Moustakas and Heldin, 2005; Zhang, 2009). Given the widespread expression of TGFβ/Activin and/or BMP receptors within a tissue, even on the surface of the same cell, and the concurrent production of numerous ligands in health and disease (Rosendahl et al., 2001, 2002), it is likely that the final response must depend on the integration of all generated signals and be influenced by the relative, quantitative and/or qualitative, characteristics of the activated canonical and/or non-canonical pathways. Therefore, besides the selective analysis of individual components of the TGFβ-superfamily signalling system, approaches that analyse a broader spectrum of signalling events and components could unveil important functional properties of the system.
Several components of the TGFβ-superfamily system, including ligands (TGFβ1, BMP4, BMP10) or receptors (TGFBR1, TGFBR2, ACVR1, BMPR2), are upregulated, either at protein and/or mRNA levels, in cells surrounding the trauma or in adult neurogenic niches following TBI in rodents (Lindholm et al., 1992; Lewen et al., 1997; Huang et al., 2010; Komuta et al., 2010; Schachtrup et al., 2010; Logan et al., 2013; Chen et al., 2018). Moreover, increased TGFβ1 and TGFβ2 protein levels have been described in human spinal cord injuries (Buss et al., 2008), and BMP7 mRNA (Setoguchi et al., 2001) and protein levels in rat spinal cord injury (Hampton et al., 2007). Despite accumulated evidence, the precise role of this signalling system, in particular, the interplay between the TGFβ and BMP branches in the context of TBI pathophysiology is still not fully understood. To address this, we have utilized the most controlled form of TBI, namely, the acute stab wound (SW) brain injury to carry out a comprehensive analysis of the TGFβ-superfamily system, identify putative cellular targets within the wound area and characterize their response to TGFβ and/or BMP. Our study demonstrates that both TGFβ and BMP pathways are activated locally upon SW injury and show that astrocytes demarcating the wound area are the selective target of BMP signalling. By analysing in vitro treated ‘reactive’ astrocytes and microglia, we unveil novel processes critical for restoration of homeostasis that may be regulated by TGFβ and/or BMP. Moreover, we provide evidence suggesting that both TGFβ1 and BMP4 modulate the plasticity of reactive astrocytes towards an ‘A2’-like, anti-inflammatory phenotype, whereas TGFβ1 modulates the plasticity of activated microglia towards ‘M0/M2’-like, quiescent and anti-inflammatory phenotypes, respectively. As such, our findings present a parallel analysis of the two signalling branches of the TGFβ-superfamily system in the context of acute SW injury and provide insights that could aid the further clarification of their role and prospectively the design of novel therapeutic strategies.
Materials and methods
Extended version is available in the Supplementary material.
Animals
Wild-type and BMP-responsive eGFP-expressing (BRE-eGFP) mice (Monteiro et al., 2008), in C57BL/6 background, were maintained in individually ventilated cages with free access to food and water, under 12 h light/dark cycle at the animal facility of BRFAA. All experimental procedures performed were approved by Institutional Ethics Committee for Use of Laboratory Animals and the Greek Ministry of Agriculture.
Surgical procedure of SW injury
Males 3–4 months old were anaesthetized with 2–3% isoflurane and positioned in a stereotaxic frame (Kopf Instruments, USA), as previously described (Xilouri et al., 2012). A unilateral lesion was performed to the right hemisphere of the cerebral cortex by inserting a 19-gauge needle at the following coordinates: +0.6 mm anteroposterior, −1.6 mm mediolateral and −3.2 mm dorsoventral from the bregma as previously described (Buffo et al., 2005; Frik et al., 2018). The needle was twisted manually and gently retracted. This was repeated thrice in total and the skin was thereafter sutured.
Immunohistochemistry
Brain tissues were perfused intracardially with 4% paraformaldehyde and processed thereafter for immunohistochemical analysis, as previously reported (Xilouri et al., 2012) with modifications described in Supplementary material.
Preparation and maintenance of mixed glia cultures
Mixed glia cultures were prepared from P1 to P3 neonatal mice as previously described (McCarthy and de Vellis, 1980; Saura et al., 2003), with modifications described in Supplementary material. Cells utilized in the present study were grown in culture for a total of ∼20–24 days before addition of recombinant TGFβ and/or BMP.
Fluorescence-activated cell sorting of cultured glia cells
Mixed glia cultures were harvested and cells were stained with antibodies against GLAST and CD11b proteins (Supplementary Table 1) for 45 min. Unstained samples and isotypic controls were also included. DAPI was added the last 5 min of incubation for dead-cell exclusion. Cells were sorted with a BD-FACSAria™-IIu as described in Supplementary material.
In vitro stimulation and immunofluorescence analysis of cultured glia cells
Mixed glia, purified astrocytes and microglia, grown for ∼20–24 days in total, were cultured in serum-free DMEM 1% Pen/Strep for the last 24 h before treatment with recombinant mouse TGFβ1, BMP4, Noggin or the SB431542 inhibitor, in the absence of FBS. Cells and culture media were collected by centrifugation (2000 rpm, RT, 2 min), snap frozen and stored at −80°C until use. Cells were cultured on poly-D-lysine (PDL)-coated coverslips for immunocytochemistry, as described in Supplementary material.
Western blot and quantitative real-time PCR analysis
For protein or RNA isolation from TBI-subjected animals, isolated brains were placed on an acrylic mouse brain matrix and 2 mm coronal-sections were prepared. Tissue areas encompassing the trauma or the respective contralateral/healthy area were further micro-dissected (Fig. 1A), snap frozen and stored at −80°C. For protein isolation, micro-dissected regions or primary glial cells were homogenized in RIPA buffer supplemented with protease and phosphatase inhibitors and western blot analysis was performed as previously reported (Xilouri et al., 2012), with modifications described in Supplementary material. Total RNA was isolated using Trizol and cDNA was synthesized as previously described (Sountoulidis et al., 2012). The primer pairs and the amplification parameters used for quantitative real-time PCR (qRT-PCR) are shown in Supplementary material.
Figure 1.

Increased levels of phosphorylated Smads, TGFβ-superfamily ligands and relevant target genes are found within the TBI wound area. (A) Schematic representation of the strategy followed for tissue dissection of ipsilateral injured areas (I) and corresponding contralateral control areas (C) for protein or RNA isolation. (B) Representative immunoblots for pSmad1/5/8, pSmad2 and β-actin (loading control) levels in protein extracts from dissected contralateral (C) and ipsilateral (I) tissues at 1, 3 and 7 dpi. (C) Quantitative analysis of phosphorylated Smad levels shown in B, depicted as ratio of ipsilateral versus contralateral levels for each time-point (n = 3–5 animals/group). (D) qRT-PCR analysis for Bmp4, Bmp7, Tgfb1, Tgfb2 and Tgfb3 mRNA levels at 3 and 7 dpi (n = 5 animals/group). (E) qRT-PCR analysis for Noggin, Id1, Smad6, Serpine1 and Pmepa1 mRNA levels, at 3 and 7 dpi (n = 5 animals/group). (F) Representative immunofluorescence images of coronal brain sections at the level of the wound area at 3 dpi stained for Noggin (white) and nuclei (DAPI, blue) (Scale bar = 150 μm). Right image is a magnified caption (Scale bar = 30 μm) of the area outlined with a magenta dotted square showing the glial morphology of Noggin-expressing cells. Data are expressed as mean ± standard error of the mean (SEM) analysed using one-way analysis of variance with Bonferroni’s post hoc test analysis (*P < 0.05, **P < 0.01, ***P < 0.001). F values and degrees of freedom (df) are shown in parentheses for each data set.
Transcriptomic analysis
One microgram purified RNA was used for preparation of each cDNA library, using the TruSeq RNA Preparation Kit (Illumina), according to manufacturer’s instructions. Library quality was evaluated with Agilent DNA Kit and concentration was measured using library-standards (KAPA Biosystems). Sequencing was performed at the Genome Center Facility of BRFAA. Bioinformatics analysis was performed using Ingenuity® Pathway Analysis (IPA®, Qiagen), MultiExperiment Viewer (MeV, TM4 Microarray Software Suite; Saeed et al., 2003), Venny 2.1 and Venn Diagram software (Pacific Northwest National Laboratory, U.S. Department of Energy).
Statistical analysis
Data are expressed as mean ± standard error of the mean (SEM). Data were analysed using one-way analysis of variance with Bonferroni’s post hoc test analysis. One asterisk (*) corresponds to statistical significance of P < 0.05, two asterisks (**) to P < 0.01, three asterisks (***) to P < 0.001 and non-significant (ns) to P > 0.05. In few indicated cases, the Newman–Keuls Multiple Comparison Test was applied with one hashtag (#) corresponding to statistical significance of P < 0.05. In Fig. 4B, differences were assessed using two-tailed unpaired t-test (*P < 0.05, **P < 0.01 and ***P < 0.001). All analyses were performed using GraphPad Prism software version 5.0 for Windows (GraphPad Software, San Diego, CA, USA).
Figure 4.

Astrocytes and microglia modulate expression of distinct sets of genes upon TGFβ1 treatment, express different components of the canonical pathways and are poised to activate different non-canonical pathways. (A) Venn diagrams representing the number of statistically significant (baseMean > 30 and FDR < 0.05) up- or down-regulated genes only in astrocytes (light blue), only in microglia (pink) or in both glial populations (purple), following stimulation with 2 ng/ml TGFβ1 for 2 h (left panel) or 24 h (right panel). The percentages indicate the gene overlap between astrocytes and microglia (n = 3 cultures/group). (B) mRNA expression levels of key components of the TGFβ-superfamily system, including type-I receptors, type-II receptors and Smads, in vehicle-treated purified microglia (magenda dots) or astrocytes (blue dots) derived from RNA-Seq data that were normalized and expressed as Fragments Per Kilobase per Million reads mapped (FPKM) (n = 6 cultures/group). **P < 0.01; ***P < 0.001; two-tailed unpaired t-test. (C) Representative immunoblot analysis of canonical and non-canonical TGFβ-signalling pathways using antibodies against pSmad2, total Smad2/3, pSmad1/5/8, total Smad1/5/8, phosphorylated AKT (pAKT), total AKT, phosphorylated P38 (pP38), total P38, phosphorylated ERK1/2 (pERK1/2), total ERK2 and β-actin (loading control) in purified microglia (MG) and astrocytes (ACS) upon 10 ng/ml TGFβ1 (on the left) or 50 ng/ml BMP4 stimulation (on the right).
Data availability
Raw data supporting the findings of this study are available from the corresponding author on request. Gene expression data are available from Sequence Read Archive (https://www.ncbi.nlm.nih.gov/sra), accession number PRJNA527753.
All materials used in this study are listed in Supplementary Table 3.
Results
Both TGFβ and BMP branches of the TGFβ-superfamily signalling system are activated upon acute TBI within the wound area
To investigate the role of the TGFβ-superfamily system and assess in parallel the activation of the TGFβ and BMP branches upon brain injury, mice were subjected to unilateral SW injury of the right cerebral cortex, which represents a moderate model of TBI. One of the advantages of the model is that the injury is restricted only in the ipsilateral side, whereas the contralateral side of each animal can be used as control (Supplementary Fig. 1A). Tissues encompassing injured (ipsilateral) and corresponding intact (contralateral) areas were surgically micro-dissected (Fig. 1A) and hallmarks of TGFβ-superfamily activation were analysed. Increased levels of phosphorylated (p) Smad1/5/8 were detected in protein extracts of the ipsilateral side at 1 and 3 days post-injury (dpi). Likewise, increased pSmad2 levels were observed at 1 dpi (Fig. 1B and C). Consistently, increased mRNA levels of ligands (Bmp4, Bmp7, Tgfb1, Tgfb2 and Tgfb3) and known TGFβ-superfamily target genes (Id1, Smad6, Serpine1 and Pmepa1) were detected in the ipsilateral side at 3 and/or 7 dpi (Fig. 1D and E and Supplementary Fig. 1B). Interestingly, the trauma area was characterized by decreased mRNA and protein levels for Noggin, the natural BMP inhibitor (Fig. 1E and F), suggesting that BMP signalling upon TBI may be driven locally by altering the balance between ligands and corresponding inhibitors.
To further verify the activation of canonical BMP-mediated signalling and characterize potential cellular targets, available transgenic animals carrying BMP-responsive eGFP alleles (BRE-eGFP) were subjected to TBI. Injury led to substantial activation of the BMP reporter in cells with glial morphology around the trauma, which reached highest levels between 3 and 7 dpi (Fig. 2A and B). Interestingly, the area occupied by BRE-eGFP+ cells coincided with the area characterized by decreased Noggin immunoreactivity (Supplementary Fig. 2A). Increased expression of the astrocytic protein GFAP was found around the trauma already at 1 dpi and peaked at 7 dpi, when intense astrogliosis, as previously described (Mathewson and Berry, 1985), was observed throughout the ipsilateral cortex (Fig. 2A). The increased astrogliosis was verified by qRT-PCR and immunoblot analysis of GFAP in dissected wound tissues (Supplementary Fig. 2B and C).
Figure 2.

The BRE-eGFP reporter is activated upon acute TBI in astrocytes that demarcate the wound area. (A) Representative tile scan confocal immunofluorescence images of the astrocytic cytoskeletal protein GFAP (white, top row) and the BRE-eGFP reporter (green, bottom row) in coronal brain sections at the level of the wound at 1, 3, 7 and 14 dpi (Scale bar = 1mm). (B) Higher magnification immunofluorescence images of the ipsilateral side, showing the time-course of BRE-eGFP reporter expression (green) around the trauma (Scale bar = 150 μm). (C) Representative immunofluorescence image of the GFAP (white) and BRE-eGFP reporter (green) expression in the ipsilateral side at 7 dpi (left panel, Scale bar = 300μm). Magenta squares and arrows denote origin of higher magnification single layer images (right panel, Scale bar = 25μm) showing BRE-eGFP-positive cells co-expressing the GFAP protein. (D) Representative immunostaining for microglial marker AIF1/IBA1 (white) and BRE-eGFP (green) in the ipsilateral side at 7 dpi (left panel, Scale bar = 300 μm) and higher magnification single layer images (right panel, Scale bar = 25μm). DAPI (blue) is used for nuclear staining. Asterisks illustrate constitutive BRE-eGFP reporter expression in ependymal cells lining the medial walls of lateral ventricles.
To characterize the BRE-eGFP+ cells, tissue sections from TBI-subjected animals were analysed by immunostaining with cell-lineage specific markers (Fig. 2C and D and Supplementary Fig. 3A). The vast majority of BRE-eGFP+ cells around the injury site co-expressed GFAP, ALDH1L1, GLUL and S100β and thus were characterized as astrocytes (Fig. 2C and Supplementary Fig. 3A). Scattered BRE-eGFP+/GFAP+cells were also detected across the ipsilateral cortex at 7 dpi (Supplementary Fig. 2D). Interestingly, none of the AIF1/IBA1+ microglia expressed the BRE-eGFP reporter, even when localized among numerous eGFP-expressing astrocytes (Fig. 2D).
Astrocytes and microglia exhibit differential responsiveness to TGFβ1 and BMP4 in vitro
To characterize the impact of TGFβ or BMP signalling on activated glia cells and assess the failure to detect in situ activation of BRE-eGFP reporter in microglia, we analysed in vitro their responsiveness to TGFβ1 and BMP4, the two ligands exhibiting highest mRNA up-regulation within the injured tissues (Fig. 1D). Pure ‘reactive’ astrocytes and microglia were isolated by sorting mixed glia cultures prepared from neonatal mice, using anti-GLAST/SLC1A3 (astrocytes) and anti-CD11b/ITGAM (microglia) antibodies (Fig. 3A and Supplementary Fig. 3B). The purified cells, after a total of ∼20–24 days in culture, were stimulated with recombinant ligands for 2 and 24 h, and their transcriptomes were analysed by RNA-Seq.
Figure 3.

Purified astrocytes stimulated in vitro with TGFβ1 or BMP4 share highly overlapping transcriptomes, whereas, microglia respond selectively to TGFβ1. (A) Isolation of pure astrocytes and microglia from mixed glia cultures utilizing anti-GLAST (astrocytes) and anti-CD11b (microglia) antibodies. Mixed glia cultures, prepared from newborn mice, comprised of astrocytes (GFAP+), microglia (AIF1/IBA1+) and oligodendrocytes (OLIG2+; representative immunofluorescence image, left panel), were maintained for ∼10–12 days prior to staining with antibodies against GLAST (blue gate) or CD11b proteins (red gate) for cell sorting. Purified astrocytes and microglia were expanded for additional ∼10-12 days before analysis. Right panel shows representative bright field and immunofluorescence images of astrocyte and microglia cultures. DAPI (blue) is used for nuclear staining. Scale bar = 50 μm. (B and C) Venn diagrams summarizing RNA-Seq analysis results of purified astrocytes treated with 2 ng/ml TGFβ1 or 50 ng/ml BMP4 for 2 h (B) or 24 h (C) (n = 3 cultures/group). (D and E) Venn diagrams summarizing RNA-Seq analysis of purified microglia treated with 2 ng/ml TGFβ1 or 50 ng/ml BMP4 for 2 h (D) or 24 h (E) (n = 3 cultures/group). The diagrams depict numbers of statistically significant (baseMean > 30 and FDR < 0.05) up- or down-regulated genes by TGFβ1 (red), BMP4 (green) or both (orange). The percentages on the black arrows indicate the overlap between the two ligand treatments. The scatterplots in B and C show the genes with statistically significant response to TGFβ1 (red), BMP4 (green) or both (orange). The x- and y-axes represent log2 fold change of differentially expressed genes.
Upon TGFβ1 stimulation of purified astrocytes, 1069 genes were significantly upregulated and 513 downregulated at 2 h, whereas 1612 were upregulated and 1297 downregulated at 24 h. Similarly, upon BMP4 treatment, 1494 genes were upregulated and 719 downregulated at 2 h and 1702 genes were upregulated and 1568 downregulated at 24 h (Fig. 3B and C). Interestingly, TGFβ1- or BMP4-treated astrocytes exhibited highly overlapping transcriptional profiles 2 h post-stimulation, since ∼47–69% of regulated genes responded to both ligands (Fig. 3B), albeit to different degrees (Supplementary Fig. 4A), whilst at 24 h, the gene expression profiles diverged exhibiting ∼35–44% overlap (Fig. 3C). Few genes were oppositely regulated by these ligands (<2% and <8%, at 2 and 24 h, respectively), indicating limited antagonism between the two branches in astrocytes.
Purified microglia responded remarkably well to TGFβ1, since 1018 genes were upregulated and 526 downregulated at 2 h, whereas 1569 were upregulated and 1697 downregulated at 24 h (Fig. 3D and E). Notably, astrocytes and microglia regulated distinct sets of genes in response to TGFβ1, exhibiting ∼11–31% overlap (Fig. 4A). Surprisingly, still in agreement with the absence of BRE-eGFP reporter expression in AIF1/IBA1+ cells, purified microglia failed to respond robustly to BMP4. Only 88 genes were upregulated and 9 were downregulated at 2 h. Similarly, 143 genes were upregulated and 125 were downregulated after 24 h stimulation with BMP4. However, none exhibited log2 fold change >1.6, only 55 and 33 displayed log2 fold change >0.4 or <−0.4, respectively, and the top upregulated genes included key negative regulators of the pathway, such as Smad6, Smad7 and Tgif1.
To derive mechanistic explanation for the differential response of the two glial populations to TGFβ1 and BMP4, key components of the system were compared in astrocytes and microglia. Analysis of relative basal mRNA levels of TGFβ-superfamily system receptors and effector Smads demonstrated significant differences among them. Microglia were characterized by higher mRNA levels for Acvrl1/Alk1, Tgfbr1/Alk5 and Tgfbr2, compared with astrocytes that expressed higher mRNA levels for Acvr1/Alk2, Bmpr1a/Alk3, Bmpr1b/Alk6, Acvr2a, Bmpr2 and Smads 1, 5, 8, 3, 4, 6 and 7 (Fig. 4B). Both cell types expressed similar mRNA levels for Smad2. Moreover, although Smad2 was phosphorylated in both glial populations following TGFβ1 treatment, Smad1/5/8 were phosphorylated upon BMP4 stimulation only in astrocytes (Fig. 4C). Consistently, total Smad1/5/8 levels were below detection in the microglia population utilized in the analysis. Therefore, levels of BMP-related Smads and receptors may account for the low microglial response to BMP4. Interestingly, different non-canonical pathways were activated, even at baseline, in the two glial populations. Microglia were characterized by higher total and phosphorylated P38/MAP-kinase, whereas astrocytes exhibited higher phosphorylated ERK1/2 and increased pAKT levels upon ligand stimulation (Fig. 4C), suggesting that TGFβ-superfamily receptors could be wired with different non-canonical downstream signalling pathways in these two cell types.
Analysis of the transcriptomic profiles of TGFβ1- or BMP4-treated astrocytes using the IPA platform demonstrated that 2 h stimulation with either ligand led to activation of several canonical pathways related to growth factor-mediated signalling (TGFβ, BMP, IGF1, Wnt/β-catenin, ERK/MAPK, PI3K/AKT, STAT3, platelet-derived growth factor (PDGF), GDNF, neurotrophin/TRK, FGF, etc.), cytoskeletal remodelling and cell migration (ephrin receptor, integrin, actin cytoskeleton and glioma invasiveness signalling; Supplementary Fig. 5A). Twenty-four hours stimulation led to activation of pathways related to tissue-repair/remodelling (Fig. 5A). The majority of identified Gene Ontology annotations (GOs) was regulated by both TGFβ1 and BMP4, with either distinct (Fig. 7C) or overlapping gene-sets modulated by either ligand (Figs 6A and B and 7A).
Figure 5.

TGFβ1 or BMP4 stimulation guides purified astrocytes towards a reparatory, neuroprotective ‘A2’-like phenotype. (A) Graph depicting CNS-related IPA canonical pathways, selected among the top regulated, from RNA-Seq analysis of purified astrocytes stimulated with 2 ng/ml TGFβ1 or 50 ng/ml BMP4 for 24 h. The x-axis displays the statistical significance [-log (P value)] of the canonical pathways displayed along the y-axis. Grey vertical line, indicated by arrow in the x-axis, shows the threshold for statistical significance (P = 0.05). Numbers at the right side of the bars represent calculated IPA z-scores predicting an increased (black numbers) or a decreased (red numbers) pathway activity. (B) Heat map depicting log2 fold changes of differentially expressed PAN reactive, ‘A1’- and ‘A2’-specific reactive astrocyte transcripts (according to Zamanian et al., 2012 and Liddelow et al., 2017), as determined by RNA-Seq analysis of purified astrocytes following 2 ng/ml TGFβ1 or 50 ng/ml BMP4 stimulation for 2 or 24 h (baseMean > 30). Asterisks depict statistical significance (FDR < 0.05). (C) Schematic representation of the expression of chemokines, cytokines, growth factors and relevant receptors, derived from RNA-Seq analysis of purified microglia and astrocytes treated with 2 ng/ml TGFβ1 for 2 h (left panel) or 24 h (right panel). Upregulated genes are shown in black, whereas the downregulated are shown in red. Black and blue arrows depict potential heterotypic or homotypic interactions, respectively, between glial populations.
Figure 7.

Genes encoding for axonal guidance and tissue-repair components are modulated in vitro, upon stimulation of purified astrocytes with TGFβ1 or BMP4, or in vivo, upon acute TBI. (A and C) Heat maps depicting log2 fold changes of differentially expressed genes encoding (A) axonal guidance molecules [with permissive or inhibitory action, as summarized in Anderson et al. (2016)] and (C) collagens (‘Hepatic Fibrosis’ pathway, IPA), derived from RNA-Seq analysis of purified astrocytes stimulated with 2 ng/ml TGFβ1 or 50 ng/ml BMP4 for 2 or 24 h (baseMean > 30 and FDR < 0.05). (B) qRT-PCR analysis of mRNA expression of axonal growth permissive molecules Tnc and Lif and axonal growth inhibitory molecules Ncan and Nrp1 (highlighted with pink frames in A) in the contralateral (C) and ipsilateral (I) dissected tissues from TBI-subjected animals at 3 and 7 dpi (n = 5 animals/group). (D) qRT-PCR analysis of Col1a1 and Col5a2 (highlighted with pink frames in C) mRNA expression in the contralateral (C) and ipsilateral (I) tissues dissected from TBI-subjected animals at 3 and 7 dpi. Data are expressed as mean ± standard error of the mean (SEM) analysed using one-way analysis of variance with Bonferroni’s post hoc test analysis (*P < 0.05, **P < 0.01, ***P < 0.001) or Newman–Keuls Multiple Comparison Test (#P < 0.05). F values and degrees of freedom (df) are shown in parentheses for each data set. (E) Representative immunofluorescence images for Collagen-I (white) and BRE-eGFP (green) expression in coronal brain sections at the level of the wound at 7 dpi (Scale bar = 100 μm). Red dashed square indicates area shown in higher magnification (Scale bar = 50 μm) of BRE-eGFP-expressing astrocytes interacting with vasculature characterized by intense Collagen-I deposition (pink arrows). DAPI (blue) is used for nuclear tracing.
Figure 6.

Lipid biosynthesis, metabolism and trafficking components are upregulated upon TBI in the wound area and in purified astrocytes upon in vitro stimulation with TGFβ1 or BMP4. Heat maps depicting log2 fold changes of differentially expressed genes encoding (A) cholesterol biosynthesis enzymes or (B) lipid trafficking and metabolism components, as determined by RNA-Seq analysis of purified astrocytes following 2 ng/ml TGFβ1 or 50 ng/ml BMP4 stimulation for 2 or 24 h (baseMean > 30 and FDR < 0.05). Black or white numerical values indicate log2 fold differences exceeding the colour scale bar limits. Pink frames indicate selected genes whose expression was verified with independent qRT-PCR analysis (Supplementary Fig. 6A and B). (C) qRT-PCR analysis of mRNA expression of Srebf1, Abca1, Lcat, Ldlr and Apoe in dissected contralateral (C) or ipsilateral (I) regions of TBI-subjected animals at 3 and 7 dpi (n = 5 animals/group). (D) Representative immunoblots and quantitative analysis of secreted APOE protein levels in cultures of purified astrocytes treated with 2 ng/ml TGFβ1, 50 ng/ml BMP4, 100 ng/ml Noggin or 10 μΜ SB431542 for 24 h (n = 6–8 cultures/group from three independent experiments). (E) Representative immunoblots of ABCA1, APOE and β-actin (loading control) in the contralateral (C) and ipsilateral (I) dissected tissues of TBI-subjected animals at 1, 3 and 7 dpi (n = 3–5 animals/group). (F) Quantitative analysis of ABCA1 and APOE protein levels shown in E, depicted as ratio of ipsilateral versus contralateral levels for each time-point. Data are expressed as mean ± standard error of the mean (SEM) analysed using one-way analysis of variance with Bonferroni’s post hoc test analysis (*P < 0.05, **P < 0.01, ***P < 0.001) or Newman–Keuls Multiple Comparison Test (#P < 0.05). F values and degrees of freedom (df) are shown in parentheses for each data set.
The apparent activation of tissue-repair and remodelling processes prompted us to investigate whether TGFβ1- or BMP4-stimulated astrocytes expressed genes associated with the ‘A2’-like astrocytic phenotype described previously (Zamanian et al., 2012; Liddelow et al., 2017). As shown in Fig. 5B, both TGFβ1 and BMP4 shifted the profile of the astrocytes towards the ‘A2’-like phenotype, and moreover, in agreement with Liddelow et al. (2017), TGFβ1 downregulated several genes associated with the ‘A1’-like phenotype.
Analysis of TGFβ1-stimulated microglia demonstrated modulation of several pathways, some of which were also regulated in astrocytes, such as integrin-, actin cytoskeleton-, glioma invasiveness-, ephrin receptor- and PDGF-signalling (Supplementary Fig. 5B). However, various inflammation-related pathways were significantly modulated selectively in microglia. Those included, among others, production of nitric oxide, nuclear factor erythroid 2-related factor 2 (NRF2)-mediated oxidative stress response, vascular endothelial growth factor (VEGF) signalling, leucocyte extravasation, interferon and toll-like receptor signalling. The negative effect of TGFβ-signalling on microglial inflammatory responses is well-documented (Spittau et al., 2013; Taylor et al., 2017; Zoller et al., 2018). Consistently, already after 2 h stimulation, interferon and toll-like receptor signalling pathways were characterized by negative IPA z-scores (predicted decreased activity) and importantly, after 24 h the vast majority of modulated pathways was characterized by predicted decreased activity. Analysis of the expression of selected genes associated with the quiescent ‘M0’-, pro-inflammatory ‘M1’- and anti-inflammatory ‘M2’-like microglia phenotypes demonstrated a down-regulation of several ‘M1’- and a shift towards an ‘M0/M2’-like phenotype by TGFβ1 (Supplementary Fig. 5C).
Comparative analysis of TGFβ1-stimulated astrocytes and microglia revealed modulation of chemokines, cytokines, growth factors and corresponding receptors and pointed out potential TGFβ-driven cross-interactions between the two populations (Fig. 5C). Specifically, Cx3cl1, Cxcl10 and Il6 mRNA levels were upregulated at 2 and/or 24 h in astrocytes and their corresponding receptors were upregulated in microglia. Pdgfs and Vegfa were upregulated in astrocytes and microglia; however, Pdgfrb was selectively upregulated in astrocytes and Flt1/Vegfr1 and Kdr/Vegfr2 in microglia. Fgf2 was upregulated in astrocytes and the corresponding receptors were upregulated at 2 h in astrocytes and at 24 h in both glial populations. The chemokines Ccl3 and Ccl4 and their receptor Ccr5 were selectively downregulated in microglia. Interestingly, several toll-like receptors were downregulated in microglia or astrocytes. BMP4 regulated the above-mentioned astrocytic genes in a manner similar to TGFβ1, indicating that the two axes do not annul each other and may regulate the glia cross-talk cooperatively.
Since bioinformatics highlighted ‘Axonal Guidance Signaling’, ‘Superpathway of Cholesterol Biosynthesis’ and ‘Hepatic Fibrosis/Hepatic Stellate Cell Activation’ as the top pathways modulated in astrocytes after 24 h stimulation with both TGFβ1 and BMP4 (Fig. 5A), further analysis was focused on these injury-related processes.
Up-regulation of lipid metabolism components in astrocytes by TGFβ-superfamily signalling or upon TBI
Remarkably, 20 out of the 24 enzymes involved in the ‘Superpathway of Cholesterol Biosynthesis’ (IPA) were upregulated in purified astrocytes upon 24 h treatment with TGFβ1 and/or BMP4, 13 of which were upregulated by both ligands (Fig. 6A). In addition, several components of lipid trafficking and metabolism, including transcription factors (Srebf1, Srebf2), transporters (Abca1, Abcg4), receptors (Ldlr, Lrp1), enzymes (Lcat, Fasn, Acaca) and other regulators (Insig1, Insig2, Npc1), were also upregulated by one or both ligands (Fig. 6B). Interestingly, few genes within this pathway (Sqle, Abca1, Abcg4, Lcat) were oppositely regulated by the two ligands (Fig. 6A and B). The response of selected genes encoding for cholesterol biosynthesis enzymes (Hmgcr, Cyp51, Idi1, Dhcr24, Sqle, Sc5d) or lipid metabolism components (Srebf1, Srebf2, Abca1, Abcg4, Ldlr, Lcat) was verified by qRT-PCRs of independent ligand-stimulated cultures (Supplementary Fig. 6A and B). In agreement with the in vitro stimulated cultures, mRNA levels for Srebf1 and Abca1 in the TBI-subjected animals were increased at 3 and 7 dpi and Abcg4 levels were decreased at 3 dpi in the ipsilateral side (Fig. 6C). Similarly, mRNA expression of Lcat was increased at the wound area at 3 and/or 7 dpi.
Since astrocytes are the main cholesterol producers and apolipoprotein-E (APOE) is the principal cholesterol carrier in adult brain, further analysis was focused on the production and secretion of APOE upon TGFβ1 or BMP4 stimulation. Increased levels of secreted APOE were detected in supernatants of astrocyte cultures treated with ligands for 24 h (Fig. 6D). Consistently, decreased APOE levels were found in the supernatants of astrocytes treated with either the TGFβ-receptor kinase inhibitor SB431542 or the BMP-inhibitor Noggin. Moreover, Apoe mRNA expression was increased in the ipsilateral side following SW at 7 dpi (Fig. 6C). At protein level, APOE was significantly decreased at 1 dpi, reached contralateral side levels at 3 dpi and then increased at 7 dpi (Fig. 6E and F). ABCA1 protein levels were also increased in the ipsilateral side, at 3 and 7 dpi.
TGFβ-superfamily system modulates processes related to axonal guidance and tissue-repair
Expression of several astrocyte-related axonal guidance molecules (Anderson et al., 2016) was modulated upon treatment of astrocytes with either TGFβ1 or BMP4 (Fig. 7A). Interestingly, TGFβ1 induced a more axonal-permissive profile, whereas BMP4 upregulated equal numbers of inhibitory and permissive molecules. Expression of representative inhibitory and permissive molecules was analysed in tissues from TBI-subjected animals. In line with the in vitro findings, Ncan, Nrp, and notably, Tnc and Lif, two transcripts reportedly highly upregulated in ‘A2’-astrocytes (Zamanian et al., 2012), were upregulated in the ipsilateral side at 3 dpi (Fig. 7B).
Numerous genes in the ‘Hepatic Fibrosis’ pathway (IPA), including collagens, growth factors, growth factor receptors and other tissue-remodelling components were modulated in astrocytes following TGFβ1 or BMP4 stimulation (Fig. 7C and Supplementary Fig. 7A). Interestingly, marginally overlapping sets of collagens were regulated by the two ligands. The majority of genes encoding growth factors and receptors was regulated early at 2 h, in contrast with other molecules in this annotation that were regulated after 24 h treatment. Representative genes upregulated in ligand-treated astrocytes were also found upregulated within the injured tissue, following TBI. Specifically, Col1a1, Col5a2, Il6 and Mmp2 were upregulated in the ipsilateral side at 3 and 7 dpi (Fig. 7D and Supplementary Fig. 7B). Notably, increased Collagen-I deposition was detected in the vasculature of injured areas and processes of BRE-eGFP+ astrocytes consistently were found closely interacting with vessel walls characterized by intense Collagen-I immunoreactivity (Fig. 7E).
Evidently, some genes belonging to the GO annotations analysed above responded differently in the in vitro stimulated astrocytes and the ipsilateral regions dissected from SW-subjected animals. Specifically, mRNA levels for Srebf2 (‘Superpathway of Cholesterol Biosynthesis’, IPA) were upregulated in vitro by TGFβ1 and/or BMP4 but were downregulated within the ipsilateral tissue at 3 and/or 7 dpi (Supplementary Fig. 6C). Likewise levels of Hmgcr and Cyp51 were not found altered significantly in the dissected TBI regions. Despite the changes in Apoe mRNA levels in the dissected tissues, this gene was not affected by ligand stimulation in vitro. Also, the in vitro observed modulation of Ctgf/Ccn2 and Igf1 (‘Hepatic Fibrosis’, IPA) could not be reproduced in acute TBI (Supplementary Fig. 7B). These differences could reflect inability of in vitro cultures of purified cells to simulate completely the complex cellular interactions that occur within the injured tissues, the possible differences in the kinetics by which certain processes unfold in vitro versus in vivo, or the assay’s detection limitations. Nevertheless, overall, the in vitro RNA-Seq data were in good agreement with the in vivo expression patterns revealed through the analysis of dissected trauma tissues.
Discussion
To assess the involvement of both TGFβ and BMP branches of the TGFβ-superfamily signalling system in TBI pathophysiology, we utilized a SW brain injury model and analysed the expression of key molecules within the injured tissues. We supplemented our in vivo study with gene expression profiling of TGFβ1- or BMP4-stimulated purified ‘reactive’ astrocytes and microglia. Our study demonstrated that upon brain injury both TGFβ and BMP axes are activated, as exemplified by increased levels of TGFβ and BMP pSmads, and mRNA for relevant ligands and target genes in the lesion area during the first dpi (Fig. 1). Activation of the TGFβ axis in astrocytes has already been associated with TBI (Schachtrup et al., 2010); however, this is the first time that activation of the BMP axis is clearly demonstrated within the injured area. Utilizing the BRE-eGFP reporter mice, we showed that activation of canonical BMP pathway was confined to astrocytes demarcating the transition between healthy and damaged tissue, a pattern reminiscent of the increased pSmad1/5/8 levels in GFAP+ astrocytes at the injury site in a rat model of demyelinating spinal cord injury (Fuller et al., 2007). Although substantial up-regulation of Tgfb1 mRNA levels was demonstrated in TBI tissues, corresponding changes in BMP-encoding transcripts were not as robust (Fig. 1D). Instead, TBI areas were characterized by reduced Noggin mRNA and protein levels (Fig. 1E, F and Supplementary Fig. 2A), suggesting that the trauma core favours activation of the BMP axis primarily through the establishment of an inhibitor-deficient microenvironment.
Up-regulation of BMP2/4, BMP7 and Noggin (with the latter being expressed almost exclusively by reactive astrocytes at the injury site) in penetrating brain and spinal cord injuries has been previously reported (Hampton et al., 2007). On the other hand, in an experimental model of stroke, Noggin expression mainly in activated microglia began to increase after 2 weeks and was further increased at 4 weeks only in the ischaemic subcortex, but the intensity was weak compared with the intensity of BMPs (Shin et al., 2012). Earlier studies with transgenic animals overexpressing constitutively Noggin in neurons provided evidence in favour of a protective role of Noggin upon ischaemia-induced brain injury (Samanta et al., 2010). However, these studies cannot be compared directly to the current study because Noggin overexpression was forced experimentally through the entire life of the animals leading most likely to readjustments of their homeostatic set points, as exemplified by the higher density of microglia in the tissues of non-manipulated animals (Samanta et al., 2010). Moreover, conditional deletion of the BMP type-II receptor (BMPR2) in NG2-expressing cells after hypoxic-ischaemic injury globally protected the brain and prevented loss of oligodendroglia (Dettman et al., 2018). Thus, Noggin may have distinct roles in the processes of glial scar formation and neuro-restoration depending on the model utilized, the mode and the timing of intervention.
The kinetics of BRE-eGFP reporter and pSmad activation suggested that the canonical TGFβ-superfamily system is not active in astrocytes immediately after injury but rather at later stages when these cells are already activated within the injured tissues. Therefore, insights regarding the way by which TGFβs and BMPs could modify the functionality of already activated astrocytes in vivo were obtained, indirectly, by treating with TGFβ1 or BMP4 purified reactive astrocytes that were derived from neonatal mixed glia cultures grown in vitro for ∼20–24 days. Earlier studies (Cahoy et al., 2008; Foo et al., 2011; Zamanian et al., 2012) have shown that astrocytes derived from such cultures share many of the characteristics of adult reactive astrocytes and in hierarchical clustering analysis they cluster closer to ‘A2’-like reactive astrocytes and away from both quiescent and LPS-reactive, ‘A1’-like, astrocytes (Zamanian et al., 2012). Interestingly, they express ∼60% of the ‘A2’-like and ∼50% of the ‘A1’-like phenotype characteristic marker genes (Zamanian et al., 2012), representing thus a suitable population to study factors that can modify the astrocytic ‘A1’/‘A2’-phenotypic plasticity. Indeed, both TGFβ1 and BMP4 shifted the profile of cultured astrocytes towards the ‘A2’ phenotype, and moreover, in agreement with Liddelow et al. (2017), TGFβ1 downregulated several genes associated with the ‘A1’ phenotype, suggesting that both TGFβ and BMP signalling systems could be important regulators of reactive astrocyte plasticity.
Transcriptomic analysis provided clues regarding the interplay between TGFβ and BMP axes and the responses they could potentially evoke in vivo. Interestingly, activation of astrocytes with either ligand affected highly overlapping sets of genes. Up to 69% of these genes responded to both TGFβ1 and BMP4 after 2 h stimulation (Fig. 3B), modulating pathways related to growth factor-mediated signalling, cytoskeleton remodelling and cell migration (Supplementary Fig. 5A). Twenty-four hours post-stimulation, when primarily processes related to tissue-repair and remodelling were modulated, the TGFβ1 and BMP4 transcriptomes diverged; however, the overall overlap was still up to ∼44% (Figs 3C and 5A).
The substantial overlap between TGFβ1- and BMP4-induced genes in astrocytes, especially at the early stage of treatment, may be explained by the reported property of TGFβ to activate BMP-Smads, through an interplay of TGFBRI/ALK5 with either ACVRL1/ALK1 (Goumans et al., 2002) or ACVR1/ALK2 (Ramachandran et al., 2018). Consistently, astrocytes express mRNA for Acvr1/Alk2 (Fig. 4B) and transient increase in pSmad1/5/8 levels was always observed in TGFβ1-treated astrocytes (Supplementary Fig. 4B). These findings support the notion that transcriptional responses evoked by TGFβ-superfamily require integrated combinatorial signalling via both Smad2/3 and Smad1/5/8 pathways (Ramachandran et al., 2018).
Despite the aforementioned overlap, the sets of TGFβ1- or BMP4-modulated genes within each GO annotation differed qualitatively and quantitatively, indicating that each ligand could stimulate qualitatively different functional outcomes. Firstly, commonly regulated genes responded to each ligand to a different degree. Hence, classical TGFβ targets such as Serpine1 responded more robustly to TGFβ1, whereas typical BMP targets such as Id1, Smad6 and Noggin responded more robustly to BMP4 stimulation (Supplementary Fig. 4A). Secondly, within each commonly regulated pathway, some genes responded to both ligands, and some were modulated selectively by either TGFβ1 (Ctgf/Ccn2, Col12a1) or BMP4 (Gata2, Bambi). The ratio of commonly and selectively modulated genes differed among pathways.
To investigate the effect of the TGFβ-superfamily signalling system on microglia, we utilized CD11b+GLAST- cells derived from 14 days old mixed glia cultures of neonatal brains that were cultured as pure microglia for additional 10–12 days (∼24 days in total). Interestingly, such cultured microglia responded remarkably well to TGFβ1, however, consistently with the failure to detect in vivo activation of the BRE-eGFP reporter in adult microglia upon TBI (Fig. 2D), failed to respond robustly to BMP4 and expressed relatively lower mRNA levels for BMP receptors and (Fig. 4B), suggesting that microglia might be programmed to be insensitive to BMP signalling.
It is currently recognized that neonatal microglia exhibit an ‘immature’ phenotype and do not express genes that characterize adult microglia (Butovsky et al., 2014). However, the cells utilized herein, probably due to their prolonged in vitro growth in the presence of serum, although they retained some characteristics of ‘immature’ microglia, such as high mRNA levels for ApoE, yet, they expressed half of the genes that constitute the ‘adult microglial’ molecular signature (40/81 genes with FPKM from 20 to 7500) and several genes that have been associated with the reportedly (Butovsky et al., 2014) ‘M0’-, ‘M1’- and ‘M2’-like phenotypes (15/38 ‘M0’, 21/52 ‘M1’ and 24/46 ‘M2’ marker genes). Moreover, 42 out of 54 genes that have been found to constitute a TGFβ dependent signature of adult microglia were also found herein modulated by TGFβ1 in the in vitro microglia cultures (32 upregulated and 10 downregulated; Supplementary Fig. 5C), suggesting that these cells may provide a useful model for studying the effect of the TGFβ-superfamily signalling system on microglial molecular plasticity. Indeed, TGFβ1 upregulated 22 out of 35 known ‘M0’, upregulated 25 out of 44 known ‘M2’ and downregulated 30 out of 44 known ‘M1’ marker genes, suggesting that TGFβ can shift the molecular profile of activated microglia away from the pro-inflammatory ‘M1’ phenotype and towards the quiescent or anti-inflammatory ‘M0/M2’ phenotypes, respectively.
TGFβ1 modulated expression of several chemokines, cytokines, growth factors and receptors in astrocytes and microglia. Up-regulation of a ligand in one cell-type and its cognate receptor(s) on the other, as exemplified by the Pdgfs/Pdgfrb, Cx3cl1/Cx3cr1 and Cxcl10/Cxcr3 pairs (Figs 5C and 8) might highlight potential TGFβ-driven cross-interactions between them. Notably, the CX3CL1/CX3CR1 axis has been associated with reduced neurotoxicity and microglial activation (Mizuno et al., 2003; Pabon et al., 2011; Febinger et al., 2015). Likewise, PDGFs have been associated with survival signals upon CNS injury (Funa and Sasahara, 2014) and the CXCL10/CXCR3 axis has been linked to microglial recruitment to the injury site and subsequent neuronal re-organization (Rappert et al., 2004; Li et al., 2006). Moreover, pairs of ligands and cognate receptors were modulated in the same population, specifically, Fgf2/Fgfr1:Fgfr2 and Pdgfb:Pdgfc/Pdgfrb were upregulated in astrocytes, Vegfa/Flt1:Kdr were upregulated and Ccl3: Ccl4/Ccr5 downregulated in microglia, highlighting thus potential homotypic interactions. Notably, activation of the FGF pathway has been associated with suppression of the ‘A1’astrocytic phenotype (Kang et al., 2014; Liddelow et al., 2017). Hence, it appears that another way by which TGFβ1 could indirectly modulate microglial reactivity is by up-regulating anti-inflammatory mediators in reactive astrocytes and their receptors in microglia.
Maximal activation of BRE-eGFP reporter occurred between 3 and 7 dpi, a period associated with initiation of tissue-repair. Therefore, particular emphasis was given to the effect of TGFβ1 or BMP4 on astrocytes at 24 h, when the top canonical pathways modulated by both ligands were the ‘Superpathway of Cholesterol Biosynthesis’, ‘Axonal Guidance Signaling’ and ‘Hepatic Fibrosis’. In adult brain, astrocytes are the major cholesterol providers (Nieweg et al., 2009) and their lipid metabolism is critical for synapse development and function (van Deijk et al., 2017). Interestingly, almost all enzymes involved in the superpathway of cholesterol biosynthesis together with various components of lipid transport and metabolism were upregulated in ligand-treated astrocytes (Fig. 6A and B). Activation of this pathway upon TBI was verified by demonstrating up-regulation of several components, such as Srebf1, Abca1, Ldlr and the extracellular cholesterol-esterifying enzyme Lcat, within the injured tissues (Fig. 6C). The increased Lcat expression could relate to the significant increase of esterified cholesterol following cortical impact injury (Roux et al., 2016). Moreover, secreted APOE was increased upon BMP4 or TGFβ1 stimulation of astrocytes in vitro, and decreased when inhibitors of TGFβ-signalling system were applied (Fig. 6D). Consistently, APOE and ABCA1 were gradually upregulated both at mRNA and protein levels, within the SW-injured tissues (Fig. 6C, E, and F). Interestingly, APOE protein levels were significantly reduced at 1 dpi, normalized at 3 dpi and increased at 7 dpi. Pro-inflammatory cytokines such as TNFα, IL1β and IFNγ suppress APOE production (Zhang et al., 2011). It is possible that an early burst of such cytokines (at 1 dpi) in the wound microenvironment could account for the observed decreased APOE protein levels at this early time-point. The anti-inflammatory and APOE-inducing function of the TGFβ-superfamily signalling that is mobilized after 3 dpi could be responsible for the late increase in both mRNA and protein levels of APOE in the injured tissues. This hypothetical scenario warrants further investigation. Collectively, our findings implicate for the first time TGFβ and BMP signalling in the regulation of cholesterol biosynthesis, APOE secretion and possibly lipoprotein formation following interaction of APOE with ABCA1 cholesterol transporter and LCAT esterifying enzyme in astrocytes.
TGFβ1 and BMP4 regulated several molecules with axonal guidance properties. Interestingly, TGFβ1 upregulated primarily axonal-permissive molecules and downregulated inhibitory ones, whereas BMP4 upregulated both permissive and inhibitory genes (Fig. 7A). Increased expression of both permissive and inhibitory axonal growth molecules was verified within the injured tissues (Fig. 7B) suggesting that astrocytes demarcating the wound could provide directionally the signals required for axonal regeneration (Jones et al., 2003; Faulkner et al., 2004; Cregg et al., 2014). Congruently, earlier studies have shown that BMP4 up-regulates neurocan (Ncan) and brevican (Bcan), and down-regulates phosphacan (Ptprz1) mRNA in spinal cord astrocytes (Fuller et al., 2007) and TGFβ1 increases mRNA and protein levels of neurocan in cortical astrocytes (Asher et al., 2000; Schachtrup et al., 2010).
Plethora of tissue-remodelling components, including collagens, growth factors and receptors were upregulated in astrocytes by TGFβ1 or BMP4. Interestingly, the two ligands regulated different collagens, supporting the notion that TGFβ and/or BMP signalling may play distinct roles during tissue-repair. Up-regulation of selected genes, regulated in vitro selectively by TGFβ1 (Col5a2, Mmp2), BMP4 (Col1a1) or both (Il6), was verified by analysing dissected wound regions (Fig. 7C and D and Supplementary Fig. 7B). Notably, the visualization of processes of BRE-eGFP+ astrocytes in close association to vessel walls with increased Collagen-I immunoreactivity (Fig. 7E), strongly suggests that BMP-activated astrocytes could play a key role in sealing disrupted vasculature and restoring blood–brain barrier.
TBI represents one of the principal causes of death and disability worldwide and ∼50% of the world’s population will have at least one TBI incidence over their lifetime (Maas et al., 2017; Quaglio et al., 2017). Despite the rapidly increasing body of literature, TBIs remain especially challenging to treat due to their heterogeneous nature and the induction of complex pathogenesis pathways that, still, are not fully understood (Saatman et al., 2008). Therefore, clarification of the molecular processes underlying TBI pathophysiology, in particular, the cross-talk between astrocytes and microglia and the mechanisms that modulate plasticity of activated glial cells will be important steps towards the development of therapeutic strategies that could harness their reparatory capacity (Sofroniew, 2009).
Our study provides a comprehensive analysis of the potential role of both branches of the TGFβ-superfamily signalling system in the context of acute focal TBI pathology and draws attention on the capacity of both TGFβ and BMP signalling to regulate key processes for tissue-injury repair and glial pro-inflammatory activity, in activated astrocytes and microglia surrounding the trauma (Fig. 8). Both TGFβ and BMP act on astrocytes and induce highly overlapping transcriptional programmes, whereas microglia respond selectively to TGFβ signalling. Thus, selective manipulation of either branch of the TGFβ-superfamily, especially in astrocytes, should be treated with caution given the potential compensatory activation or inhibition of the other. The SW injury utilized herein represents probably the simplest form of TBI that does not simulate the biomechanical aspects of human TBI. However, the wealth of information obtained herein could be further utilized to study more complex models that simulate closer human TBI pathophysiology, ideally with genetically modified animals and inducible ablation of specific components of the TGFβ-superfamily signalling system (receptors, ligands or inhibitors) specifically in astrocytes or microglia and thus aid prospectively to the design of therapeutic strategies for TBI and similar neuro-inflammatory conditions.
Figure 8.
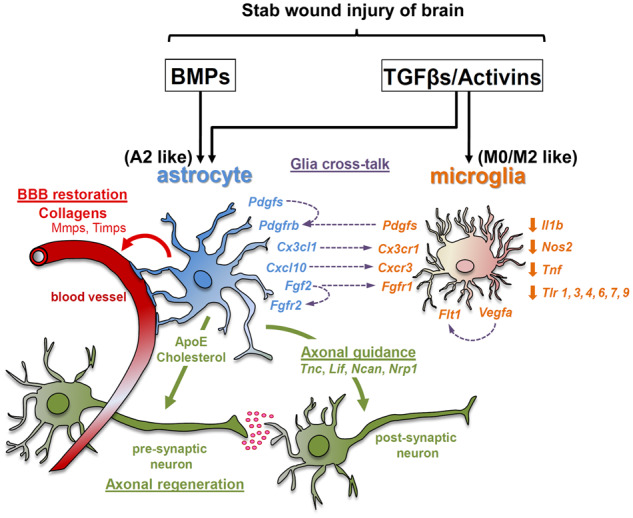
Schematic representation summarizing the TGFβ-superfamily-regulated glia cell responses in the lesion area, upon SW brain injury derived from the current study. BBB = blood–brain barrier.
Supplementary material
Supplementary material is available at Brain Communications online.
Supplementary Material
Acknowledgements
Drs. E. Rigana and S. Pagkakis, Imaging Facility, BRFAA, are gratefully acknowledged for support in confocal microscopy, Drs. A. Moustakas, E. Andreakos and K. Thanopoulou for critical reading of the manuscript and Drs. S. Georgopoulos and I. Serafimidis for kindly providing anti-APOE antibody and Fc-Noggin, respectively.
Funding
This work was supported by a grant from the National Parkinson Foundation (NPF-UP202), and the General Secretariat of Research and Development, Hellenic Ministry of Education, Research and Religious Affairs, programme ARISTEIA II (#3451).
Competing interests
The authors report no competing interests.
Glossary
- ALK
anaplastic lymphoma kinase
- BMP
Bone morphogenetic protein
- BRE
BMP-responsive elements
- dpi
days post-injury
- eGFP
enhanced green fluorescent protein
- ERK
extracellular signal-regulated kinase
- FPKM
fragments per kiloase million
- GLAST
glutamate-aspatat transporter
- GO
gene ontology
- IPA
ingenuity pathway analysis
- NRF2
nuclear factor erythroid 2-related factor 2
- PDL
poly-D-lysine
- PDGF
platelet-derived growth factor
- PI3K
phosphatidylinositol-3-kinase
- qRT-PCR
quantitative real-tme polymerse chain reac
- SW
Stab wound
- VEGF
vascular endothelial growth factor
- TBI
traumatic brain injury
- TGF
transforming growth factor beta
References
- Anderson MA, Burda JE, Ren Y, Ao Y, O’Shea TM, Kawaguchi R, et al. Astrocyte scar formation aids central nervous system axon regeneration. Nature 2016; 532: 195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asher RA, Morgenstern DA, Fidler PS, Adcock KH, Oohira A, Braistead JE, et al. Neurocan is upregulated in injured brain and in cytokine-treated astrocytes. J Neurosci 2000; 20: 2427–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas SK, Mantovani A.. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol 2010; 11: 889–96. [DOI] [PubMed] [Google Scholar]
- Budi EH, Duan D, Derynck R.. Transforming growth factor-beta receptors and Smads: regulatory complexity and functional versatility. Trends Cell Biol 2017; 27: 658–72. [DOI] [PubMed] [Google Scholar]
- Buffo A, Vosko MR, Erturk D, Hamann GF, Jucker M, Rowitch D, et al. Expression pattern of the transcription factor Olig2 in response to brain injuries: implications for neuronal repair. Proc Natl Acad Sci USA 2005; 102: 18183–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burda JE, Bernstein AM, Sofroniew MV.. Astrocyte roles in traumatic brain injury. Exp Neurol 2016; 275 (Pt 3): 305–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buss A, Pech K, Kakulas BA, Martin D, Schoenen J, Noth J, et al. TGF-beta1 and TGF-beta2 expression after traumatic human spinal cord injury. Spinal Cord 2008; 46: 364–71. [DOI] [PubMed] [Google Scholar]
- Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, et al. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat Neurosci 2014; 17: 131–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bylicky MA, Mueller GP, Day RM.. Mechanisms of endogenous neuroprotective effects of astrocytes in brain injury. Oxid Med Cell Longev 2018; 2018: 1.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci 2008; 28: 264–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Van Gulden S, McGuire TL, Fleming AC, Oka C, Kessler JA, et al. BMP-responsive protease HtrA1 is differentially expressed in astrocytes and regulates astrocytic development and injury response. J Neurosci 2018; 38: 3840–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung WS, Clarke LE, Wang GX, Stafford BK, Sher A, Chakraborty C, et al. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature 2013; 504: 394–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cregg JM, DePaul MA, Filous AR, Lang BT, Tran A, Silver J.. Functional regeneration beyond the glial scar. Exp Neurol 2014; 253: 197–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dettman RW, Birch D, Fernando A, Kessler JA, Dizon M.. Targeted knockdown of bone morphogenetic protein signaling within neural progenitors protects the brain and improves motor function following postnatal hypoxia-ischemia. Dev Neurosci 2018; 40: 23–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewan MC, Rattani A, Gupta S, Baticulon RE, Hung YC, Punchak M, et al. Estimating the global incidence of traumatic brain injury. J Neurosurg 2018; 130: 1039–408. [DOI] [PubMed] [Google Scholar]
- Donat CK, Scott G, Gentleman SM, Sastre M.. Microglial activation in traumatic brain injury. Front Aging Neurosci 2017; 9: 208.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faulkner JR, Herrmann JE, Woo MJ, Tansey KE, Doan NB, Sofroniew MV.. Reactive astrocytes protect tissue and preserve function after spinal cord injury. J Neurosci 2004; 24: 2143–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Febinger HY, Thomasy HE, Pavlova MN, Ringgold KM, Barf PR, George AM, et al. Time-dependent effects of CX3CR1 in a mouse model of mild traumatic brain injury. J Neuroinflammation 2015; 12: 154.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foo LC, Allen NJ, Bushong EA, Ventura PB, Chung WS, Zhou L, et al. Development of a method for the purification and culture of rodent astrocytes. Neuron 2011; 71: 799–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frik J, Merl-Pham J, Plesnila N, Mattugini N, Kjell J, Kraska J, et al. Cross-talk between monocyte invasion and astrocyte proliferation regulates scarring in brain injury. EMBO Rep 2018; 19. pii: e45294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller ML, DeChant AK, Rothstein B, Caprariello A, Wang R, Hall AK, et al. Bone morphogenetic proteins promote gliosis in demyelinating spinal cord lesions. Ann Neurol 2007; 62: 288–300. [DOI] [PubMed] [Google Scholar]
- Fumagalli M, Lecca D, Abbracchio MP.. Role of purinergic signalling in neuro-immune cells and adult neural progenitors. Front Biosci 2011; 16: 2326–41. [DOI] [PubMed] [Google Scholar]
- Funa K, Sasahara M.. The roles of PDGF in development and during neurogenesis in the normal and diseased nervous system. J Neuroimmune Pharmacol 2014; 9: 168–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z, Zhu Q, Zhang Y, Zhao Y, Cai L, Shields CB, et al. Reciprocal modulation between microglia and astrocyte in reactive gliosis following the CNS injury. Mol Neurobiol 2013; 48: 690–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goumans MJ, Valdimarsdottir G, Itoh S, Rosendahl A, Sideras P, ten Dijke P.. Balancing the activation state of the endothelium via two distinct TGF-beta type I receptors. EMBO J 2002; 21: 1743–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton DW, Asher RA, Kondo T, Steeves JD, Ramer MS, Fawcett JW.. A potential role for bone morphogenetic protein signalling in glial cell fate determination following adult central nervous system injury in vivo. Eur J Neurosci 2007; 26: 3024–35. [DOI] [PubMed] [Google Scholar]
- Helmy A, Carpenter KL, Menon DK, Pickard JD, Hutchinson PJ.. The cytokine response to human traumatic brain injury: temporal profiles and evidence for cerebral parenchymal production. J Cereb Blood Flow Metab 2011; 31: 658–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang RQ, Cheng HL, Zhao XD, Dai W, Zhuang Z, Wu Y, et al. Preliminary study on the effect of trauma-induced secondary cellular hypoxia in brain injury. Neurosci Lett 2010; 473: 22–7. [DOI] [PubMed] [Google Scholar]
- Jassam YN, Izzy S, Whalen M, McGavern DB, El Khoury J.. Neuroimmunology of traumatic brain injury: time for a paradigm shift. Neuron 2017; 95: 1246–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones LL, Margolis RU, Tuszynski MH.. The chondroitin sulfate proteoglycans neurocan, brevican, phosphacan, and versican are differentially regulated following spinal cord injury. Exp Neurol 2003; 182: 399–411. [DOI] [PubMed] [Google Scholar]
- Kang W, Balordi F, Su N, Chen L, Fishell G, Hebert JM.. Astrocyte activation is suppressed in both normal and injured brain by FGF signaling. Proc Natl Acad Sci USA 2014; 111: E2987–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karve IP, Taylor JM, Crack PJ.. The contribution of astrocytes and microglia to traumatic brain injury. Br J Pharmacol 2016; 173: 692–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komuta Y, Teng X, Yanagisawa H, Sango K, Kawamura K, Kawano H.. Expression of transforming growth factor-beta receptors in meningeal fibroblasts of the injured mouse brain. Cell Mol Neurobiol 2010; 30: 101–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewen A, Soderstrom S, Hillered L, Ebendal T.. Expression of serine/threonine kinase receptors in traumatic brain injury. Neuroreport 1997; 8: 475–9. [DOI] [PubMed] [Google Scholar]
- Li H, Gang Z, Yuling H, Luokun X, Jie X, Hao L, et al. Different neurotropic pathogens elicit neurotoxic CCR9- or neurosupportive CXCR3-expressing microglia. J Immunol 2006; 177: 3644–56. [DOI] [PubMed] [Google Scholar]
- Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017; 541: 481–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindholm D, Castren E, Kiefer R, Zafra F, Thoenen H.. Transforming growth factor-beta 1 in the rat brain: increase after injury and inhibition of astrocyte proliferation. J Cell Biol 1992; 117: 395–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loane DJ, Kumar A.. Microglia in the TBI brain: the good, the bad, and the dysregulated. Exp Neurol 2016; 275 (Pt 3): 316–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan TT, Villapol S, Symes AJ.. TGF-beta superfamily gene expression and induction of the Runx1 transcription factor in adult neurogenic regions after brain injury. PLoS One 2013; 8: e59250.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maas AIR, Menon DK, Adelson PD, Andelic N, Bell MJ, Belli A, et al. Traumatic brain injury: integrated approaches to improve prevention, clinical care, and research. Lancet Neurol 2017; 16: 987–1048. [DOI] [PubMed] [Google Scholar]
- Majdan M, Plancikova D, Brazinova A, Rusnak M, Nieboer D, Feigin V, et al. Epidemiology of traumatic brain injuries in Europe: a cross-sectional analysis. Lancet Public Health 2016; 1: e76–e83. [DOI] [PubMed] [Google Scholar]
- Mathewson AJ, Berry M.. Observations on the astrocyte response to a cerebral stab wound in adult rats. Brain Res 1985; 327: 61–9. [DOI] [PubMed] [Google Scholar]
- McCarthy KD, de Vellis J.. Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol 1980; 85: 890–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon DK, Schwab K, Wright DW, Maas AI.. Position statement: definition of traumatic brain injury. Arch Phys Med Rehabil 2010; 91: 1637–40. [DOI] [PubMed] [Google Scholar]
- Michell-Robinson MA, Touil H, Healy LM, Owen DR, Durafourt BA, Bar-Or A, et al. Roles of microglia in brain development, tissue maintenance and repair. Brain 2015; 138: 1138–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazawa K, Miyazono K.. Regulation of TGF-beta family signaling by inhibitory Smads. Cold Spring Harb Perspect Biol 2017; 9: a022095.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno T, Kawanokuchi J, Numata K, Suzumura A.. Production and neuroprotective functions of fractalkine in the central nervous system. Brain Res 2003; 979: 65–70. [DOI] [PubMed] [Google Scholar]
- Monteiro RM, de Sousa Lopes SM, Bialecka M, de Boer S, Zwijsen A, Mummery CL.. Real time monitoring of BMP Smads transcriptional activity during mouse development. Genesis 2008; 46: 335–46. [DOI] [PubMed] [Google Scholar]
- Moustakas A, Heldin CH.. Non-Smad TGF-beta signals. J Cell Sci 2005; 118: 3573–84. [DOI] [PubMed] [Google Scholar]
- Moustakas A, Souchelnytskyi S, Heldin CH.. Smad regulation in TGF-beta signal transduction. J Cell Sci 2001; 114: 4359–69. [DOI] [PubMed] [Google Scholar]
- Myer DJ, Gurkoff GG, Lee SM, Hovda DA, Sofroniew MV.. Essential protective roles of reactive astrocytes in traumatic brain injury. Brain 2006; 129: 2761–72. [DOI] [PubMed] [Google Scholar]
- Nieweg K, Schaller H, Pfrieger FW.. Marked differences in cholesterol synthesis between neurons and glial cells from postnatal rats. J Neurochem 2009; 109: 125–34. [DOI] [PubMed] [Google Scholar]
- Pabon MM, Bachstetter AD, Hudson CE, Gemma C, Bickford PC.. CX3CL1 reduces neurotoxicity and microglial activation in a rat model of Parkinson’s disease. J Neuroinflammation 2011; 8: 9.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfrieger FW, Ungerer N.. Cholesterol metabolism in neurons and astrocytes. Prog Lipid Res 2011; 50: 357–71. [DOI] [PubMed] [Google Scholar]
- Quaglio G, Gallucci M, Brand H, Dawood A, Cobello F.. Traumatic brain injury: a priority for public health policy. Lancet Neurol 2017; 16: 951–2. [DOI] [PubMed] [Google Scholar]
- Ramachandran A, Vizan P, Das D, Chakravarty P, Vogt J, Rogers KW, et al. TGF-beta uses a novel mode of receptor activation to phosphorylate SMAD1/5 and induce epithelial-to-mesenchymal transition. Elife 2018; 7. pii: e31756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappert A, Bechmann I, Pivneva T, Mahlo J, Biber K, Nolte C, et al. CXCR3-dependent microglial recruitment is essential for dendrite loss after brain lesion. J Neurosci 2004; 24: 8500–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosendahl A, Checchin D, Fehniger TE, ten Dijke P, Heldin CH, Sideras P.. Activation of the TGF-beta/activin-Smad2 pathway during allergic airway inflammation. Am J Respir Cell Mol Biol 2001; 25: 60–8. [DOI] [PubMed] [Google Scholar]
- Rosendahl A, Pardali E, Speletas M, Ten Dijke P, Heldin CH, Sideras P.. Activation of bone morphogenetic protein/Smad signaling in bronchial epithelial cells during airway inflammation. Am J Respir Cell Mol Biol 2002; 27: 160–9. [DOI] [PubMed] [Google Scholar]
- Roux A, Muller L, Jackson SN, Post J, Baldwin K, Hoffer B, et al. Mass spectrometry imaging of rat brain lipid profile changes over time following traumatic brain injury. J Neurosci Methods 2016; 272: 19–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saatman KE, Duhaime AC, Bullock R, Maas AI, Valadka A, Manley GT.. Classification of traumatic brain injury for targeted therapies. J Neurotrauma 2008; 25: 719–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeed AI, Sharov V, White J, Li J, Liang W, Bhagabati N, et al. TM4: a free, open-source system for microarray data management and analysis. Biotechniques 2003; 34: 374–8. [DOI] [PubMed] [Google Scholar]
- Samanta J, Alden T, Gobeske K, Kan L, Kessler JA.. Noggin protects against ischemic brain injury in rodents. Stroke 2010; 41: 357–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saura J, Tusell JM, Serratosa J.. High-yield isolation of murine microglia by mild trypsinization. Glia 2003; 44: 183–9. [DOI] [PubMed] [Google Scholar]
- Schachtrup C, Ryu JK, Helmrick MJ, Vagena E, Galanakis DK, Degen JL, et al. Fibrinogen triggers astrocyte scar formation by promoting the availability of active TGF-beta after vascular damage. J Neurosci 2010; 30: 5843–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setoguchi T, Yone K, Matsuoka E, Takenouchi H, Nakashima K, Sakou T, et al. Traumatic injury-induced BMP7 expression in the adult rat spinal cord. Brain Res 2001; 921: 219–25. [DOI] [PubMed] [Google Scholar]
- Shi Y, Massague J.. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 2003; 113: 685–700. [DOI] [PubMed] [Google Scholar]
- Shin JA, Kang JL, Lee KE, Park EM.. Different temporal patterns in the expressions of bone morphogenetic proteins and noggin during astroglial scar formation after ischemic stroke. Cell Mol Neurobiol 2012; 32: 587–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sica A, Mantovani A.. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest 2012; 122: 787–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidoryk-Wegrzynowicz M, Wegrzynowicz M, Lee E, Bowman AB, Aschner M.. Role of astrocytes in brain function and disease. Toxicol Pathol 2011; 39: 115–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci 2009; 32: 638–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV, Vinters HV.. Astrocytes: biology and pathology. Acta Neuropathol 2010; 119: 7–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sountoulidis A, Stavropoulos A, Giaglis S, Apostolou E, Monteiro R, Chuva de Sousa Lopes SM, et al. Activation of the canonical bone morphogenetic protein (BMP) pathway during lung morphogenesis and adult lung tissue repair. PLoS One 2012; 7: e41460.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spittau B, Wullkopf L, Zhou X, Rilka J, Pfeifer D, Krieglstein K.. Endogenous transforming growth factor-beta promotes quiescence of primary microglia in vitro. Glia 2013; 61: 287–300. [DOI] [PubMed] [Google Scholar]
- Szepesi Z, Manouchehrian O, Bachiller S, Deierborg T.. Bidirectional microglia-neuron communication in health and disease. Front Cell Neurosci 2018; 12: 323.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor RA, Chang CF, Goods BA, Hammond MD, Mac Grory B, Ai Y, et al. TGF-beta1 modulates microglial phenotype and promotes recovery after intracerebral hemorrhage. J Clin Invest 2017; 127: 280–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Deijk AF, Camargo N, Timmerman J, Heistek T, Brouwers JF, Mogavero F, et al. Astrocyte lipid metabolism is critical for synapse development and function in vivo. Glia 2017; 65: 670–82. [DOI] [PubMed] [Google Scholar]
- Xilouri M, Kyratzi E, Pitychoutis PM, Papadopoulou-Daifoti Z, Perier C, Vila M, et al. Selective neuroprotective effects of the S18Y polymorphic variant of UCH-L1 in the dopaminergic system. Hum Mol Genet 2012; 21: 874–89. [DOI] [PubMed] [Google Scholar]
- Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG, et al. Genomic analysis of reactive astrogliosis. J Neurosci 2012; 32: 6391–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Wu LM, Wu J.. Cross-talk between apolipoprotein E and cytokines. Mediators Inflammation 2011; 2011: 1.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YE. Non-Smad pathways in TGF-beta signaling. Cell Res 2009; 19: 128–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoller T, Schneider A, Kleimeyer C, Masuda T, Potru PS, Pfeifer D, et al. Silencing of TGFbeta signalling in microglia results in impaired homeostasis. Nat Commun 2018; 9: 4011.. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data supporting the findings of this study are available from the corresponding author on request. Gene expression data are available from Sequence Read Archive (https://www.ncbi.nlm.nih.gov/sra), accession number PRJNA527753.
All materials used in this study are listed in Supplementary Table 3.