

Abstract
Background
Human relaxin‐2 is a peptide hormone capable of pleiotropic effects in several organ systems. Its recombinant formulation (serelaxin) has been demonstrated to reduce infarct size and prevent excessive scar formation in animal models of cardiac ischemia‐reperfusion injury. B7‐33, a synthetically designed peptide analogous to B‐chain of relaxin‐2, invokes signaling at relaxin family peptide receptor 1 (cognate receptor for relaxin‐2) by preferentially phosphorylating the mitogen‐activated protein kinase extracellular signal‐regulated kinase 1/2. We sought to investigate the effects of B7‐33 treatment post ischemia‐reperfusion injury in mice.
Methods and Results
Adult male CD1 mice were subjected to ischemia‐reperfusion via ligation of left anterior descending artery for 30 minutes, followed by 24 hours or 7 days of reperfusion. Echocardiography was performed to assess cardiac function, and cardiac tissue was stained to determine infarct size at 24 hours. B7‐33 significantly reduced infarct size (21.99% versus 45.32%; P=0.02) and preserved fractional shortening (29% versus 23%; P=0.02) compared with vehicle. The difference in fractional shortening further increased at 7 days post myocardial infarction (29% versus 20% for B7‐33 and vehicle groups, respectively). In vitro, primary cardiomyocytes were isolated from adult hearts and subjected to simulated ischemia‐reperfusion injury (simulated ischemia reoxygenation). B7‐33 (50 and 100 nmol/L) improved cell survival and reduced the expression of GRP78 (glucose regulated protein), an endoplasmic reticulum stress marker. Subsequently, B7‐33 (100 nmol/L) reduced tunicamycin (2.5 μg/mL) induced upregulation of GRP78 in an extracellular signal‐regulated kinase 1/2–dependent manner.
Conclusions
B7‐33 confers acute cardioprotection and limits myocardial infarction–related adverse remodeling in mice by attenuating cardiomyocyte death and endoplasmic reticulum stress as well as preserving cardiac function.
Keywords: adverse cardiac remodeling, echocardiography, endoplasmic reticulum stress, myocardial infarction, relaxin
Subject Categories: Contractile function, Fibrosis, Ischemia, Myocardial Infarction, Remodeling
Nonstandard Abbreviations and Acronyms
- ASC
apoptosis‐associated speck‐like protein containing a caspase recruitment domain
- FS
fractional shortening
- LVID
left ventricular internal diameter
- RXFP1
relaxin family peptide receptor 1
- SIRO
simulated ischemia reoxygenation
- TUN
tunicamycin
Clinical Perspective
What Is New?
The protective effects of B7‐33, a functionally selective relaxin family peptide receptor 1 agonist, against ischemia‐reperfusion injury are demonstrated in the heart post infarction.
A novel association between the mitogen‐activated protein kinase signaling elicited by B7‐33 and the unfolded protein response in the postinfarcted heart is identified.
What Are the Clinical Implications?
Despite the benefits of timely reperfusion, there is a paucity of therapeutics that are directly indicated for thwarting adverse remodeling following ischemia‐reperfusion injury to the heart.
The hormone relaxin has been identified to play a role in mitigating the complex pathophysiological characteristics of the postinfarcted heart, and the signaling diversity elicited by alternative ligands at the relaxin receptor 1 (relaxin family peptide receptor 1) represents the potential of unexplored therapeutic strategies.
In this article, we outline our findings involving B7‐33, a relaxin family peptide receptor 1 ligand, in conferring cardioprotection post myocardial infarction.
Introduction
Myocardial infarction (MI) is characterized by cessation of blood supply to the cardiac tissue, leading to damage and death of cardiomyocytes within the affected region. Despite the significant reduction in in‐hospital mortality because of advances in revascularization techniques,1 reintroduction of oxygen initiates a cascade of maladaptive signaling within the ischemic tissue. The resultant pathologic injury, termed ischemia‐reperfusion (IR) injury, facilitates proinflammatory changes within the tissue during early stages of infarct healing.2
The pleiotropic hormone relaxin (human relaxin 2) has been shown to confer cardioprotection in several animal models of heart disease.2 Relaxin is the cognate ligand for relaxin family peptide receptor 1 (RXFP1), a G‐protein–coupled receptor. In most cell types, it generates a biphasic cAMP signal, along with activation of the extracellular signal‐regulated kinase (Erk) 1/2 pathway.3 Interestingly, RXFP1 has been shown to exhibit ligand‐specific biased signaling; the allosteric agonist ML290 biases the receptor toward a cGMP (as opposed to the canonical cAMP) response in cardiac fibroblasts.4 Recently, a synthetic variant of the B‐chain of relaxin (B7‐33) has been shown to cause RXFP1‐mediated Erk 1/2 activation, without generating cAMP in fibroblasts.5 This signaling paradigm has potential benefits because cAMP signaling during the progression of heart failure may cause deleterious consequences. To this end, β‐blocker therapy has been shown to thwart ventricular hypertrophy, dysfunction, and fibrosis through inhibition of β2‐adrenergic receptor–induced cAMP activity.6 Furthermore, the activation of Erk 1/2 triggered by several protective strategies, including ischemic preconditioning, has been shown to reduce infarct size and prevent cell death during the reperfusion period.7 Among several forms of injury mediated by ischemia, endoplasmic reticulum (ER) stress contributes to the pathophysiological characteristics because of cell death pathways initiated by unfolded protein response (UPR). Mitogen‐activated protein kinase signaling, including that transduced via Erk 1/2 activation, mitigates the inflammatory response downstream of UPR in in vitro systems.8, 9 We sought to investigate the benefits of short‐ and long‐term B7‐33 administration in the context of MI, while focusing on the potential intersection between B7‐33–induced Erk 1/2 signaling and UPR.
Methods
In adherence to Transparency and Openness Promotion Guidelines, raw data supporting the findings of this study can be made available on reasonable request to corresponding author.
Animals
Adult male (aged 6–8 weeks) CD1 mice were purchased from Charles River, and were allowed to acclimate in a temperature‐controlled vivarium for up to a week before any intervention, with access to water and food. All experiments were performed with strict adherence to Guidelines for the Care and Use of Laboratory Animals, as updated by the National Institute of Health (eighth edition, 2011). All of our animal techniques are approved by Virginia Commonwealth University Institutional Animal Care and Use Committee.
Drugs and Chemicals
Triphenyltetrazolium chloride and tunicamycin were purchased from Sigma‐Aldrich. Trypan blue solution (0.4%) was obtained from Thermo Fisher Scientific. PD98059 (Erk 1/2 inhibitor) was purchased from Cell Signaling Technologies. Phthalo blue dye was purchased from Quantum Ink (Louisville, KY). B7‐33 was kindly provided by our collaborators from University of Melbourne (Victoria, Australia). Reagents used in myocyte perfusion and digestion buffers (NaCl, KCl, MgSO4, CaCl2, NaHCO3, 2,3‐butanedione monoxime, glucose, protease XIV from streptomyces griseus, and taurine) were purchased from Sigma‐Aldrich. Collagenase II was obtained from Worthington Biochemical Corporation (Lakewood, NJ).
Experimental Protocol
In Vivo
Myocardial IR protocol
Adult male CD1 mice were anesthetized with pentobarbital IP (70 mg/kg). Sedation was confirmed on lack of responsiveness to toe pinch. After intubation, thoracotomy was performed to visualize the left anterior descending coronary artery. A 7.0 silk ligature was tied around the artery, and a polyethylene tube (PE10) was used to occlude blood flow for the 30‐minute ischemic period. Animals were assigned to sham, vehicle, or treatment (B7‐33) groups, and the surgeon was blinded to the assignment throughout the entire procedure. Five minutes before the onset of reperfusion, vehicle (0.9% saline) (34 mice) or B7‐33 (38 mice) (0.25 mg/kg) IP was administered. Sample size was estimated on the basis of previous studies that followed a similar experimental design and evaluated comparable functional and molecular parameters. After 30 minutes, the polyethylene tube was removed, and the color change in ventricular tissue (from pale to pink) was recorded to ensure proper induction of reperfusion. When reperfusion was not successfully achieved, animals were excluded from the study after confirming the presence of significant wall thinning and global dilatation via echocardiography at 24 hours (6 mice total: 4 from B7‐33, and 2 from vehicle). In mice assigned to sham group (9 mice), thoracotomy was performed without induction of the left anterior descending ligature. Postoperatively, analgesia (buprenorphine SR LAB, 0.5 mg/kg; SC) and antibiotic (gentamicin, 0.7 mg/kg) were administered, in compliance with our animal surgery protocol. Mice were monitored for up to 7 days, and were given either daily doses of B7‐33 (twice a day, 0.25 mg/kg per dose, once every 12 hours) or saline until the time of euthanasia. The protocol for general in vivo method is outlined in Figure 1.
Figure 1. Schematic illustrating the experimental protocol for in vivo studies.
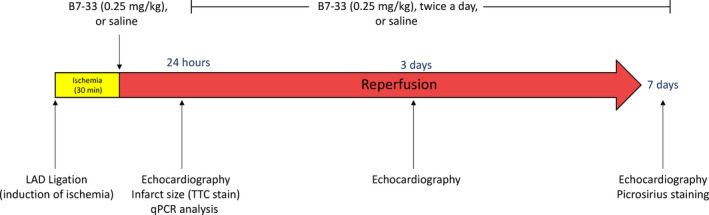
Left anterior descending coronary (LAD) artery in mice was occluded for 30 minutes, followed by either 24 hours or 7 days of reperfusion. Mice were treated at the onset of reperfusion with either B7‐33 or saline. In the longer‐term group of mice (7 days), mice received regular (twice a day) administration of B7‐33 or saline until time of euthanasia. qPCR indicates quantitative polymerase chain reaction; and TTC, triphenyltetrazolium chloride.
Echocardiography
Cardiac function was assessed via echocardiography before surgery, and at 24 hours, 3 days, and 7 days postoperatively using the Vevo2100 imaging system (VisualSonics Inc, Toronto, Canada). The general procedure for acquiring short‐axis images, estimating fractional shortening (FS), and obtaining left ventricular (LV) internal diameter (systolic and diastolic) was detailed previously.1 The epicardium and endocardium were traced, and the VevoStrain software was used to measure radial strain, radial strain rate (as a function of time), and Max dV/dT (maximum instantaneous rate of change in volume, during relaxation). Once the radial strain of the anterior apical, anterior mid, anterior basal, posterior apical, posterior mid, and posterior basal segments was assessed, the SD from average was calculated to estimate dyssynchrony of radial strain.
Infarct size assessment
After 24 hours of reperfusion, mice were sedated with a terminal dose of pentobarbital (100 mg/kg). The heart was subsequently excised along with the aortic root and cannulated for subsequent perfusion in Langendorff mode. Briefly, the heart was perfused with Krebs‐Henseleit buffer, followed by 10% triphenyltetrazolium chloride. Then, the ligature around coronary artery was retightened, and aorta was perfused with phthalo blue dye to delineate the nonrisk area. Hearts were then removed from the apparatus and frozen before slicing them into several apical and basal sections. After fixing the sections in 10% formaldehyde for 6 hours, the area of infarct size as a percentage of at‐risk area was calculated using ImageJ software (W.S. Rasband, ImageJ, US National Institutes of Health, Bethesda, MD, https://imagej.nih.gov/ij/index.html), as we described previously.1
Quantification of LV scar
After 7 days of reperfusion, in the long‐term vehicle or treatment groups, hearts were excised from anesthetized mice and stored in 10% formaldehyde for at least 48 hours. LV cross‐sections were then paraffin embedded, and sections were subsequently analyzed for estimation of scar size via picrosirius red staining, as we previously reported.10 Multiple sections from each sample were analyzed for infarct and peri‐infarct associated scar and expressed as a percentage of total LV area per section, and the outputs were averaged to estimate mean scar size per sample. Image analysis was performed using Image Pro Plus software (Media Cybernetics, Rockville, MD) by an investigator blinded to treatment allocation.
In Vitro
Cardiomyocyte and fibroblast isolation
Our protocol for isolating primary cardiomyocytes from adult mice, and performing simulated ischemia reoxygenation (SIRO) in isolated cells was followed as outlined previously.11 Hearts were isolated from anesthetized mice, followed by perfusion with Ca2+ free myocyte isolation buffer via Langendorff apparatus. Then, collagenase II and protease XIV were used to digest the extracellular matrix. The digested tissue was centrifuged at 300g for 2 minutes to collect the myocyte pellet. The supernatant was saved to culture fibroblasts. The myocyte pellet was allowed to reconstitute in calcium reintroduction buffers before plating in laminin (Thermo Fisher Scientific) coated dishes. Myocytes were subsequently incubated in myocyte medium (MEM‐NEAA and 10% fetal bovine serum, 1% penicillin/streptomycin). Cardiac fibroblasts were isolated from the supernatant, as described above. The pellet was then left to adhere for 2 hours at 37°C with 5% CO2 using DMEM/F‐12 with 10% fetal bovine serum and plated on 1% porcine type‐B gelatin (Sigma Aldrich) precoated 30‐mm dishes.12 Before experimentation, fibroblasts were cultured in DMEM/F‐12 with 10% fetal bovine serum for at least 2 passages while monitoring for morphological changes until reaching 70% of cellular confluency.
SIRO protocol
Myocyte medium was aspirated from freshly plated myocytes, and the cells were allowed to equilibrate in an ischemia buffer (consisting of NaCl, NaHCO3, NaH2PO4, CaCl2, MgCl2, sodium lactate, KCl, and deoxyglucose). Plated myocytes were placed in a 1% O2 hypoxia chamber to induce simulated ischemia for 40 minutes. After the ischemic period, cells were placed back in normoxia and reperfused with fresh myocyte media (with or without B7‐33) until sample collection/analysis. For cardiac fibroblasts, the protocol was modified to allow for 4 hours of hypoxia, followed by 12 hours of reperfusion with control or B7‐33 infused media. Fibroblast viability was assessed with the MTT cell proliferation assay kit (ab211091; Abcam, Cambridge, UK) at the end of the reperfusion period.
Western blotting
Western blotting to quantify protein expression was done as explained previously in our literature.1 Briefly, frozen tissue samples or live cells were incubated in a radioimmunoprecipitation assay buffer (Cell Signaling Technologies) infused with protease and phosphatase inhibitors. Lysates were ultrasonicated and centrifuged at 12 000g for 10 minutes at 4°C. Total protein was quantified via Bradford assay using the Quick Start Bradford Protein Assay (Bio‐Rad). Subsequently, 50 μg per sample was separated via SDS‐PAGE on 4% to 20% acrylamide gel, and then transferred onto nitrocellulose membranes. The membranes were blocked by incubation with 5% nonfat dry milk dissolved in Tris‐buffered saline. Primary antibodies were dissolved in 5% BSA in Tris‐buffered saline for overnight incubation to probe for phosphorylated (Thr202/Tyr204) and total Erk 1/2 (Cell Signaling Technologies), GRP78 (Cell Signaling Technologies), and ASC (apoptosis‐associated speck‐like protein containing a caspase recruitment domain; Sigma Aldrich).
Real‐time polymerase chain reaction
mRNA was extracted from frozen tissue samples using the Qiagen miRNeasy kit (Qiagen, Hilden, Germany), and the concentration was estimated via nanodrop analyzer (Thermo Fisher Scientific). Genomic DNA was digested, and reverse transcription was performed with iScript gDNA clear cDNA synthesis kit (Bio‐Rad). Real‐time polymerase chain reaction was performed using SSoAdvanced Universal SYBR Green Supermix (Bio‐Rad), with the following sequences for forward and reverse primers (β‐actin: CTAAGGCCAACCGTGAAAAG [forward] and ACCAGAGGCATACAGGGACA [reverse]; CCAAT/enhancer‐binding protein‐homologous protein (Chop): CCCAGGAAACGAAGAGGAAGAA [forward] and ATGTGCGTGTGACCTCTGTT [reverse]; Grp78: CTATTCCTGCGTCGGTGTGT [forward] and GCCCTGATCGTTGGCTATGA [reverse]; toll‐like receptor 4: TGGTTGCAGAAAATGCCAGG [forward] and ATTAGGAACTACCTCTATGCAGGG [reverse]; tissue inhibitor of metalloproteases [Timp] 1: CTCGGACCTGGTCATAAGGG [forward] and ACGCTGGTATAAGGTGGTCTC [reverse]; Timp2: CACGCTTAGCATCACCCAGA [forward] and GAGTGATCTTGCACTCACAGC [reverse]). Data were recorded and analyzed on Bio‐Rad CFX96 to quantify gene expression.
Statistical Analysis
Data for infarct size, LV fibrosis, quantitative polymerase chain reaction, Western blotting, echocardiography parameters, and cell survival experiments were assessed for normality via Shapiro‐Wilk normality test. Normally distributed data were summarized as averages±SEM, and nonnormally distributed data (LV fibrosis) were summarized as medians with interquartile ranges. Unpaired T test was used for comparing 2 normal distributions, and ANOVA was chosen for ≥3 groups. Comparisons were deemed significant if P value for rejecting null hypothesis was <0.05. For post hoc analysis, Holm‐Sidak multiple comparisons test was used for pairwise comparisons. For nonnormally distributed data (LV fibrosis), Mann‐Whitney U test was implemented. GraphPad Software Inc (version 8) was used for generating data and statistical analysis. All authors have supervised and assume responsibility for the progress and presentation of the current work.
Results
B7‐33 Improves Cardiac Function 24 Hours Post MI
Twenty‐four hours post IR surgery, echocardiography was performed to compare several functional parameters, including FS and LV internal diameter (LVID) at systole and diastole through analysis of M‐mode images. Before IR, FS and LVID at systole and diastole were measured in healthy CD1 male mice to exclude cardiac abnormalities. Mice that received B7‐33 at reperfusion had significantly higher FS (Figure 2A) and lower LVID at systole (Figure 2B) compared with vehicle‐treated mice at 24 hours. LVID at diastole did not significantly vary from sham IR group levels compared with B7‐33 and vehicle‐treated groups (Figure 2D). Infarct size was assessed via triphenyltetrazolium chloride staining, and quantified as a percentage of area at risk distal to the left anterior descending ligature. Area at risk did not differ significantly between the groups (not shown). Infarct size was significantly reduced in B7‐33 versus vehicle‐treated animals 24 hours post MI (Figure 2E). Speckle tracking analysis was performed on long‐axis B‐mode images acquired 24 hours post MI (Figure 3A and 3B). Baseline radial strain dyssynchrony and Max dV/dT were also measured before IR. Animals treated with B7‐33 demonstrated significantly higher Max dV/dT compared with vehicle animals (Figure 3C). Radial dyssnchrony was also significantly lower in the treated group (Figure 3D).
Figure 2. B7‐33 improves cardiac function 24 hours post MI.
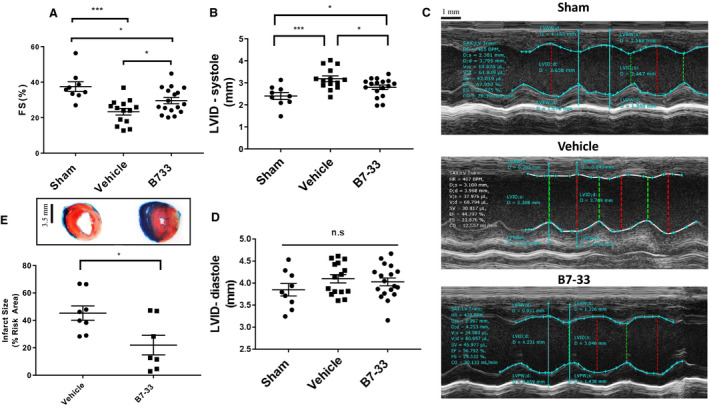
A, Fractional shortening (FS) is preserved in mice treated with B7‐33 (0.25 mg/kg) 24 hours post myocardial infarction (29.57±1.74% vs 23.34±1.84%), compared to vehicle. B, Left ventricular internal diameter (LVID) is significantly elevated in vehicle‐treated mice (3.17±0.13 mm) vs B7‐33–treated mice (2.83±0.11 mm). C, Representative sketches of M‐mode acquisitions and subsequent measurements (bar=1 mm). D, No significant differences in LVID at diastole among the experimental groups (for A‐D, n=9 for sham, and n=14–18 for vehicle and B7‐33 groups). E, Infarct size, expressed as a percentage of risk area, is significantly lower in B7‐33 group (21.99±7.17%; n=7) vs vehicle group (45.32±5.28%; n=8) (bar=3.5 mm). For A, B, and D, 1‐way ANOVA was used to determine significance, and if P<0.05, Holm‐Sidak test was used for post hoc analysis. For E, unpaired T test was used. *P<0.05, ***P<0.001.
Figure 3. Analysis of long‐axis echocardiography views 24 hours post MI.
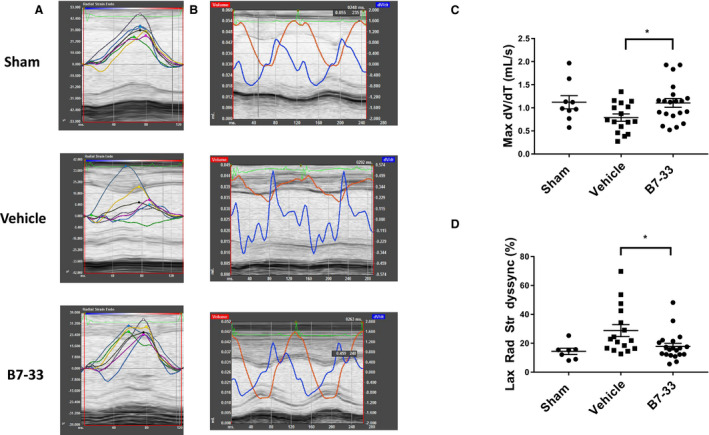
A, Representative tracings from experimental groups demonstrating radial velocity vs time, with individual colors representing the 6 long‐axis left ventricular segments to calculate radial strain dyssynchrony. B, Representative tracings from experimental groups depicting dV/dT as a function of time, with the local maxima corresponding to peak relaxation velocity, estimated via speckle tracking analysis. C, Preserved Max dV/dT in B7‐33–treated group (1.11±0.09 mL/s for B7‐33 vs 0.79±0.08 mL/s for vehicle) 24 hours post myocardial infarction (MI). D, Reduced left ventricular radial strain dyssynchrony in B7‐33–treated group 24 hours post MI (17.51±2.17% vs 28.77±4.13% for vehicle; P<0.05). Dyssynchrony was calculated from SD of left ventricular segmental radial strains, and expressed as a percentage of average radial strain. For C and D, n=9 for sham and n=15 to 21 for vehicle and B7‐33 groups. For C and D, 1‐way ANOVA was used to determine significance, and if P<0.05, Holm‐Sidak test was used for post hoc analysis. *P<0.05.
mRNA isolated from cardiac tissue samples 24 hours post MI showed an increase in the expression of Chop (mediates apoptosis during ER stress),13 ASC and toll like receptor 4 (Figure 4A through 4C). The latter 2 are involved in priming of the inflammasome, a required event preceding its activation.14
Figure 4. Real‐time polymerase chain reaction analysis at 24 hours, and assessment of cardiac function at 3 & 7 days post MI.
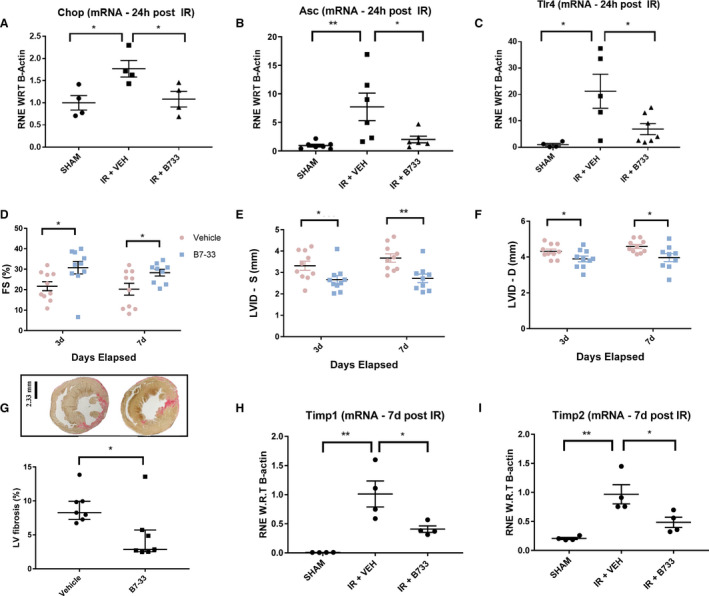
A through C, Treatment with B7‐33 significantly lowers Chop (CCAAT/enhancer‐binding protein‐homologous protein), Asc (apoptosis‐associated speck‐like protein containing a caspase recruitment domain), and toll‐like receptor 4 (Tlr4) mRNA in extracts from cardiac tissue, 24 hours post myocardial infarction (MI) (for A through C, n=9–10). D, Preserved fractional shortening (FS) 3 and 7 days post MI in B7‐33–treated group (30.73±2.20% and 28.29±1.63% vs 21.66±3.05% and 20.25±2.92% for B7‐33– and vehicle [VEH]‐treated group, respectively). E, Left ventricular internal diameter at systole (LVID‐S) at 3 and 7 days (2.66±0.18 and 2.72±0.2 mm vs 3.31±0.21 and 3.70±0.19 mm for B7‐33 and VEH, respectively). F, LVID at diastole (LVID‐D) for 3 and 7 days (3.90±0.15 and 3.96±0.22 mm vs 4.32±0.12 and 4.58±0.12 mm for B7‐33 and VEH groups, respectively) was significantly lower in B7‐33–treated groups (for D through F, n=4–7 per group). G, LV fibrosis is significantly lower in B7‐33–treated animals (n=7; 2.86% [2.52%‐5.71%]) than in VEH‐treated animals (n=7; 8.27% [7.27%‐9.95%]; bar=2.33 mm). H and I, Treatment with B7‐33 significantly lowers tissue inhibitor of metalloproteases (Timp) 1 and Timp2 mRNA expression in cardiac tissue 7 days post ischemia‐reperfusion (IR) surgery (n=4 for per group). Data expressed as relative normalized expression with respect to (RNE W.R.T) B‐actin. For A through C, H, and I, 1‐way ANOVA was used to determine significance, and if P<0.05, Holm‐Sidak test was used for post hoc analysis. For D through F, unpaired T test was used. For G, Mann‐Whitney U test was used to account for the nonnormal distribution. *P<0.05, **P<0.01.
B7‐33 Improves Functional Parameters and Mitigates Adverse Remodeling 7 Days Post MI
At the end of the 7‐day period, cardiac function was compared among vehicle and B7‐33 treated mice. Functionally, LV FS was preserved at 3 and 7 days post MI (Figure 4D) in B7‐33–treated animals in comparison to vehicle group. LVID at systole was significantly lower at 3 and 7 days with treatment (Figure 4E). LVID at diastole demonstrated similar trends at 3 and 7 days (Figure 4F). Paraffin‐fixed LV cross‐sections from mice at the end of the 7‐day period were assessed for fibrosis via picrosirius staining. Animals treated with B7‐33 had a significantly lower scar size compared with vehicle‐treated animals (Figure 4G). Quantitative polymerase chain reaction analysis of cardiac tissue samples shows significantly decreased expression of the profibrotic markers Timp1 and Timp2 (Figure 4H and 4I) 7 days post MI in B7‐33 group.
B7‐33 Protects Primary Cardiomyocytes and Fibroblasts Against SIRO
To establish the in vitro dose for the treatment of primary myocytes with B7‐33, freshly isolated cells were treated with 10, 50, and 100 nmol/L doses of B7‐33. Trypan blue staining was used to estimate the percentage of dead myocytes after SIRO, in the presence of varying doses of B7‐33. Compared with control, cells treated with either 50 or 100 nmol/L of B7‐33 had reduced trypan blue positive cells (Figure 5A). To test whether the reduction in cell death was through B7‐33 mediated phosphorylation of Erk 1/2, the dual specificity mitogen‐activated protein kinase (MEK1 and MEK2) inhibitor PD98059 (50 μmol/L) was used to reduce phosphorylated Erk 1/2 levels via pretreatment of primary cardiomyocytes for 1 hour before SIRO. The mitigation of SIRO‐induced cell death by B7‐33 is significantly abolished when coincubated with PD98059 (Figure 5A). The viability of primary cardiac fibroblasts after SIRO was assessed through the MTT assay, and cells treated with 10, 50, or 100 nmol/L B7‐33 showed significantly higher relative absorbance compared with the control group (Figure 5B).
Figure 5. Simulated ischemia reoxygenation (SIRO) of primary cardiomyocytes and fibroblasts.
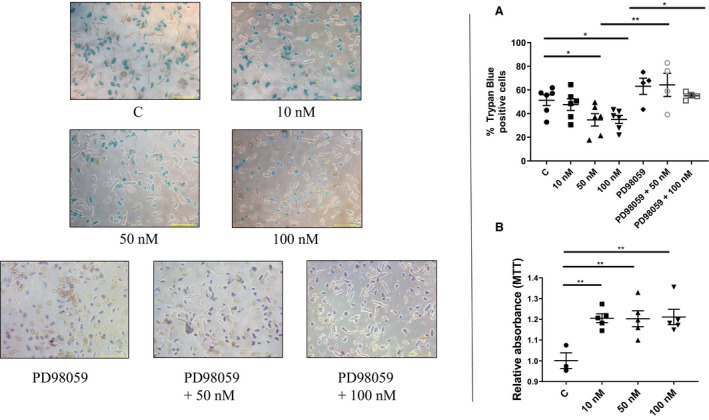
A, Primary cardiomyocytes treated with either 50 or 100 nmol/L of B7‐33 in myocyte media following simulated ischemia have a lower trypan blue positive percentage of cells (control [51.33±4.53%] vs 50 nmol/L [34.72±5.20%] or 100 nmol/L [35.03±3.43%] of B7‐33) (n=4–6 experiments/group). Pretreating cardiomyocytes with extracellular signal‐regulated kinase (Erk) 1/2 inhibitor PD98059 followed by coincubation with B7‐33 abolishes the protective effects at both 50 nmol/L (64.28±9.73%) and 100 nmol/L (54.35±2.23%) concentrations of B7‐33 (bar=100 μm). B, Fibroblast viability is higher, compared with control, when treated with 10, 50, or 100 nmol/L of fibroblast media (n=3–5 experiments/group) post SIRO. For A and B, 1‐way ANOVA was used to determine significance, and if P<0.05, Holm‐Sidak test was used for post hoc analysis. *P<0.05, **P<0.01.
To evaluate the signaling effects of B7‐33 on cardiomyocytes, protein was extracted and phosphorylation of Erk 1/2 was assessed via Western blot after 15 minutes of exposure. Phosphorylation ratio increased with higher doses of B7‐33, reaching significance at 100 nmol/L dose (Figure 6A). For subsequent myocyte experimentation, 100 nmol/L dose of B7‐33 was chosen. Primary myocytes subjected to SIRO were treated with B7‐33 for 24 hours during the reoxygenation phase, and protein isolated from cell lysates was analyzed. Treatment with B7‐33 reduced expression of ASC 6 hours post SIRO (Figure 6B), and GRP78, an ER chaperone that functions as a key mediator in several UPR‐associated signaling cascades, was reduced in B7‐33–treated myocytes 24 hours post SIRO (Figure 6C).
Figure 6. Western blot analysis of primary cardiomyocytes treated with B7‐33.
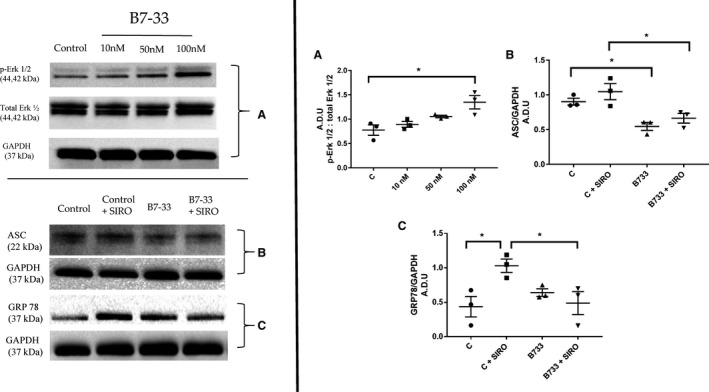
In primary cardiomyocytes, increased phosphorylation of extracellular signal‐regulated kinase (Erk) 1/2 (p‐Erk 1/2) in cells treated with B7‐33, reaching significance at 100 nmol/L (A); reduced expression of ASC (apoptosis‐associated speck‐like protein containing a caspase recruitment domain) in cells 6 hours post simulated ischemia reoxygenation (SIRO), on treatment with B7‐33 (B); and reduced expression of GRP78 in cells 24 hours post SIRO, on treatment with B7‐33 (C) (for A through C, n=3 experiments per group). Western blots are quantified and expressed as arbitrary densitometric units (A.D.U). For A through C, 1‐way ANOVA was used to determine significance, and if P<0.05, Holm‐Sidak test was used for post hoc analysis. *P<0.05. C indicates control.
B7‐33 Reduces Tunicamycin‐Induced ER Stress in Primary Cardiomyocytes
The reduction in expression of IR injury–related ER stress associated markers in vivo and in vitro on treatment with B7‐33 prompted further inquiry into studying the association between ER stress and relaxin signaling. An alternative model of injury was sought because IR induces dysfunction via multiple interlinked causes (sterile inflammation, reactive oxygen species production, mitochondrial disruption), along with UPR. Therefore, we used tunicamycin, a bioactive compound that inhibits N‐linked glycosylation of proteins, to study ER stress exclusively.13 Primary cardiomyocytes were incubated with either 2.5 or 7.5 μg/mL tunicamycin for 24 hours. Western blot analysis after the incubation period with tunicamycin significantly reduced phosphorylation of Erk 1/2 at 2.5 and 7.5 μg/mL doses (Figure 7A). Furthermore, treatment with tunicamycin showed a significant increase in GRP78 expression in myocytes at both the concentrations (Figure 7B). Coincubation with 100 nmol/L B7‐33 significantly reduced GRP78 expression at 2.5 μg/mL dosage of tunicamycin (Figure 7B). To test the hypothesis that B7‐33–induced phosphorylation of Erk 1/2 is essential to limit ER stress, primary cardiomyocytes were pretreated with PD98059 for 1 hour before tunicamycin and/or B7‐33 exposure for 24 hours. PD98059 significantly reduced Erk 1/2 phosphorylation despite coincubation with B7‐33 (Figure 8A and 8B). Inhibition of Erk 1/2 phosphorylation through PD98059 abolished the protective effect of B7‐33 in reducing tunicamycin‐induced GRP78 expression (Figure 8A and 8C).
Figure 7. Western blot analysis of tunicamycin‐induced endoplasmic reticulum (ER) stress in protein lysates from primary cardiomyocytes.
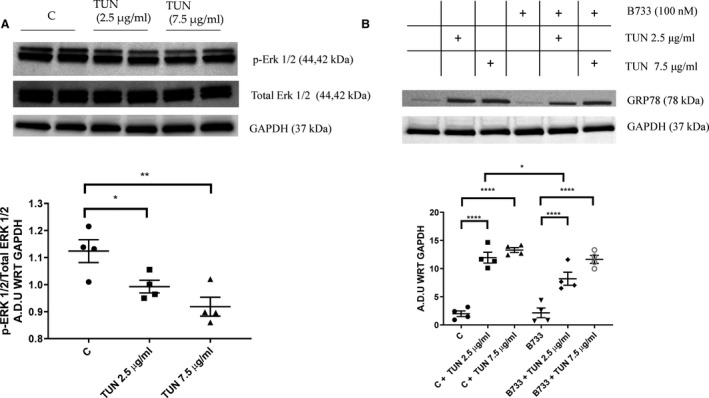
Exposure of primary cardiomyocytes to tunicamycin (TUN) for 24 hours significantly reduces phosphorylation of extracellular signal‐regulated kinase (Erk) 1/2 (p‐Erk 1/2) at both 2.5 and 7.5 μg/mL concentrations of TUN (A); and significantly increases expression of GRP78 at both doses of TUN (B). B7‐33 (100 nmol/L) significantly lowers GRP78, compared with control (C), on coincubation with 2.5 μg/mL TUN (for A and B, n=4–6 experiments/group). Western blots are quantified and expressed as arbitrary densitometric units with respect to (A.D.U WRT) the expression of GAPDH. For A and B, 1‐way ANOVA was used for significance testing, and if P<0.05, Holm‐Sidak test was used for post hoc analysis. *P<0.05, **P<0.01, ****P<0.0001.
Figure 8. Treatment of primary cardiomyocytes with tunicamycin (TUN) with/without PD98059 and/or B7‐33.
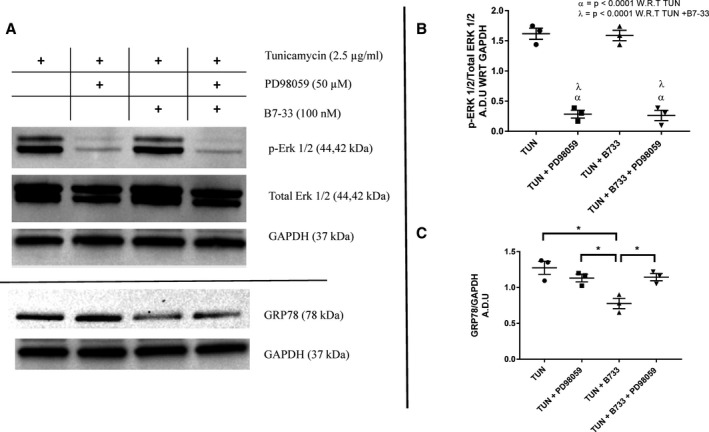
A, Representative Western blots of primary cardiomyocytes treated with tunicamycin (TUN) with/without PD98059 and/or B7‐33. B, Exposure to PD98059 (50 μmol/L) significantly lowers phosphorylation of extracellular signal‐regulated kinase (Erk) 1/2 (p‐Erk 1/2) 24 hours post incubation in primary cardiomyocytes treated with TUN with/without B7‐33. No significant difference between p‐Erk 1/2 levels was found between TUN and TUN+B7‐33 groups. C, Coincubating TUN with PD98059 abrogates the reduction in GRP78 observed when treated with B7‐33 (for B and C, n=3 experiments/group). Western blots are quantified and expressed as arbitrary densitometric units with respect to (A.D.U WRT) the expression of GAPDH. For B and C, 1‐way ANOVA was used for significance testing, and if P<0.05, Holm‐Sidak test was used for post hoc analysis. *P<0.05.
Discussion
Human relaxin‐2 is extensively involved in mammalian pregnancy because of its modulation of maternal hemodynamics by lowering total peripheral resistance and increasing cardiac output.15 The vasodilatory effects of the hormone are primarily transduced through NO signaling in smooth muscle cells. Because of these pharmacologic properties, recombinant relaxin‐2 (serelaxin; Novartis Pharmaceuticals) was considered a viable option for the treatment of acute heart failure. The phase 3 trials, RELAX‐AHF (Relaxin for the Treatment of Acute Heart Failure)16 and RELAX‐AHF217 used a short‐term (48‐hour) infusion strategy to assess clinical end points pertinent to acute heart failure. Specifically, cardiovascular‐related mortality at 180 days and worsening heart failure at 5 days were considered primary end points in the latter, more recent trial (RELAX‐AHF2).17 Serelaxin failed to achieve these outcomes, in contrast to findings from the former trial.16 The recently published article on RELAX‐AHF217 attributes this inconsistency to either chance or variability in patient populations, because RELAX‐AHF2 enrolled patients with worse renal function and incorporated a higher threshold for natriuretic peptide levels. Nevertheless, the authors question the efficacy of a 48‐hour infusion protocol in addressing long‐term end points for acute heart failure. Another plausible explanation could be attributed to recent findings suggesting that the effects of relaxin may be compromised in the context of heart failure in patients who are already receiving angiotensin receptor blockers.18 The utility of relaxin‐associated therapeutics remains potentially unharnessed, given the wealth of preclinical data implicating relaxin signaling in mitigating progression toward heart failure in various preclinical models.2 Translationally, this potential could be realized in causes where overt damage to the myocardium occurs in a symptomatically or clinically detectable manner, such as in the case of MI. Our laboratory previously investigated the effect of serelaxin in conferring cardioprotection against acute IR injury. We demonstrated that relaxin‐induced cardioprotection is dependent on endothelial NO synthase activity, and showed that the hormone suppresses inflammasome activity in cardiac tissue through endothelial NO synthase–dependent mechanism.1 Sterile inflammation, through the priming and activation of Nucleotide‐binding oligomerization domain, Leucine rich Repeat and Pyrin domain containing 3 (NLRP3) inflammasome, has been conclusively shown to induce activated interleukin‐1β production and cardiomyocyte pyroptosis (through activated caspase‐1) in the heart.14 Other studies show reduced mitochondrial swelling during IR injury on treatment with relaxin.19 This is especially relevant, because reactive oxygen species released by dysfunctional mitochondria contribute further to inflammasome activity.14
The peptide structure of relaxin resembles that of other insulin‐like peptides: two chains (A and B) linked through disulfide bonds. The single B‐chain analogue B7‐33 was synthesized on the basis of the premise that the lack of disulfide linkages facilitates lower production costs and increased affordability.5, 20 When RXFP1 was expressed in a heterologous HEK‐293T system, B7‐33 failed to induce a cAMP response when compared with analogous concentrations of relaxin‐2.5 In vitro exposure to B7‐33 increased phosphorylated Erk 1/2 levels within 10 minutes in rat renal myofibroblasts.5 We observed similar results in primary adult cardiomyocytes within 15 minutes of exposure. The preferential bias toward Erk 1/2 signaling, mediated by the βγ subunits of RXFP1,2 has important physiological implications; the presence of excessive cAMP in the ischemic heart could potentially increase myocardial workload, further exacerbating contractile demand on myocytes from remote/viable myocardium.6 On the other hand, Erk 1/2–mediated activation of NO synthases has potent antifibrotic effects through the involvement of cGMP‐induced matrix metalloproteinase (2 and 9) upregulation.21 Therefore, RXFP1‐mediated Erk 1/2 phosphorylation transduces multiple protective mechanisms, and B7‐33 is well placed to recapitulate these effects in lieu of relaxin‐2 therapy.
In this current study, B7‐33 was administered after 30 minutes of ischemia, immediately before the onset of reperfusion. A short‐term, IP dosage of 0.25 mg/kg was chosen on the basis of a previous in vivo study (Hossain et al5). Although this study used 0.5 mg/kg per day for treatment (which we replicated in our 7‐day studies), we used 0.25 mg/kg at reperfusion to achieve equimolar levels of relaxin concentration used in Hossain et al, because relaxin has roughly twice the molecular weight of B7‐33. B7‐33 has been shown to achieve a similar pharmacodynamic profile as relaxin‐2 in cells (cardiac fibroblasts) endogenously expressing RXFP1,5 and is likely to possess similar half‐life to relaxin22 because of common structural features. We were unable to quantify the bioavailability of B7‐33 post injection, because the ELISA kit (R&D Systems) for relaxin‐2 failed to recognize B7‐33 in our preliminary studies. However, as discussed previously,1 it is likely that a small peptide, such as B7‐33, reaches circulation within minutes of intraperitoneal administration. B7‐33 reduced infarct size, preserved FS, and reduced radial strain dyssynchrony 24 hours post MI. The reduction in infarct size (21% versus 45% in saline‐treated group) with B7‐33 is roughly comparable to the reduction in infarct size on reperfusion therapy with serelaxin (15.2% versus 46.5% in saline‐treated group) in our previous study.1 Similarly, FS 24 hours post MI on B7‐33 treatment (29.57%) roughly corresponds to FS in mice treated with serelaxin (28%).1 Therefore, B7‐33 recapitulates the protective effects conferred by serelaxin in mice subjected to the same MI protocol. In the current study, LVID at systole was significantly smaller in the treatment group (because of preserved contractility), but no differences were found in LVID at diastole at 24 hours. However, LVID at diastole diverged between the groups at 3‐ and 7‐day time points, perhaps because of increased dilatation attributable to progressive adverse remodeling in the vehicle group. Therefore, B7‐33–induced protection was not solely limited to infarct‐sparing effects, as therapeutic benefits from daily administrations were realized over the course of remodeling. Cell type–specific responses were further studied when cardiomyocytes and fibroblasts were subjected to SIRO. We chose a longer duration of ischemia for fibroblasts because they are more resilient to ischemic damage compared with primary cardiomyocytes.23 Fibroblast viability was increased, at all concentrations of B7‐33 (10, 50, and 100 nmol/L), after SIRO. This is interesting, considering the long‐term antifibrotic effects of B7‐33. Fibroblast proliferation in the early stages of infarct healing (as it coincides temporally with our in vitro experiment) might be vital,23 but the long‐term effect of B7‐33 exposure to remodeling fibroblasts is yet to be fully characterized. Relaxin signaling has been shown to inhibit phenotypic differentiation of these cells into myofibroblasts on exposure to transforming growth factor‐β.24 Interestingly, a recent study demonstrates that relaxin‐induced inhibition of myofibroblast transition on transforming growth factor‐β stimulation is dependent on Wnt signaling. This recently characterized arm of relaxin signaling occurs independent to cAMP generation.25 Therefore, the interaction of this pathway with B7‐33–induced activation of RXFP1 is of potential interest in future studies. In our study, LV sections stained for scar size show the antifibrotic effects of B7‐33, and corroborate previous findings, where reduced interstitial fibrosis was detected in B7‐33–treated animals subjected to MI (permanent ligation) and isoproterenol induced heart failure.5 Analysis of LV samples 7 days post MI shows reduced expression of the profibrotic markers Timp 1 and 2 on treatment with B7‐33. The inhibitory effects of relaxin signaling on Timp expression,26, 27 and antifibrotic modulation of matrix metalloproteinase/TIMP ratio by relaxin,21 have been previously examined.
The complex interplay between mitochondria and ER in the ischemic heart28, 29 led us to investigate alternate, previously unexamined modalities of protection through relaxin therapy. IR injury damages mitochondria, and leads to production of reactive oxygen species through damaged complex I and III within the electron transport chain when oxygen is reintroduced.30 Mitochondrial reactive oxygen species is central to inflammasome activation,14 and can contribute to UPR.28 Given the mitoprotective features of relaxin therapy, we tried to elucidate whether B7‐33 impacted ER stress response post IR injury. Relaxin‐2 was previously shown to inhibit glucose‐induced apoptosis in neonatal rat myocytes by inhibiting ER stress.31 In our study, B7‐33 reduced the expression of GRP78, a chaperone involved in transducing UPR‐associated pathways, 24 hours post SIRO in cardiomyocytes. As misfolded proteins accumulate in ER lumen, GRP78 is disengaged from the inner ER membrane as it activates the protein kinase RNA‐like endoplasmic reticulum kinase (PERK), inositol‐requiring enzyme (IRE1), and activating transcription factor (ATF6) arms of the response.28 The role and significance of ER stress signaling in ischemic heart disease is of intense scrutiny. Although the net effect could be restorative and reduce the burden of misfolded proteins, UPR can also trigger proapoptotic and inflammatory cascades, causing cell death.29 For instance, endothelin‐1–induced overexpression of GRP78 increased hypoxia tolerance in neonatal rat myocytes,32 but use of tauroursodeoxycholic acid reduced apoptosis in rats33 post MI, and reduced adverse modeling through inhibition of GRP78 in a pressure overload model of heart failure.34 In our model, B7‐33 could potentially be acting upstream of GRP78 recruitment as misfolded proteins accumulate. Nevertheless, this remained unclarified because the data were confounded by the possibility that B7‐33–mediated GRP78 reduction was a consequence of overall reduction in cell death through other protective pathways. To circumvent this issue, we studied the effect of induced ER stress in isolation by incubating cardiomyocytes with a nonlethal dose of tunicamycin. B7‐33 reduced tunicamycin‐induced upregulation of GRP78, indicating a role for RXFP1 in UPR, when activated.
B7‐33–mediated reduction of GRP78 expression was abrogated when cardiomyocytes were coincubated with PD98059 and tunicamycin, potentially implicating mitogen‐activated protein kinase signaling in the protective effect. As shown elsewhere, activation of Erk 1/2 in NIH3T3 cells was protective against ER stress‐induced apoptosis.9 In MCF‐7 breast cancer cells, ER stress induction led to rapid activation of Erk 1/2, an event shown to divert cell fate toward removal of misfolded proteins and survival, while curbing proapoptotic signaling.35 In our study, tunicamycin led to overall reduction in phosphorylated Erk 1/2 levels at 24 hours. Although B7‐33 led to a short‐term increase in phosphorylated Erk 1/2, this activation was transient as the levels normalized at 24 hours. This corroborates the findings seen in rat renal myofibroblasts, where the Erk 1/2 response with B7‐33 stimulation was muted within minutes of exposure.5 The transient upregulation observed in our primary cardiomyocytes potentially underscores the importance of downstream mediators in reducing the burden of misfolded proteins.
In summary, B7‐33 confers short‐term cardioprotection by reducing infarct size and preserving cardiac function 24 hours post IR injury, and prevents further adverse remodeling. In vitro assessment using B7‐33 demonstrates remarkable protection in cardiomyocytes and fibroblasts, while implicating Erk 1/2 activation in mediating the protection against IR injury‐induced ER stress. Therefore, B7‐33 could potentially be used as a novel therapy for attenuation of MI and prevention of MI‐related adverse cardiac remodeling and failure.
Limitations
The results from our current study, although potentially impactful, are limited by the nature of our animal model. Although the murine model recapitulates the structural and functional deficits observed in human MI, multifactorial influences (including environment, genetics, and existing pharmacotherapy) underscoring the pathophysiological characteristics of adverse remodeling and heart failure in humans are manifestly absent in mice. Characterizing the therapeutic utility of B7‐33 necessitates extensive safety and efficacy trials in humans, and this requires formulation of strategies ensuring large‐scale production and distribution of B7‐33. Although the prospect of commercializing B7‐33 is still in its infancy, the reduced complexity and fewer purification steps (compared with recombinant relaxin) could potentially streamline the process in the long‐term. In the current study, CD1 mice were used because of a wider genetic diversity stemming from outbreeding within mice populations.36 The exclusive use of male mice limits the scope of this current work. However, expression of RXFP1 in female rodents has been shown to be altered over the course of estrus cycle in other tissues (skeletal).37 To reduce the confounding effect of reproductive hormone‐induced changes in RXFP1, male mice were chosen in this study. Analysis in both male and female animal populations would be necessary for evaluating the potential benefits of treatment with B7‐33 in the future.
Sources of Funding
This study was supported by the American Heart Association (18PRE33990001) and the Wright Center for Clinical and Translational Research Center (Wright Scholar) to Dr Devarakonda, National Health and Medical Research Council Australia (GNT1122170) to Dr Hossain, and the National Institutes of Health (R01HL142281, R21AG053654, and R01HL133167) to Dr Salloum.
Disclosures
None.
(J Am Heart Assoc. 2020;e015748 DOI: 10.1161/JAHA.119.015748.)
For Sources of Funding and Disclosures, see page 13.
References
- 1. Valle Raleigh J, Mauro AG, Devarakonda T, Marchetti C, He J, Kim E, Filippone S, Das A, Toldo S, Abbate A, et al. Reperfusion therapy with recombinant human relaxin‐2 (serelaxin) attenuates myocardial infarct size and NLRP3 inflammasome following ischemia/reperfusion injury via eNOS‐dependent mechanism. Cardiovasc Res. 2017;2:1–11. [DOI] [PubMed] [Google Scholar]
- 2. Devarakonda T, Salloum FN. Heart disease and relaxin: new actions for an old hormone. Trends Endocrinol Metab. 2018;29:338–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sarwar M, Samuel CS, Bathgate RA, Stewart DR, Summers RJ. Serelaxin‐mediated signal transduction in human vascular cells: bell‐shaped concentration‐response curves reflect differential coupling to G proteins. Br J Pharmacol. 2015;172:1005–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kocan M, Sarwar M, Ang SY, Xiao J, Marugan JJ, Hossain MA, Wang C, Hutchinson DS, Samuel CS, Agoulnik AI, et al. ML290 is a biased allosteric agonist at the relaxin receptor RXFP1. Sci Rep. 2017;7:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hossain MA, Kocan M, Yao ST, Royce SG, Nair VB, Siwek C, Patil NA, Harrison IP, Rosengren KJ, Selemidis S, et al. A single‐chain derivative of the relaxin hormone is a functionally selective agonist of the G protein‐coupled receptor, RXFP1. Chem Sci. 2016;7:3805–3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ali DC, Naveed M, Gordon A, Majeed F, Saeed M, Ogbuke MI, Atif M, Zubair HM, Changxing L. β‐Adrenergic receptor, an essential target in cardiovascular diseases. Heart Fail Rev. 2019;25:343–354. [DOI] [PubMed] [Google Scholar]
- 7. Heusch G. Molecular basis of cardioprotection signal transduction in ischemic pre‐, post‐, and remote conditioning. Circ Res. 2015;116:674–699. [DOI] [PubMed] [Google Scholar]
- 8. Wang Z, Zhang H, Xu X, Shi H, Yu X, Wang X, Yan Y, Fu X, Hu H, Li X, et al. bFGF inhibits ER stress induced by ischemic oxidative injury via activation of the PI3K/Akt and ERK1/2 pathways. Toxicol Lett. 2012;212:137–146. [DOI] [PubMed] [Google Scholar]
- 9. Darling NJ, Balmanno K, Cook SJ. ERK1/2 signalling protects against apoptosis following endoplasmic reticulum stress but cannot provide long‐term protection against BAX/BAK‐independent cell death. PLoS One. 2017;12:1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Torrado J, Cain C, Mauro AG, Romeo F, Ockaili R, Chau VQ, Nestler JA, Devarakonda T, Ghosh S, Das A, et al. Sacubitril/valsartan averts adverse post‐infarction ventricular remodeling and preserves systolic function in rabbits. J Am Coll Cardiol. 2018;72:2342–2356. [DOI] [PubMed] [Google Scholar]
- 11. Toldo S, Das A, Mezzaroma E, Chau VQ, Marchetti C, Durrant D, Samidurai A, Van Tassell BW, Yin C, Ockaili RA, et al. Induction of microRNA‐21 with exogenous hydrogen sulfide attenuates myocardial ischemic and inflammatory injury in mice. Circ Cardiovasc Genet. 2014;7:311–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nevers T, Salvador AM, Velazquez F, Ngwenyama N, Carrillo‐Salinas FJ, Aronovitz M, Blanton RM, Alcaide P. Th1 effector T cells selectively orchestrate cardiac fibrosis in nonischemic heart failure. J Exp Med. 2017;214:3311–3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shen M, Wang LIN, Guo X, Xue Q, Huo C, Li X, Fan LI, Wang X. A novel endoplasmic reticulum stress—induced apoptosis model using tunicamycin in primary cultured neonatal rat cardiomyocytes. Mol Med Rep. 2015;12:5149–5154. [DOI] [PubMed] [Google Scholar]
- 14. Toldo S, Mezzaroma E, Mauro AG, Salloum F, Van Tassell BW, Abbate A. The inflammasome in myocardial injury and cardiac remodeling. Antioxid Redox Signal. 2014;22:1146–1161. [DOI] [PubMed] [Google Scholar]
- 15. Bathgate RAD, Halls ML, van der Westhuizen ET, Callander GE, Kocan M, Summers RJ. Relaxin family peptides and their receptors. Physiol Rev. 2013;93:405–480. [DOI] [PubMed] [Google Scholar]
- 16. Teerlink JR, Cotter G, Davison BA, Felker GM, Filippatos G, Greenberg BH, Ponikowski P, Unemori E, Voors AA, Adams KF, et al. Serelaxin, recombinant human relaxin‐2, for treatment of acute heart failure (RELAX‐AHF): a randomised placebo‐controlled trial. Lancet. 2013;381:29–39. [DOI] [PubMed] [Google Scholar]
- 17. Metra M, Teerklink J, Cotter G, Davison B, Felker G, Filippatos G, Greenberg BH, Pang PS, Ponikowski P, Voors AA, et al. Effects of serelaxin in patients with acute heart failure. N Engl J Med. 2019;381:716–726. [DOI] [PubMed] [Google Scholar]
- 18. Chow BSM, Kocan M, Shen M, Wang Y, Han L, Chew JY, Wang C, Bosnyak S, Mirabito‐Colafella KM, Barsha G, et al. AT1R‐AT2R‐RXFP1 functional crosstalk in myofibroblasts: impact on the therapeutic targeting of renal and cardiac fibrosis. J Am Soc Nephrol. 2019;30:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Masini E, Bani D, Bello MG, Bigazzi M, Mannaioni PF, Sacchi TB. Relaxin counteracts myocardial damage induced by ischemia‐reperfusion in isolated guinea pig hearts: evidence for an involvement of nitric oxide. Endocrinology. 1997;138:4713–4720. [DOI] [PubMed] [Google Scholar]
- 20. Marshall SA, O'Sullivan K, Ng HHH, Bathgate RAD, Parry LJ, Hossain MA, Leo CH. B7‐33 replicates the vasoprotective functions of human relaxin‐2 (serelaxin). Eur J Pharmacol. 2017;807:190–197. [DOI] [PubMed] [Google Scholar]
- 21. Chow BSM, Chew EGY, Zhao C, Bathgate RAD, Hewitson TD, Samuel CS. Relaxin signals through a RXFP1‐pERK‐nNOS‐NO‐cGMP‐dependent pathway to up‐regulate matrix metalloproteinases: the additional involvement of iNOS. PLoS One. 2012;7:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bani D, Masini E, Bello MG, Bigazzi M, Sacchi TB. Relaxin protects against myocardial injury caused by ischemia and reperfusion in rat heart. Am J Pathol. 1998;152:1367–1376. [PMC free article] [PubMed] [Google Scholar]
- 23. Shinde AV, Frangogiannis NG. Fibroblasts in myocardial infarction: a role in inflammation and repair. J Mol Cell Cardiol. 2014;70:74–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sassoli C, Chellini F, Pini A, Tani A, Nistri S, Nosi D, Zecchi‐Orlandini S, Bani D, Formigli L. Relaxin prevents cardiac fibroblast‐myofibroblast transition via notch‐1‐mediated inhibition of TGF‐β/Smad3 signaling. PLoS One. 2013;8:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Martin B, Gabris B, Barakat AF, Henry BL, Giannini M, Reddy RP, Wang X, Romero G, Salama G. Relaxin reverses maladaptive remodeling of the aged heart through Wnt‐signaling. Sci Rep. 2019;9:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Williams EJ, Benyon RC, Trim N, Hadwin R, Grove BH, Arthur MJP, Unemori EN, Iredale JP. Relaxin inhibits effective collagen deposition by cultured hepatic stellate cells and decreases rat liver fibrosis in vivo. Gut. 2001;49:577–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Palejwala S, Stein DE, Weiss G, Monia BP, Tortoriello D, Goldsmith LT. Relaxin positively regulates matrix metalloproteinase expression in human lower uterine segment fibroblasts using a tyrosine kinase signaling pathway. Endocrinology. 2001;142:3405–3413. [DOI] [PubMed] [Google Scholar]
- 28. Hotamisligil G, Davis RJ. Cell signaling and stress responses. Cold Spring Harb Perspect Biol. 2016;8:1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liu M, Dudley SC. Role for the unfolded protein response in heart disease and cardiac arrhythmias. Int J Mol Sci. 2016;17:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen Q, Yin G, Stewart S, Hu Y, Lesnefsky EJ. Isolating the segment of the mitochondrial electron transport chain responsible for mitochondrial damage during cardiac ischemia. Biochem Biophys Res Commun. 2010;397:656–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang X, Ma X, Zhao M, Zhang B, Chi J, Liu W, Chen W, Fu Y, Liu Y, Yin X. H2 and H3 relaxin inhibit high glucose‐induced apoptosis in neonatal rat ventricular myocytes. Biochimie. 2015;108:59–67. [DOI] [PubMed] [Google Scholar]
- 32. Pan Y, Lin L, Ren A, Pan X, Chen H, Tang C, Yuan W. HSP70 and GRP78 induced by endothelin‐1 pretreatment enhance tolerance to hypoxia in cultured neonatal rat cardiomyocytes. J Cardiovasc Pharmacol. 2004;44:117–120. [DOI] [PubMed] [Google Scholar]
- 33. Rivard AL, Steer CJ, Kren BT, Rodrigues CMP. Administration of tauroursodeoxycholic acid (TUDCA) reduces apoptosis following myocardial infarction in rat. Am J Chin Med. 2007;35:279–295. [DOI] [PubMed] [Google Scholar]
- 34. Rani S, Sreenivasaiah PK, Kim JO, Lee MY, Kang S, Kim YS, Ahn Y, Park WJ, Cho C, Kim H. Tauroursodeoxycholic acid (TUDCA) attenuates pressure overload‐induced cardiac remodeling by reducing endoplasmic reticulum stress. PLoS One. 2017;12:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hu P, Han Z, Couvillon AD, Exton JH. Critical role of endogenous Akt/IAPs and MEK1/ERK pathways in counteracting endoplasmic reticulum stress‐induced cell death. J Biol Chem. 2004;279:49420–49429. [DOI] [PubMed] [Google Scholar]
- 36. Aldinger KA, Sokoloff G, Rosenberg DM, Palmer AA, Millen KJ. Genetic variation and population substructure in outbred CD‐1 mice: implications for genome‐wide association studies. PLoS One. 2009;4:2–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dehghan F, Soori R, Dehghan P, Gholami K, Muniandy S, Azarbayjani MA, Yusof A. Changes in knee laxity and relaxin receptor isoforms expression (RXFP1/RXFP2) in the knee throughout estrous cycle phases in rodents. PLoS One. 2016;11:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]