

Abstract
Glioblastoma is a fatal disease in which most targeted therapies have clinically failed. However, pharmacological reactivation of tumour suppressors has not been thoroughly studied as yet as a glioblastoma therapeutic strategy. Tumour suppressor protein phosphatase 2A is inhibited by non-genetic mechanisms in glioblastoma, and thus, it would be potentially amendable for therapeutic reactivation. Here, we demonstrate that small molecule activators of protein phosphatase 2A, NZ-8-061 and DBK-1154, effectively cross the in vitro model of blood–brain barrier, and in vivo partition to mouse brain tissue after oral dosing. In vitro, small molecule activators of protein phosphatase 2A exhibit robust cell-killing activity against five established glioblastoma cell lines, and nine patient-derived primary glioma cell lines. Collectively, these cell lines have heterogeneous genetic background, kinase inhibitor resistance profile and stemness properties; and they represent different clinical glioblastoma subtypes. Moreover, small molecule activators of protein phosphatase 2A were found to be superior to a range of kinase inhibitors in their capacity to kill patient-derived primary glioma cells. Oral dosing of either of the small molecule activators of protein phosphatase 2A significantly reduced growth of infiltrative intracranial glioblastoma tumours. DBK-1154, with both higher degree of brain/blood distribution, and more potent in vitro activity against all tested glioblastoma cell lines, also significantly increased survival of mice bearing orthotopic glioblastoma xenografts. In summary, this report presents a proof-of-principle data for blood–brain barrier—permeable tumour suppressor reactivation therapy for glioblastoma cells of heterogenous molecular background. These results also provide the first indications that protein phosphatase 2A reactivation might be able to challenge the current paradigm in glioblastoma therapies which has been strongly focused on targeting specific genetically altered cancer drivers with highly specific inhibitors. Based on demonstrated role for protein phosphatase 2A inhibition in glioblastoma cell drug resistance, small molecule activators of protein phosphatase 2A may prove to be beneficial in future glioblastoma combination therapies.
Keywords: PME-1, CIP2A, DT-061, E98, tricyclic neurological drugs
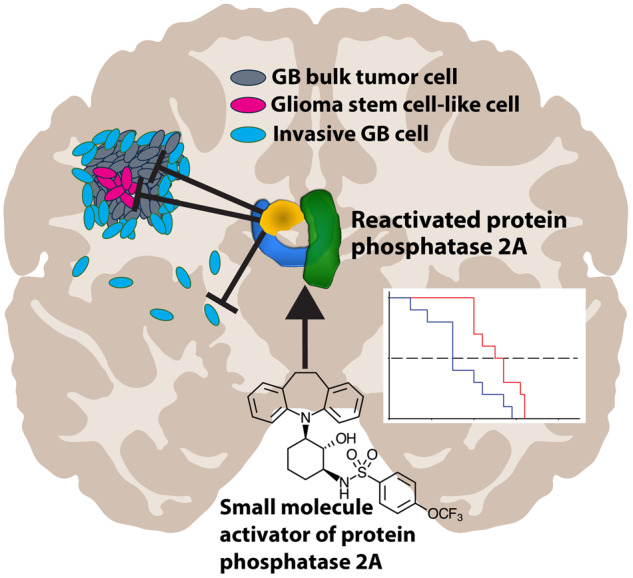
This study introduces the therapeutic potential of orally bioavailable protein phosphatase 2A reactivating small molecules, in an intracranial infiltrative glioblastoma model. In vitro, small molecule activators of protein phosphatase 2A are effective against 14 different glioblastoma cell lines, regardless of their subtype, genetic background or stemness properties. Conceptually, the results reveal drug-elicited tumour suppressor reactivation for glioblastoma.
Graphical Abstract
Graphical Abstract.
Introduction
One of the hallmarks of glioblastoma (GB) is dysregulated phosphorylation-dependent pathways, which are drivers of its malignant progression (Dunn et al., 2012). Nevertheless, all tested kinase inhibitors have failed to prolong the overall survival of GB patients in clinical trials (Mooney et al., 2019; Tomiyama and Ichimura, 2019). One of the reasons is that the blood–brain barrier (BBB) prevents the otherwise potentially effective kinase inhibitors from reaching the brain at high enough concentrations (Harder et al., 2018). Another potential reason is non-mutational plasticity induced by kinase inhibitors in GB cells (van den Heuvel et al., 2017). Notably, phosphorylation of GB driver pathways is not only regulated by kinases, but also by phosphatases (Narla et al., 2018; Tomiyama et al., 2019). Amongst them, protein phosphatase 2A (PP2A) is a serine/threonine phosphatase that regulates multiple oncogenic kinase signalling pathways, as well as apoptotic mechanisms (Perrotti and Neviani, 2013; Kauko and Westermarck, 2018). Interestingly, although PP2A is an established tumour suppressor (Perrotti and Neviani, 2013; Narla et al., 2018), the genes coding for PP2A complex components are rarely mutated in clinical GB samples (Kaur et al., 2016). Instead, GB cells overexpress endogenous inhibitor proteins of PP2A (PAIPs)(Kauko and Westermarck, 2018), such as PME-1, CIP2A and ARPP19 (Jiang et al., 2016; Kaur et al., 2016; Qin et al., 2018). Inhibition of these PP2A inhibitor proteins restricts GB cell growth, and PME-1-mediated PP2A inhibition drives widespread kinase inhibitor resistance in GB (Kaur et al., 2016). Because PP2A is non-genetically inactivated in GB by the PAIPs, it could be a suitable target for tumour suppressor reactivation therapies (Westermarck, 2018; Tomiyama et al., 2019).
A series of small molecule activators of PP2A (SMAPs) were recently derived from tricyclic neurological drugs such as Chlorpromazine and Clomipramine (Kastrinsky et al., 2015), which have a long association with anti-proliferative properties in vitro (Sachlos et al., 2012; Sangodkar et al., 2012), and with anti-cancer effects in patients, based on epidemiology studies (Dalton et al., 2006; Zingone et al., 2017). However, although these drugs obviously cross BBB, attempts to therapeutically exploit the anti-glioma properties of these tricyclics themselves have failed due to their potent inherent effects on G protein-coupled receptor and amine transporter pharmacology. Both activities are responsible for central nervous system (CNS)—and cardiovascular toxicity when used at doses where anti-tumour effects are manifest (Murren et al., 1996; Jahchan et al., 2013). Based on photoaffinity labelling studies, radiolytic footprinting and mutagenesis of the binding site, the target of SMAPs has been identified as the interface of the A and C subunits in the PP2A complex (Sangodkar et al., 2017). The SMAPs have shown efficacy as orally available monotherapy in animal models in solid non-CNS cancer types (Sangodkar et al., 2017; Kauko et al., 2018), but neither their BBB penetration properties, nor potential as CNS cancer drug has not been studied as yet.
Materials and methods
Compounds and cell lines
The synthesis of SMAPs is described in detail in public patent application PCT/US2015/019674. For in vitro work NZ-8-061 and DBK-1154 were dissolved in DMSO and for in vivo work compounds were formulated in 10% N,N-dimethylacetamide (DMA—Sigma, cat# 27012), 10% Kolliphor HS 15 (solutol—Sigma, cat# 42966) and 80% sterile water, and kept in room temperature and protected from light.
Established human GB cell lines U87MG, A172, U118, E98-FM-Cherry (Claes et al., 2008) were cultured in DMEM (Sigma-Aldrich) whereas T98G was cultured in Eagle MEM (Sigma-Aldrich). Human fibroblasts (kind gift of Johanna Ivaska, Turku Bioscience, Turku, Finland) were cultured in DMEM (Sigma-Aldrich). All growth mediums were supplemented with 10% heat-inactivated foetal bovine serum (Biowest) and fibroblasts medium was additionally supplemented with 20% foetal bovine serum, 2 mmol/l l-glutamine and penicillin (50 units/ml)/streptomycin (50 µg/ml). Cultured were maintained in a humified atmosphere of 5% CO2 at 37°C.
Patient-derived glioma stem cell cultures were obtained from HGCC biobank (Xie et al., 2015). BT3CD133+ gliospheres were originated from surgical samples (Kuopio University Hospital, Finland) during years 2010–11 and sorted for their CD133 expression from BT3 cells as described in Le Joncour et al. (2019a). Cells were cultivated as adherent cultures in serum-free neural stem cell (NSC) media containing an equal mix of DMEM/F12 (1:1) with glutamax (31331-093, Fisher Scientific) and neurobasal (21103-049, Fisher Scientific) media with 1X B-27 (12587001, Fisher Scientific) 1× N-2 (17502001, Fisher Scientific) supplements, 1% penicillin-streptomycin (P4333, Sigma-Aldrich), bFGF (10 ng/ml, 100-18B, Peprotech) and EGF (10 ng/ml, AF-100-15, Peprotech) (NSC+/+ medium). Cells were cultured on Primaria vessels (Fisher Scientific) coated with 1% laminin (L2020-1MG, Sigma-Aldrich) solution in sterile PBS at 37°C with 5% CO2.
Colony formation assay
Optimized number of cells (3 × 103 to 10 × 103) were seeded in 12-well plates (Sigma-Aldrich) and allowed to attach. After 24 h, cells were treated with determined concentration of chemical compounds. After 72 h, medium was replaced with non-drug containing medium and the cells were left for another 72 h or until the control well was confluent. Cell colonies were fixed with ice-cold methanol and stained with 0.2% crystal violet solution in 10% ethanol for 15 min at room temperature. Plates were dried and scanned with Epson Perfection V700 Photo scanner. Quantifications were performed with ImageJ by using Colony area (Guzman et al., 2014) plugin. Data were normalized and presented as a per cent of the control.
Cell viability assay
Optimized number of U87MG, A172, U118, T98G and E98 cells (3 × 103 to 5 × 103) were seeded in white 96-well plates (Perkin Elmer) and allowed to attach. After 24 h, cells were treated with determined concentration of chemical compounds for 24–72 h. Cell viability was measured using a CellTitre Glo assay (Promega) according the manufacturer’s instructions. Luminescence was read with BioTek Synergy H1 plate reader (BioTek, Country). Bioluminescence was normalized and presented as a per cent of the control.
Cell viability for glioma stem cells was seeded into 96-well plates 5 × 103 cells/well and allowed to attach overnight. Next day medium was changed into a medium containing drugs. After 72 h of treatment medium was changed into fresh NSC+/+ containing 10% of Alamar blue solution (10099022, Fisher Scientific). Fluorescent signal was measured after 210 min of incubation in 37°C using Wallac Victor 1420 plate reader (Perkin Elmer). Fluorescence was normalized and presented as a per cent of the control.
Animal experiments
The animal experiments for this study were carried out according to the Animal Experiment Board in Finland (ELLA) for the care and use of animals under the licenses 4161/04.10.07/2015 and 9241/2018. Six- to 8-week-old female athymic BALB/cOlaHsd-Foxn1nu (Envigo, France). The animals were kept under specific pathogen-free conditions in individually ventilated cages in the animal care facility. Mice were kept on a 12-h light/dark cycle with access to the autoclaved water and irradiated chow ad libitum. Mice were allowed to adapt to the facility for 1 week before starting the experiments.
The intracranial E98 GB mouse model was done by preparing a cell suspension of 1.5 × 105 cells in 5 µl of PBS which was inoculated into the brain of anaesthetized (isoflurane) mice. Injection was done as followed. A small incision (0.5 mm) was made to skin middle of the skull and a small hole (Ø 0.2 mm) was drilled above the injection site. Coordinates for the injection site from bregma: 1 mm posterior, 2 mm to right, −3 mm depth from the skull. Injection was done slowly within 5 min and the needle was allowed to stay in the brain for 5 min after the injection. After the injection, wound was sutured and mice were allowed to wake up in controlled environment. After 10 days, mice were imaged with bioluminescence and based on the bioluminescence signal from the tumour the mice were randomized to two equal groups, control and treated. Randomization was done with web-based programme (Laajala et al., 2016). Drug treatment of the mice was started after the randomization. The NZ-8-061 experiment had 8 mice per group and the DBK-1154 had 10 mice per group. NZ-8-061 and DBK-1154 were formulated in 10% N,N-dimethylacetamide (DMA—Sigma, cat# 27012), 10% Kolliphor HS 15 (solutol—Sigma, cat# 42966), 80% sterile water. In the studies, NZ-8-061 was given 30 mg/kg and DBK-1154 100 mg/kg and dosed twice a day. Higher dose for DBK-1154 is explained because of its lower bioavailability through oral dosing.
In the experiment’s tumour growth was monitored approximately twice a week by bioluminescence imaging using Xenogen IVIS Spectrum (Caliper Life Sciences) until the end of the experiment. For imaging, mice were injected with 150 mg/kg mouse weight XenoLight d-luciferin substrate (Caliper Life Sciences). The imaging was performed under isoflurane gas anaesthesia. Images were quantified with Living image®.
In the experiments, animals were monitored closely and upon showing discomfort or weight loss (15%), they were sacrificed, and their brains were harvested euthanized by cervical dislocation. Brains were then removed and either fixed in formalin or frozen in isopentane on dry ice. Formalin-fixed samples were embedded in paraffin, sectioned and histological or immunohistochemical staining were performed with HE (Fluka, 03971), Vimentin (Dako, M0725), Ki67 (Dako, M7240). Ki67 stainings were quantified with ImmunoRatio programme (jvsmicroscope.uta.fi/immunoratio/).
Statistical analysis
The significance level of differences between the mean values of two groups of data was evaluated as follows. For in vivo bioluminescence follow-up, in vitro BBB diffusion model, cell viability comparison and caspase 3/7 comparison a two-tailed unpaired Student’s t-test assuming unequal variances among the sample means was used. Ki67 comparison was determined with a two-tailed Mann–Whitney test. Survival was determined by Gehan–Breslow–Wilcoxon test. Differences with probability value P < 0.05 were described as statistically significant.
Additional information is provided in Supplementary methods.
Results
Development of BBB permeable small molecule reactivators of PP2A
We sought to improve the potency and oral bioavailability of RTC-5 and RTC-30 (Kastrinsky et al., 2015), earlier members of the SMAP series derived from tricyclics, by constraint of the linear spacer moiety between the tricyclic and sulfonamide, resulting in structures exemplified by DBK-1154 and NZ-8-061 (Fig. 1A and Supplementary Fig. 1). However, it was unclear whether the polarity introduced to tricyclics by the sulfonamide and hydroxyl moieties (Fig. 1A) would compromise BBB permeability, and eventually CNS availability, of the SMAPs as compared with tricyclics. We, therefore, started by investigating the in vitro BBB passage of NZ-8-061 (a.k.a DT-061) that has been widely used in cancers outside the CNS (Sangodkar et al., 2017; Kauko et al., 2018; McClinch et al., 2018). Quantified by HPLC-MS/MS, NZ-8-061 was found to cross the artificial BBB (Le Joncour et al., 2019b), consisting of murine brain microcapillary endothelial cells and astrocytes (Fig. 1B and C). Furthermore, 24-h pre-treatment with NZ-8-061 did not modify diffusion of a low-molecular weight fluorescent probe, the sodium fluorescein (Na-Fl), indicating that passage of NZ-8-061 was not a bystander effect due to its effects on BBB model permeability (Fig. 1D).
Figure 1.

BBB penetrance and mouse in vivo pharmacokinetics of NZ-8-061. (A) An overview of development of SMAPs, NZ-8-061 and DBK-1154. The synthesis is described in detail in Supplementary Fig. 1. (B) Schematic presentation of in vitro BBB model, which consists of murine endothelial cells and astrocytes. (C) NZ-8-061 passage through the in vitro BBB model after addition of 15 µM dosage on the upper chamber at indicated timepoints. Data shown are means from two replicates ± SD. (D) Sodium-fluorescein diffusion through the in vitro BBB after 24 hour of pre-treatment with 15 µM NZ-8-061 on the upper chamber. Fluorescence signal of sodium fluorescein was measured from lower chamber after 15 min. Data shown are means from two replicates ± SD. (E) Mouse in vivo pharmacokinetic parameters (T½ h, Tmax h, Cmax ng/ml, area under curve hr.ng/ml, CL ml/h/kg, %F and blood–brain ratio) after 1 or 100 mg/kg dosage via p.o. or i.v. of NZ-8-061.
To study brain penetration of NZ-8-061 in vivo, we performed a pharmacokinetic study by either administering 1 mg/kg i.v. or a bolus oral dose of 100 mg/kg. The in vivo pharmacokinetic parameters are shown in Fig. 1E. NZ-8-061 shows 100% oral bioavailabilty based on dose-adjusted fraction absorbed (%F) and moderate clearance as judged from half-life (T1/2) in plasma with T1/2 of 3 h after I.V. dose. Peak plasma concentration after oral dose is around 14 µM and combined with moderate clearance and high area under curve, shows significant, and sustained systemic exposure. Importantly, based on HPLC-MS/MS analysis from the whole-brain homogenate, NZ-8-061 partitions into brain with a brain/plasma ratio of 1:1 at 6 h post-drug administration (Fig. 1E).
Together these results identify NZ-8-061 as an orally bioavailable, and BBB-permeable drug candidate for GB treatment.
NZ-8-061 potently inhibits the viability of GB cells with heterogenous genetic background
Frequency of genetic mutations or deletions in any of the genes coding for core PP2A complex components in GB clinical isolates is negligible (Kaur et al., 2016). However, we found that all studied GB cell lines expressed higher levels of PP2A inhibitor proteins (PAIPs); PME-1, CIP2A, SET and ARPP-19 (Kauko and Westermarck, 2018), as compared with non-tumour fibroblasts (Fig. 2A). These results provide a non-genetic candidate mechanism for PP2A deregulation in GB cells.
Figure 2.

NZ-8-061 potently inhibits viability of molecularly heterogenous GB cells in vitro. (A) Western blot of endogenous PP2A inhibitor proteins (CIP2A, SET, PME-1 and ARPP-19) and (B) GB stem cell markers (Sox2 and Nestin) in indicated GB cell lines. BT3-CD133+ (BT3 patient-derived cells sorted for CD133) cells were used as a control for GB stem cell markers, and human fibroblasts were used as a negative control cell line in both. Please see Supplementary material of full uncut western blots. (C) The dose-dependent effect of NZ-8-061 on the viability of indicated GB cell lines after 72 h. Data shown are means from six replicates ± SD. IC50 values were calculated with GraphPad Prism 8. (D, E) Colony growth inhibition after NZ-8-061 treatment in GB cell lines with indicated concentrations. IC50 values for each cell line are shown. Data shown are means from four replicates ± SD. IC50 values were calculated with GraphPad Prism 8. (F) Expression levels endogenous PP2A inhibitor proteins (CIP2A, SET, PME-1 and ARPP-19) in patient-derived primary glioma cells. Please see Supplementary material for full uncut western blots. (G) Cell viability inhibition after NZ-8-061 (5 µM) treatment in patient-derived primary glioma cells. Data shown are means from six replicates ± SD.
Genomic heterogeneity and stemness characteristics are known to affect GB cell therapy responses. Genomic characteristics of the U87MG, A172, U118 and T98G cell lines were examined using the Cancer Cell Line Encyclopedia (CCLE) (Barretina et al., 2012) database, whereas for the E98 cells published data were used (Claes et al., 2008; Navis et al., 2015). Collectively, the cell lines displayed a variety of known genomic alterations in GB (Supplementary Fig. 2A). However, the only common genomic change across all cell lines was copy number loss of CDKN2A (Supplementary Fig. 2A). Related to stemness properties, most of the established cell lines, except for E98, did not express glioma stem cell markers SOX2 or Nestin (Fig. 2B).
We previously demonstrated notable kinase inhibitor resistance of T98G cells across different kinase families (Kaur et al., 2016). To further examine kinase inhibitor responses of the used cell lines, we extracted the IC50 values for selected inhibitors from Genomics of Drug Sensitivity in Cancer database (https://www.cancerrxgene.org/). No information for E98 cell line was available. Consistently with their heterogenous mutation profiles (Supplementary Fig. 2A), each of the cell line has very variable sensitivity against different kinase inhibitors (Supplementary Fig. 2B). We experimentally validated Gefitinib resistance of three of the cell lines (Supplementary Fig. 2C).
These genetically diverse cell lines, displaying differential drug sensitivities, were then screened for their response to PP2A reactivation by NZ-8-061 in a cell viability assay. NZ-8-061 did induce a dose-dependent reduction in cell viability in all of the tested cell lines (Fig. 2C). Opposite to their kinase inhibitor responses, neither drastic differences nor correlations of response with their genetic background, were found among the tested cell lines based on their IC50s. As a pharmacologic control, U87MG, U118 or A172 cells were also treated with increasing doses of TRC-766, a structurally similar but biologically inactive derivate of SMAPs (Supplementary Fig. 3A) (Sangodkar et al., 2017; McClinch et al., 2018). While TRC-766 still binds PP2A, it is unable to reactivate PP2A even at a concentration of 20 µM in vitro (Sangodkar et al., 2017). Notably, TRC-766 did not affect the viability of GB cells at concentrations up to 40 µM (Supplementary Fig. 3B). This strongly supports a dependence on PP2A reactivation for NZ-8-061-elicited killing of GB cells. Using a colony growth assay, NZ-8-061 treatment resulted in dose-dependent growth inhibition in all of the cell lines tested and in approximately the same concentration range as seen in the cell viability assay (Fig. 2D and E). Of note, micromolar dosing of SMAP is consistent with micromolar concentration of its molecular target PP2A in cancer cells, and required as serum binding of these compounds decreases their apparent potency in standard cell culture medium.
To extend these observations, we tested the therapeutic potential of NZ-8-061 in a series of thoroughly validated patient-derived primary glioma cells (Xie et al., 2015). Notably, chosen cells represented all three molecular subtypes of GB according to Verhaak and colleagues (Wang et al., 2017) (Supplementary Fig. 4A and B). Each of the patient-derived glioma cell line, cultured in serum-free NSC media for the maintenance of stemness properties, expressed Nestin and Sox2, as well as PAIPs (Supplementary Fig. 4C and Fig. 2F). Furthermore, they have a diverse genetic background (Supplementary Fig. 5A and B), and kinase inhibitor responses (Supplementary Fig. 6). All nine tested primary glioma cell lines displayed near to complete suppression of cell viability when treated with 5 µM NZ-8-061 (Fig. 2G). The lower working concentration of NZ-8-061 as compared with standard GB cell line cultures could be explained by the lack serum in the NSC media, thus increasing the apparent potency of NZ-8-061.
Together these data demonstrate that SMAPs, as exemplified by NZ-8-061, have wide-spectrum therapeutic effect in vitro across human GB cells; regardless of their genetic background, disease subtype or stemness properties.
Preclinical activity of NZ-8-061 in an infiltrative intracranial GB model
To analyse in vivo therapeutic potential of oral dosing of NZ-8-061, we used an intracranial GB tumour model with luciferase-expressing/bioluminescent E98 cells (Claes et al., 2008). Among the cell lines, E98 cells were selected as a model due to their aggressivity; i.e. stem-like properties (Fig. 2B), and infiltrative growth pattern in mouse brain recapitulating the human malignant glioma histology (Claes et al., 2008) (Fig. 3A). E98 was thought to be a representative cell line for assessing SMAP response in vivo also due to their average to low NZ-8-061 responsiveness in vitro (Fig. 2C and E). Prior to the treatment, mice were randomized into two groups based on the tumour bioluminescence signal and using a protocol for optimized design and analysis of preclinical intervention studies in vivo (Laajala et al., 2016). NZ-8-061 was thereafter orally dosed twice a day at 30 mg/kg. Based on pharmacokinetics and tissue distribution (Fig. 1E), the brain exposure of NZ-8-061 was estimated to transiently reach 10 µM, which is roughly comparable to the IC50 for colony formation assay with NZ-8-061 in vitro (Fig. 2E).
Figure 3.

Therapeutic potential of oral dosing of NZ-8-061 as monotherapy in an infiltrative intracranial GB mouse model. (A) Example picture of infiltrative growth of intracranial human E98-FM-Cherry cell line xenograft. The mouse brain tissue was stained with human-specific vimentin antibody. (B) Bioluminescence follow-up of the intracranial human E98-FM-Cherry cell line xenograft growth during vehicle or 30 mg/kg NZ-8-061 treatment. When tumours were visible with bioluminescence, mice were randomized for either vehicle or NZ-8-061 groups. Data shown are means from eight mice ± SEM, P-values by Student’s t-test. (C) Representative images of Ki-67 staining from four vehicle and four NZ-8-061-treated endpoint tumours from B. (D) Quantification of Ki-67 positivity from C. Data shown are means of % of Ki-67 positive tumour cells from 7 to 8 tumours/group ± SD, *P-value by Mann–Whitney test.
Intracranial tumour growth was followed by bioluminescence measurements every third day. Notably, NZ-8-061 induced tumour growth stasis at day 12 and displayed a significant reduction in tumour size at later timepoints (Fig. 3B). The therapeutic response was validated by significant inhibition in tumour cell Ki67 expression in the endpoint tumours when comparing treated and non-treated groups (Fig. 3C and D). Importantly, analysis of contralateral brain hemisphere without injected tumour cells from NZ-8-061 treated mice, did not show after 21 days any apparent signs of tissue toxicity based on H&E staining, or analysis of apoptosis activity by TUNEL staining (Supplementary Fig. 7). Consistent with previously published data (Sangodkar et al., 2017; Kauko et al., 2018), NZ-8-061 treated mice did not also show any systemic signs of toxicity, or weight loss (Supplementary Fig. 8A).
DBK-1154, with higher degree of brain/blood distribution, and more efficient in vitro activity, increases survival of mice bearing orthotopic GB tumours
Despite its significant in vivo efficacy in reducing intracranial tumour growth with roughly IC50 dosing (Fig. 3B–D), NZ-8-061 monotherapy failed to improve the mouse survival (Supplementary Fig. 8B). We, therefore, next tested the BBB permeability and GB cell-killing properties of DBK-1154. DBK-1154, with a dibenzoazepine tricyclic, has a hydrocarbon bridge versus an oxygen bridge in NZ-8-061 (Fig. 1A), making DBK-1154 somewhat more lipophilic. The calculated LogP (log octanol-water partition coefficient), a measure of lipophilicity, is higher for DBK-1154 (cLogP 7.0) than NZ-8-061 (cLogP 6.6). In addition, total polar surface area is higher for NZ-8-061 versus DBK-1154 (88 vs 79 A2). Lower total polar surface area and higher cLogP generally correlate with higher CNS distribution (Kelder et al., 1999).
In vivo pharmacokinetic parameters for DBK-1154 in mouse are shown in Fig. 4A and in Supplementary Fig. 8C. DBK-1154 is orally bioavailable; however, it showed significantly lower systemic plasma exposure compared with NZ-8-061. Importantly, in addition to in vitro BBB permeability (Supplementary Fig. 9A and B), the in vivo evaluation of distribution of DBK-1154 into the CNS showed a 2.3-fold higher concentration in brain tissue versus plasma (Fig. 4A).
Figure 4.

Oral dosing of DBK-1154 increases survival of mouse with intracranial infiltrative GB xenograft. (A) Mouse in vivo pharmacokinetic parameters (T½ h, Tmax h, Cmax ng/ml, area under curve h.ng/ml, CL ml/h/kg, %F and blood–brain ratio) after 100 mg/kg dosage via p.o. of DBK-1154. (B) Time-dependent Caspase-3/7 activation in E98 cells, after either DT-061 or DBK-1154 treatment (13 µM). Data shown are means from six replicates ± SD. (C) Time-dependent inhibition of cell viability in E98 cells, after either DT-061 or DBK-1154 treatment (13 µM). Data shown are means from six replicates ± SD. (D) Colony growth reduction after DBK-1154 treatment in GB cell lines in indicated concentrations. IC50 comparison between DBK-1154 and NZ-8-061 is shown in the insert. Data shown are means from four replicates ± SD. (E) Cell viability inhibition after DBK-1154 (5 µM) treatment in patient-derived primary glioma cells cultured in serum-free NSC medium. Data shown are means from six replicates ± SD. (F) Bioluminescence comparison on the day 14 during the orthotopic E98 in vivo model between vehicle or 100 mg/kg DBK-1154 treatment. Data shown are means from 8 vehicle and 10 DBK-1154-treated mice ± SEM, *P-value by Student’s t-test. (G) Survival of mice with orthotopic E98 xenografts after vehicle or 100 mg/kg DBK-1154 treatment, *P < 0.05 by Gehan–Breslow–Wilcoxon test. Mice were randomized to two groups of 10 mice each based on bioluminescence signal before starting the treatment. Median survival was increased with DBK-1154 treatment from 15 to 26 days.
Based on the IC50-value, higher potential of DBK-1154 was seen in addition to cell viability assay (Supplementary Fig. 9C), also in a long-term colony growth assay in which the IC50 for DBK-1154 was almost 2-fold lower than that of NZ-8-061 in both cell lines (Fig. 4D, insert). In addition, DBK-1154 was qualitatively different from NZ-8-061 in terms of its apoptosis-inducing potential. With single 13 µM dosing, only DBK-1154 induced Caspase-3/7 cleavage at 48 h (Fig. 4B). This was reflected in a significant time-dependent difference in inhibition of cell viability also starting at 48 h (Fig. 4C). The higher potency of DBK-1154, when compared with NZ-8-061, was also seen across patient-derived primary glioma cells in which DBK-1154 induced complete inhibition of cell viability (Fig. 4E). Importantly, using the same cell culture conditions, most of the tested kinase inhibitors did not reach even 50% reduction in cell viability with up to 10 µM concentration in all five of the tested patient-derived primary glioma cells (Supplementary Fig. 6).
Next, we proceeded to test DBK-1154 in the same intracranial E98 model as with NZ-8-061. To compensate for the lower oral bioavailability of DBK-1154, it was dosed at 100 mg/kg, twice daily, using the same homogeneous formulation, with expected transient brain exposure of about 8 µM twice a day. This level of CNS exposure is roughly comparable with the IC50 for colony formation for DBK-1154 in vitro (Fig. 4D). DBK-1154 therapy caused tumour growth reduction after day 5 (Supplementary Fig. 9D), and reaching statistically significant difference at day 14 (Fig. 4F). Furthermore, while NZ-8-061 monotherapy did not increase the survival of the intracranial tumour-bearing mice (Supplementary Fig. 8B), the higher in vitro efficiency and in vivo brain penetrance of DBK-1154 translated to a significant, almost 2-fold longer overall mouse survival. The median survival was 15 days for the control group, and 26 days for the DBK-1154 treated group (P = 0.012) (Fig. 4G). Together these data indicate that of the studied SMAPs, DBK-1154 has a greater potential as a GB therapy candidate molecule.
Considering the potential future development of DBK-1154 derivatives for GB therapy, DBK-1154 was evaluated in an acute rat pilot (non-good laboratory practice) toxicology study at doses up to 800 mg/kg daily. Consistently with previous SMAP in vivo studies (Sangodkar et al., 2017; Kauko et al., 2018; McClinch et al., 2018), no significant body weight loss, deaths or adverse behavioural or neurological effects were observed. The major morphologic finding was hepatocellular hypertrophy (panlobular) in all test article-treated groups, with increased severity as the dose level increased. This observation was likely related to compound specific pregnane X receptor agonist activity, and was considered an adaptive rather than toxic effect. These findings indicate for a clear therapeutic window between normal and cancer cells in vivo.
Discussion
Collectively, these results provide proof-of-principle evidence for preclinical in vivo efficacy, and acceptable safety profile of SMAPs as novel candidate class of BBB penetrable, tumour suppressor reactivation therapeutics. Notably, the efficacy of SMAPs did not depend on the subtype of GB, or the genomic alterations, in either established GB cell lines or in patient-derived primary glioma cells. Moreover, SMAPs were found to be superior to a range of kinase inhibitors in their capacity to kill patient-derived primary glioma cells. These results provide the first indications that PP2A reactivation might be able to challenge the current paradigm in GB therapies which has been strongly focused on targeting specific genetically altered cancer drivers with highly specific inhibitors (Verhaak et al., 2010; Barretina et al., 2012; Brennan et al., 2013). PP2A is known to simultaneously target a number of cancer driver pathways and pro-apoptotic mechanisms (Perrotti and Neviani, 2013; Kauko and Westermarck, 2018). We envision that these wide-spectrum effects may explain the very robust preclinical survival effects across kinase inhibitor-resistant cell lines harbouring heterogenous genetic drivers. The clear limitation of the study was that the in vivo therapeutic efficacy studies with SMAPs were limited to only one cell model; highly invasive E98 cells (Fig. 3A) with stemness characteristics (Fig. 2B). Therefore, therapeutic effects of SMAPs need to be tested in the future by using cell models representing all major GB classes.
Another serious obstacle for development of GB therapies is inadequate exposure of most developed molecules in the CNS due to the BBB (Harder et al., 2018). Here, we show that both SMAPs have retained the BBB penetration properties of tricyclics from which they were derived. This was not obvious as SMAPs are significantly modified from the original tricyclics by replacement of a tertiary amine salt with an aryl sulfonamide and addition of a hydroxyl moiety. The observation that out of the studied SMAPs, DBK-1154 preferably partitions into the brain, is consistent with its lower total polar surface area and higher cLopP. Consequently, this better brain partitioning can partly explain the significant almost 2-fold prolongation of survival of mice with DBK-1154 versus NZ-8-061. In addition to GB, poor small molecule BBB penetration is a serious problem also for the therapy of brain metastasis from many non-CNS cancer types. As these are more common than primary brain tumours such as GB, the presented proof-of-principle data for usefulness of SMAPs as CNS therapeutics might be relevant for much larger patient population than only GB patients.
In addition to their different in vivo brain penetrance properties, NZ-8-061 seems to have cytostatic effects at the doses tested, whereas DBK-1154 most probably induces both, decreased proliferation and cell death. On the other hand, a SMAP derivative DBK-766 defective in PP2A reactivation (Sangodkar et al., 2017; McClinch et al., 2018), failed to suppress GB cell viability as expected. The intracellular pathways involved in the apoptosis induction by DBK-1154 in vitro were not addressed in this report, but clearly remains as an important future question to be addressed. As such, the results provide a clear indication that chemical structure of DBK-1154 could serve as a scaffold for further development of even more potent SMAPs for GB therapy. Following this rationale, a medicinal chemistry programme identifying DBK-1154 analogues with improved metabolic stability and oral bioavailability is currently in progress.
PP2A activity is known to modulate kinase inhibitor responses in haematological and solid cancers (Neviani et al., 2013; Kaur et al., 2016; Kauko et al., 2018). In vivo, combination studies with SMAPs demonstrated significant tumour regression in KRAS-mutant lung cancer xenograft model with MEK inhibitors (Kauko et al., 2018). Therefore, the results reported here not only serve as a proof-of-principle for the feasibility of SMAPs as novel GB therapeutics, but also pave the way for future combination studies with kinase inhibitors, and potentially also other type of therapies in brain cancers. Suppression of PP2A activity is also one of the pathogenic mechanisms underlying several neurodegenerative diseases, including Alzheimer’s (Shentu et al., 2018) and Parkinson’s diseases; thus, our data demonstrating BBB penetration and in vivo therapeutic effects of SMAPs in the intracranial model described here may also prompt the use of SMAPs in other CNS pathologies.
Data availability statement
Data are available from the corresponding author J.W. (jukka.westermarck@bioscience.fi) upon request.
Supplementary Material
Acknowledgements
Authors want to acknowledge Finnish Brain Tumor Research Association (FiBTRA) members for constructive comments to the work. Taina Kalevo-Mattila is acknowledged for excellent technical support. We thank Turku Center for Disease Modelling (TCDM) and Made Consulting for expert help with mouse experiments. The personnel of Bioanalytic laboratory at Institute of Biomedicine, University of Turku is thanked for their valuable help with HPLC-MS/MS analysis.
Funding
Project was funded by Jane and Aatos Erkko foundation, Sigrid Juselius Foundation and Finnish Cancer Foundation.
Competing interests
The Icahn School of Medicine at Mount Sinai has filed patents covering DBK-1154 and NZ-8-061 on behalf of M.O. and D.B.K. (International Application Numbers: PCT/US15/19770, PCT/US15/19764; and US Patent: US 9,540,358 B2). Other authors declare no competing interests.
Glossary
Abbreviations
- BBB =
blood–brain barrier
- CCLE =
Cancer Cell Line Encyclopedia
- CNS = c
entral nervous system
- GB =
glioblastoma
- LogP =
log octanol-water partition coefficient
- Na-Fl =
sodium fluorescein
- PAIPs =
endogenous inhibitor proteins of PP2A
- PP2A =
protein phosphatase 2A
- SMAPs =
small molecule activators of PP2A
- T1/2 =
half-life
References
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012; 483: 603–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The somatic genomic landscape of glioblastoma. Cell 2013; 155: 462–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claes A, Schuuring J, Boots-Sprenger S, Hendriks-Cornelissen S, Dekkers M, van der Kogel AJ, et al. Phenotypic and genotypic characterization of orthotopic human glioma models and its relevance for the study of anti-glioma therapy. Brain Pathol 2008; 18: 423–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalton SO, Johansen C, Poulsen AH, Norgaard M, Sorensen HT, McLaughlin JK, et al. Cancer risk among users of neuroleptic medication: a population-based cohort study. Br J Cancer 2006; 95: 934–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn GP, Rinne ML, Wykosky J, Genovese G, Quayle SN, Dunn IF, et al. Emerging insights into the molecular and cellular basis of glioblastoma. Genes Dev 2012; 26: 756–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman C, Bagga M, Kaur A, Westermarck J, Abankwa D.. ColonyArea: an ImageJ plugin to automatically quantify colony formation in clonogenic assays. PLoS One 2014; 9: e92444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harder BG, Blomquist MR, Wang J, Kim AJ, Woodworth GF, Winkles JA, et al. Developments in blood-brain barrier penetrance and drug repurposing for improved treatment of glioblastoma. Front Oncol 2018; 8: 462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahchan NS, Dudley JT, Mazur PK, Flores N, Yang D, Palmerton A, et al. A drug repositioning approach identifies tricyclic antidepressants as inhibitors of small cell lung cancer and other neuroendocrine tumors. Cancer Discov 2013; 3: 1364–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang T, Zhao B, Li X, Wan J.. ARPP-19 promotes proliferation and metastasis of human glioma. Neuroreport 2016; 27: 960–6. [DOI] [PubMed] [Google Scholar]
- Kastrinsky DB, Sangodkar J, Zaware N, Izadmehr S, Dhawan NS, Narla G, et al. Reengineered tricyclic anti-cancer agents. Bioorg Med Chem 2015; 23: 6528–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauko O, O’Connor CM, Kulesskiy E, Sangodkar J, Aakula A, Izadmehr S, et al. PP2A inhibition is a druggable MEK inhibitor resistance mechanism in KRAS-mutant lung cancer cells. Sci Transl Med 2018; 10: eaaq1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauko O, Westermarck J.. Non-genomic mechanisms of protein phosphatase 2A (PP2A) regulation in cancer. Int J Biochem Cell Biol 2018; 96: 157–64. [DOI] [PubMed] [Google Scholar]
- Kaur A, Denisova OV, Qiao X, Jumppanen M, Peuhu E, Ahmed SU, et al. PP2A inhibitor PME-1 drives kinase inhibitor resistance in glioma cells. Cancer Res 2016; 76: 7001–11. [DOI] [PubMed] [Google Scholar]
- Kelder J, Grootenhuis PD, Bayada DM, Delbressine LP, Ploemen JP.. Polar molecular surface as a dominating determinant for oral absorption and brain penetration of drugs. Pharm Res 1999; 16: 1514–9. [DOI] [PubMed] [Google Scholar]
- Laajala TD, Jumppanen M, Huhtaniemi R, Fey V, Kaur A, Knuuttila M, et al. Optimized design and analysis of preclinical intervention studies in vivo. Sci Rep 2016; 6: 30723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Joncour V, Filppu P, Hyvonen M, Holopainen M, Turunen SP, Sihto H, et al. Vulnerability of invasive glioblastoma cells to lysosomal membrane destabilization. EMBO Mol Med 2019a; 11. pii: e9034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Joncour V, Karaman S, Laakkonen PM.. Predicting in vivo payloads delivery using a blood-brain tumor-barrier in a dish. J Vis Exp 2019b; 146. [DOI] [PubMed] [Google Scholar]
- McClinch K, Avelar RA, Callejas D, Izadmehr S, Wiredja D, Perl A, et al. Small-molecule activators of protein phosphatase 2A for the treatment of castration-resistant prostate cancer. Cancer Res 2018; 78: 2065–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mooney J, Bernstock JD, Ilyas A, Ibrahim A, Yamashita D, Markert JM, et al. Current approaches and challenges in the molecular therapeutic targeting of glioblastoma. World Neurosurg 2019; 129: 90–100. [DOI] [PubMed] [Google Scholar]
- Murren J, Durivage H, Buzaid A, Reiss M, Flynn S, Carter D, et al. Trifluoperazine as a modulator of multidrug resistance in refractory breast cancer. Cancer Chemother Pharmacol 1996; 1: 65–70. [DOI] [PubMed] [Google Scholar]
- Narla G, Sangodkar J, Ryder CB.. The impact of phosphatases on proliferative and survival signaling in cancer. Cell Mol Life Sci 2018; 75: 2695–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navis AC, van Lith SA, van Duijnhoven SM, de Pooter M, Yetkin-Arik B, Wesseling P, et al. Identification of a novel MET mutation in high-grade glioma resulting in an auto-active intracellular protein. Acta Neuropathol 2015; 130: 131–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neviani P, Harb JG, Oaks JJ, Santhanam R, Walker CJ, Ellis JJ, et al. PP2A-activating drugs selectively eradicate TKI-resistant chronic myeloid leukemic stem cells. J Clin Invest 2013; 123: 4144–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrotti D, Neviani P.. Protein phosphatase 2A: a target for anticancer therapy. Lancet Oncol 2013; 14: e229–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin S, Li J, Si Y, He Z, Zhang T, Wang D, et al. Cucurbitacin B induces inhibitory effects via CIP2A/PP2A/Akt pathway in glioblastoma multiforme. Mol Carcinog 2018; 57: 687–99. [DOI] [PubMed] [Google Scholar]
- Sachlos E, Risueno RM, Laronde S, Shapovalova Z, Lee JH, Russell J, et al. Identification of drugs including a dopamine receptor antagonist that selectively target cancer stem cells. Cell 2012; 149: 1284–97. [DOI] [PubMed] [Google Scholar]
- Sangodkar J, Dhawan NS, Melville H, Singh VJ, Yuan E, Rana H, et al. Targeting the FOXO1/KLF6 axis regulates EGFR signaling and treatment response. J Clin Invest 2012; 122: 2637–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangodkar J, Perl A, Tohme R, Kiselar J, Kastrinsky DB, Zaware N, et al. Activation of tumor suppressor protein PP2A inhibits KRAS-driven tumor growth. J Clin Invest 2017; 127: 2081–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shentu YP, Huo Y, Feng XL, Gilbert J, Zhang Q, Liuyang ZY, et al. CIP2A causes Tau/APP phosphorylation, synaptopathy, and memory deficits in Alzheimer's disease. Cell Rep 2018; 24: 713–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomiyama A, Ichimura K.. Signal transduction pathways and resistance to targeted therapies in glioma. Semin Cancer Biol 2019; 58: 118–29. [DOI] [PubMed] [Google Scholar]
- Tomiyama A, Kobayashi T, Mori K, Ichimura K.. Protein phosphatases-A touchy enemy in the battle against glioblastomas: a review. Cancers (Basel) 2019; 11: 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Heuvel C, Navis AC, de Bitter T, Amiri H, Verrijp K, Heerschap A, et al. Selective MET kinase inhibition in MET-dependent glioma models alters gene expression and induces tumor plasticity. Mol Cancer Res 2017; 15: 1587–97. [DOI] [PubMed] [Google Scholar]
- Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010; 17: 98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Hu B, Hu X, Kim H, Squatrito M, Scarpace L, et al. Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell 2017; 32: 42–56.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermarck J. Targeted therapies don't work for a reason; the neglected tumor suppressor phosphatase PP2A strikes back. FEBS J 2018; 285: 4139–45. [DOI] [PubMed] [Google Scholar]
- Xie Y, Bergstrom T, Jiang Y, Johansson P, Marinescu VD, Lindberg N, et al. The human glioblastoma cell culture resource: validated cell models representing all molecular subtypes. eBioMedicine 2015; 2: 1351–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zingone A, Brown D, Bowman ED, Vidal O, Sage J, Neal J, et al. Relationship between anti-depressant use and lung cancer survival. Cancer Treat Res Commun 2017; 10: 33–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available from the corresponding author J.W. (jukka.westermarck@bioscience.fi) upon request.