

Abstract
Heart failure (HF) is a global pandemic with significant mortality and morbidity. Despite current medications, 50% of individuals die within 5 years of diagnosis. Of these deaths, 30–50% will be a result of sudden cardiac death from ventricular arrhythmias. This review discusses two stress-induced mechanisms, phosphorylation from chronic β-adrenoceptor (β-AR) stimulation and thiol modifications from oxidative stress, and how they modulate the cardiac ryanodine receptor type 2 (RyR2) and foster an arrhythmogenic phenotype. Calcium (Ca2+) is the ubiquitous secondary messenger of excitation–contraction coupling and provides a common pathway for contractile dysfunction and arrhythmia genesis. In a healthy heart, Ca2+ is released from the sarcoplasmic reticulum (SR) by RyR2. The open probability of RyR2 is under the dynamic influence of co-proteins, ions, and kinases that are in strict balance to ensure normal physiological functioning. In HF, chronic β-AR activity and production of reactive oxygen species and reactive nitrogen species provide two stress-induced mechanisms uncoupling RyR2 control, resulting in pathological diastolic SR Ca2+ leak. This increased cytosolic [Ca2+] promotes Ca2+ extrusion via the local Na+/Ca2+ exchanger, resulting in net sarcolemmal depolarization, delayed after depolarization and ventricular arrhythmia. Experimental models researching oxidative stress and phosphorylation have aimed to identify how post-translational modifications to the RyR2 macromolecular complex, and the associated Na+/Ca2+ cycling proteins, result in pathological Ca2+ handling and diastolic leak. However, the causative molecular changes remain controversial and undefined. Through understanding the molecular mechanisms that produce an arrhythmic phenotype, novel therapeutic targets to treat HF and prevent its malignant course can be identified.
Keywords: heart failure, arrhythmia, ryanodine receptor, phosphorylation, redox, beta-adrenoceptors
Burden of Heart Failure and Arrhythmia
Heart failure (HF) with reduced ejection fraction (HFrEF) is a global pandemic, affecting ∼26 million people worldwide.1 In Australia, the prevalence of HF is 1.0–2.0% of the population, consistent with North America and Europe.2 HF is more prevalent in the elderly, with the mean age of patients admitted with a primary diagnosis of HF ranging from 70 to 75 years of age.1 Approximately 11% of the world’s population is over the age of 60 years, and this is predicted to reach 22% by 2050.3 Consequently, the prevalence of HF will increase.
HFrEF is a chronic disease with a malignant prognosis. Impaired contractility is the central pathophysiological feature, which determines quality of life and the risk of sudden cardiac death (SCD). In 1993, the reported median survival following HF diagnosis was 1.7 years for men and 3.2 years for women.4 Corresponding 1 and 5 year survival rates were 57 and 25% in men and 64 and 38% in women, respectively. Of these deaths, 30–50% were the result of SCD from ventricular arrhythmias.4 Patients at greatest risk of SCD are those with a left ventricular function (LVF) ≤ 35% despite optimal guideline-directed medical therapy (GDMT). There is consensus within global guidelines that these individuals derive benefit from an implantable cardioverter–defibrillator (ICD). These devices can abort malignant ventricular arrhythmias by delivering high-voltage shocks. Despite the development of disease modifying medications and utilization of ICDs, present registries report an all-cause 1 year mortality of 23.6%.5 However, controversy exists within the published literature. Contemporary studies recruiting nonischemic HF patients with LVF ≤ 35% revealed no significant benefit in total mortality at 5.6 years with ICD therapy.6 However, prespecified subgroup analysis showed evidence of a prognostic benefit in patients younger than 68 years. Thus, the potential utility of ICDs depend on the patient’s risk of sudden arrhythmic death relative to nonsudden death. Older patients are twice as likely to die of causes other than SCD.7 Taken together, young patients with nonischemic cardiomyopathy remain at high risk for SCD despite GDMT. Identifying cellular mechanisms implicated in the development of contractile dysfunction and generation of arrhythmia may provide novel medication targets with the potential to benefit millions of people suffering from HF.
Cardiac Excitation–Contraction Coupling
In order to understand the complex interplay of cardiac contraction and development of ventricular arrhythmia, it is important to have an appreciation of cardiac excitation–contraction coupling (ECC): the process of translating an electrical action potential (AP) into mechanical output.
Action Potential
The ventricular AP is a consequence of sequential activation and inactivation of ion channels conducting inward sodium (Na+) and potassium (K+) depolarizing currents and outward K+ repolarizing currents (Figure 1).8 Depolarization increases intracellular calcium concentration [Ca2+]i, which is central to regulating troponin C (TnC), exposing actin to the myosin heads, resulting in force generation.9,10
Figure 1.

Action potential of ventricular myocardium. Illustration of the different phases of a ventricular myocyte action potential and the relationship of the different ionic currents involved in its generation.
As an AP propagates, it evokes depolarization of adjacent cells by increasing the resting membrane potential (Em) to a threshold that activates voltage-gated Na+ (Nav) channels (INa-fast, Nav1.5). The inward sodium current (INa) sets up a positive feedback loop creating depolarization and the rapid upstroke of an AP, characteristic of phase 0. Phase 1, or early repolarization, results from inactivation of Nav channels and activation of the fast transient voltage-gated outward K+ current (Ito). The AP then enters a long plateau with a steady Em, phase 2, a balance between outward flow of K+ and inward voltage-gated Ca2+ current (ICa,L) through L-type calcium channels (LTCC).8 This influx of Ca2+ is the primary trigger for ECC. Eventually, the LTCC is inactivated, and the outward flow of K+ predominates, facilitating repolarization (phase 3). Multiple types of voltage-gated K+ currents and non-voltage-gated inwardly rectifying K+ currents contribute to repolarization, including IKr [IK(rapid)], IKs [IK(slow)], and IKu [IK(ultrarapid)].8 Phase 4 reflects the cell at rest. Here, the membrane is most permeable to K+ and the flow is driven by an ionic concentration gradient, generated by Na+/K+-ATPase pumps. Non-voltage-gated inward rectifying K+ channels (IKi) primarily contribute to the maintenance of a resting diastolic potential of −89 mV.8,11 The stability of the resting membrane potential affects cardiac excitability and AP morphology. The ion channels discussed are composed of many pore-forming subunits and channel accessories which underlie the channels’ contribution to the myocardial AP. Mutations in genes encoding these subunits have been found to underpin several genetic cardiac arrhythmia syndromes.8,12,13
Sarcolemma
The sarcolemma is a highly specialized plasma membrane (Figure 2) which includes ion channels, transporters and pumps that are responsible for ionic fluxes involved in ECC and calcium homeostasis. The sarcolemma exhibits both regional specialization, important for spatial and temporal summation of local Ca2+ transients, and global orchestration of cell-wide Ca2+ transients.9 First, it forms transverse tubules (T tubules), invaginations that extend deep into the interior of the cardiomyocyte, which couple with the terminal cisternae of the sarcoplasmic reticulum (SR), the intracellular Ca2+ reservoir.14 Second, cardiomyocytes interdigitate and are closely opposed at intercalated discs. Here, cells form mechanical connections and gap junctions to provide a low-resistance electrical pathway that allows the AP to propagate, coordinating the heart to function as an electrical syncytium.9,14
Figure 2.

Schematic diagram of a cardiomyocyte. Each cardiomyocyte is surrounded by the meshwork of the sarcolemma which forms invaginations called T tubules. At the intercalated disc, desmosomes and gap junctions can be found. Reproduced with permission from ref (15). Copyright 1984 Williams & Wilkins.
Calcium-Induced Calcium Release
The efflux of Ca2+ from the SR is crucial for the activation of contraction as independently Ca2+ influx via LTCC is too small and insufficient to engage the contractile machinery. Calcium-induced calcium release (CICR) is the most widely accepted mechanism of coordinated SR Ca2+ release16−18 (Figure 3). The voltage-dependent LTCCs are found clustered on membrane microdomains on T tubules, which are closely opposed to a RyR2 found on the membrane of the SR. Groupings of LTCCs and RyR2s in so-called Ca2+ release units (CRU) or couplons are the initiation sites for where each AP may cause transient flows of Ca2+ from the SR to occur. This spatial arrangement of Ca2+ cycling proteins is important to the efficiency of ECC.19 LTCCs are activated when the transmembrane potential increases to −55 mV allowing an inward ICa,L.9 RyR2 within the corresponding couplon releases Ca2+ from the SR lumen to the cytosol. This local increase in concentration of intracellular Ca2+ from a single RyR2 is termed a calcium spark.20 Subsequently, neighboring RyR2 are activated via the local influx of Ca2+ (>10 μM), and the spatial temporal summation of these sparks forms a Ca2+ wave which propagates through the cardiomyocyte. Once the cytosolic [Ca2+] reaches a threshold, it acts as a switch, binding troponin C and allowing the contractile machinery to engage.10,17 The amount of Ca2+ released from the SR determines the Ca2+ transient amplitude which correlates well with strength of contraction.21
Figure 3.

Excitation–contraction coupling and control of Ca2+ transients in ventricular myocytes. Action potential stimulates intracellular Ca2+ influx (ICa,L) via voltage-gated L-type calcium channels. Ryanodine receptors (RyR) in the sarcoplasmic reticulum (SR) are activated by the local increases in cytosolic Ca2+ releasing the SR’s stored Ca2+, a process called calcium-induced calcium release. Ca2+ binds to troponin C, and the myofilaments contract. Ca2+ is sequestered into the SR via the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA), and to the extracellular space via the Na+/Ca2+ exchanger (NCX). Reproduced with permission ref (17). Copyright 2002 Springer Nature Limited.
Intracellular Calcium Handling in Healthy Cardiomyocytes
ECC is a delicate balance of highly orchestrated intracellular signaling pathways converting electrical activation into mechanical force. Ca2+ is the ubiquitous second messenger pertaining to electrophysiology and mediating contraction. It is becoming clear that abnormalities in Ca2+ homeostasis play a key role in the sine qua non of systolic HF, reduced contraction, and the generation of arrhythmias from pathological diastolic SR Ca2+ release.17,21,22
In normal physiology, cellular Ca2+ must remain in a steady state between contraction and relaxation. This is attained by cycling between the cytoplasm and SR.18 In systole, intracellular Ca2+ must rise significantly from resting level, achieved by the RyR2 and LTCC. In relaxation, prior to the next AP, cytosolic [Ca2+] must return to baseline and SR Ca2+ content must be replenished, achieved by a combination of (i) sequestration into the SR via sarcoplasmic reticulum Ca2+-ATPase (SERCA2a) and (ii) removal from the cytosol by sarcolemmal Na+/Ca2+ exchanger (NCX) which is equivalent to the small amount of Ca2+ that entered through LTCC.17,18 The co-localization of these Ca2+ extrusion pathways allows them to interact and compete for Ca2+ within a specialized microenvironment.23
Cardiac Ryanodine Receptor Type 2
RyR2 is a 2.2 MDa homotetramer which comprises four 560 kDa subunits extending from the SR lumen to the cytosol.24,25 It is the principle cardiac SR Ca2+ release channel. Three isoforms of RyR are expressed in striated muscle: RyR1 predominates in skeletal muscle, RyR2 predominates in cardiomyocytes, and RyR3 predominates in developing skeletal muscle. Peng et al.25 has illustrated the domain organization at a resolution of 3.8 Å (Figure 4). The primary structure includes a large cytoplasmic region, six transmembrane regions, and a small SR luminal region. Under physiological conditions, RyR2 activation is primarily regulated by Ca2+. However, RyR2 also acts as a scaffold for regulatory subunits to assemble upon, forming the SR Ca2+ release complex. Consequently, RyR2 regulation is dynamic, influenced by the integrated effects of functional ligand binding.25,26
Figure 4.

Cryo-EM structure of RyR2. (A) Domain organization of a RyR2 depicting 8 of the cytoplasmic structural domains attached to the channel domain–the N-terminal domain (NTD), the P1 domain, the three SPRY domains (SPRY 1, 2, and 3), the Jsol/handle (handle), the helical/Bsol domain (helical), and the central (central) domain. (B) Structure of the RyR2 in a closed formation at a resolution of 4.2 Å. The tetrameric structure reveals the large cytoplasmic region and domain colors are the same scheme as in panel A. (C) Structural comparison of RyR2 (yellow) with rRyR1 and the position of FKBP12 binding. Reproduced with permission from ref (25). Copyright 2016 American Association for the Advancement of Science.
Calcium
Regulation of RyR2 gating by intracellular Ca2+ is central to the controlled release of SR Ca2+. The RyR2 open probability (Po) has a steep dependence on cytoplasmic [Ca2+], with Hill coefficients between 2 and 4.27 At low levels of cytoplasmic [Ca2+] (∼100–200 nM), RyR2 channels are inactive, with a low Po. Ca2+ influx, through LTCC, increase cytoplasmic [Ca2+] within the diadic cleft to submicromolar concentrations.9 Ca2+ binds RyR2 at high-affinity activation sites (A-site).28 This facilitates SR Ca2+ release, resulting in local positive feedback, CICR. The Po reaches a nadir as cytoplasmic [Ca2+] reaches 10 μM. Any further increase in [Ca2+] will result in a decreasing Po, suggesting that Ca2+ binds to low-affinity in-activation sites.29
In total, four Ca2+ regulation sites have been proposed: two activation sites located on cytoplasmic (A-site) and luminal (L-site) domains and two cytoplasmic inhibitory sites (I1 and I2).27 Des George et al. used cryo-EM reconstruction to localize the A-site Ca2+-binding site at the interdomain interfaces of the RyR1 central domain and C-terminal domains.30 Evidence for luminal activation has proved challenging. While it has been recognized that increased SR [Ca2+] can trigger Ca2+ release, it was not until 1994 that Sitsapesan and Williams observed direct luminal activation.31 Laver et al. have proposed both direct luminal activation and a secondary but stabilizing feed-through mechanism.32 As SR Ca2+ content increases, Ca2+ binds specific luminal activation sites (L-site). The subsequent passage of Ca2+ accesses and binds the cytoplasmic A-site, stabilizing the open state. RyR2 can be inactivated by the I1 (low-affinity) I2 (high-affinity) sites. The I1 inactivation site helps produce the bell-shaped curve of activation and inactivation to cytoplasmic [Ca2+] at micro- and millimolar concentrations, respectively.28,32 Magnesium (Mg2+) can act as a competitive antagonist of Ca2+ at both the A- and I1-sites, directly inhibiting RyR2 activation and increasing the required Ca2+ activation concentration.33
Adenine Nucleotides
Adenosine-derived nucleosides, especially ATP, are physiological regulators of RyR2. They enable greater increases in channel activity relative to smaller changes in cytosolic Ca2+. The degree to which they increase Po is dependent on the number of phosphates present on the ribose ring. ATP produces the greatest increase in Po, and AMP produces the smallest increase.30 Ca2+ and ATP act synergistically, with maximal Po occurring when both are present.24 Mg2+ may also form a complex with ATP: MgATP, the predominate form of biologically active ATP. As previously described, increasing [Mg2+] inhibits RyR2 but it may also alter the ratio ATP/MgATP in favor of MgATP, decreasing RyR2 activity.24 What remains unclear is whether ATP or MgATP regulates activity.
FKBP12 and FKBP12.6
Calstabin 1 and 2, also known as FKBP12 and FKBP12.6, are two Ca2+-channel-binding and stabilizing proteins. FKBP12 and FKBP12.6 preferentially bind RyR1 and RyR2, respectively, interacting with a hydrophobic cluster in the SPRY1 domain and junctional solenoid, stabilizing the link between the cytoplasmic region and the pore of RyR2 in a closed conformation.16,30 This prevents Ca2+ leak and regulates the dynamic interaction of RyRs, promoting coupled gating and improving the efficiency of ECC.34,35 Genetic deletion of FKBP12.6 or pharmacological removal with either FK506 or rapamycin alters the biophysical properties of RyR2 and induces significant subconductance openings.35 In these models the resultant increase in SR Ca2+ release has been associated with ventricular arrhythmias.34
Calmodulin (CaM)
CaM is a 16.7 kDa cytosolic Ca2+-binding protein that regulates RyR2 by direct binding. CaM binds RyR2 stoichiometrically, 4 CaMs per tetrameric RyR2, at high affinity subunits (RyR2 amino acids 3581–3610).36 CaM inhibits RyR2 opening at all [Ca2+]. In diastole, it is reported that CaM stabilizes and maintains RyR2 in the closed state, helping terminate Ca2+ release.37 Mutations in the highly conserved CaM-binding region severely reduce and prevent CaM–RyR2 binding.36 In animal models this has been associated with severe hypertrophic cardiomyopathy and catecholaminergic polymorphic ventricular tachycardia (CPVT).38 In nonischemic HF, CaM–RyR2 binding affinity is reduced, contributing to the increased diastolic Ca2+ leak, like mice carrying the human CPVT mutations (R2474S). Oxidative stress, which will be discussed in detail later, has been the proposed mechanism for reducing the binding affinity of CaM.39 LTCCs are another important binding partner for CaM. Ca2+ that enters via LTCCs binds CaM which in turn, allows it to interact with the LTCC to increase Ca2+ entry to initiate ECC.21
Triadin, Junctin, and Calsequestrin
A complex consisting of triadin, junctin, and calsequestrin (CSQ) act as a Ca2+ sensor on the RyR2 luminal side. They participate in intra-SR Ca2+ buffering and modulate release. Triadin and junctin are membrane-bound proteins that interact directly with RyRs, but also serve as connecting proteins, associating with CSQ.24 CSQ is a low affinity, high-capacity Ca2+ storage protein which dissociates from RyR2 as luminal Ca2+ increases, increasing RyR2 Po and normalizing SR Ca2+ content. Cardiomyopathies and arrhythmic pathologies associated with an imbalance in Ca2+ handling have been linked to a deficiency in SR luminal CSQ and a lack of, or overexpression of triadin and junctin.24,40
L-Type Ca2+ Channel
LTCCs are responsible for the voltage dependent inward ICa,L transient responsible for the plateau phase of an AP and triggering CICR. The maximal amplitude of the ICa,L is delayed due to the channels time dependent nature, and product of high channel conductance with low driving force (Em – ECa).41 LTCC termination is primarily mediated by cytosolic Ca2+-dependent inactivation via CaM. As local [Ca2+] increases from ICa,L and SR release, Ca2+ binds CaM which binds to the carboxy terminus of the LTCC.42 This local negative feedback highlights the importance of the co-location of key calcium handling proteins and how local [Ca2+] can derive an effect over global cellular Ca2+ content. LTCC are also regulated by protein kinase A (PKA) and Ca2+/calmodulin-dependent protein kinase II (CaMKII). They increase ICa,L amplitude and sensitivity, with activation occurring at more negative Em.41
Sarcoplasmic Reticulum Ca2+-ATPase
Removal of cytosolic Ca2+ and restoration of the SR Ca2+ reservoir is primarily achieved by SERCA2a. SERCA2a is crucial for diastolic Ca2+ cycling, promoting efficient muscle relaxation and, by restoring the SR Ca2+reservoir, it has an important inotropic contribution for the following contraction. Schultz et al. developed SERCA2a knockout mice to understand its role in Ca2+ homeostasis and cardiac contractility.43 Homozygous null (SERCA2a–/–) mice died early in development, while heterozygous (SERCA2a–/+) mice, unlike the wild-type (WT) mice, rapidly developed cardiomyopathy when the heart was stressed by pressure overload. In contrast, overexpression was well-tolerated, leading to increased SR Ca2+ restoration, increasing contractile strength and improving lusitropy (relaxation). After myosin ATPase, SERCA2a is the largest consumer of ATP, and studies suggest it is the ATPase most sensitive to a reduction in supply of ATP.44 HF induces a state of increased ATP demand from reduced pump function and increased sympathetic activity which may affect ECC coupling and contractile function.45 SERCA2a activity is regulated by two molecules: phospholamban (PLN) and sarcolipin (SLN). PLN is a phosphoprotein that regulates SERCA2a pump activity by changing its affinity for Ca2+ depending on cytosolic [Ca2+].45 PLN can be phosphorylated by PKA and CaMKII at Ser-16 and Thr-17, respectively, after activation of either β1-AR or β2-ARs,46,47 thus releasing SERCA2a from PLN inhibition. Preventing phosphorylation at Ser-16 increases PLN affinity, suppressing SERCA2a function and is correlated with reduced Ca2+ uptake and cardiac contractility.48 Sarcolipin, when coexpressed with SERCA2a, decreases Ca2+ affinity, inhibiting the SERCA2a pump.45
Na+/Ca2+ Exchanger
Cardiac NCX is located primarily at the T tubule with recent high-resolution imaging suggesting partial co-localization with RyR2.49 NCX is the main effector of cardiac myocyte Ca2+ extrusion. It exchanges Ca2+ with a Na+/Ca2+ stoichiometry of 3:1 producing an ionic charge from a gain of one net charge. Under the control of local Em and intracellular and extracellular [Na+] and [Ca2+], NCX can both extrude Ca2+ and work in reverse for Ca2+ influx. The initial upstroke of the AP increases the Em such that the NCX favors Ca2+ influx. As the RyR2 is activated to release Ca2+ there is a local rise in intracellular [Ca2+] versus global intracellular [Ca2+], causing NCX Ca2+ efflux. As repolarization proceeds, Ca2+ extrusion continues to be thermodynamically favored. The intracellular and SR Ca2+ content can be raised by modest increases in [Na+]i, inhibiting Ca2+ extrusion. While an increased SR Ca2+ content can increase contractile strength, conditions of Ca2+ overload may promote spontaneous Ca2+ release and formation of Ca2+ waves. This Ca2+ current may activate NCX in its forward-mode, extruding Ca2+. This generates a depolarizing current termed a delayed after depolarization (DAD), which if large enough can produce a triggered beat by activating the rapid Na+ inward current.50,51
Post-Translational Modifications
At basal resting conditions when Ca2+ influx remains constant, efflux must remain unchanged within cardiomyocytes to ensure a steady state.18 The channels, pumps, and transporters discussed are regulated by multiple ligands and complex signaling cascades. They modulate the activity and amplitude of Ca2+ transients in response to changing demands for cardiac output. A further level of control is provided by post-translational modifications which target vulnerable residues on both ion channels and associated ligands. Alterations in sympathetic tone and redox balance drive phosphorylation and thiol modifications, two post-translational modifications which are highly regulated in the normal heart, fine-tuning Ca2+ handling. Unfortunately, in pathological states, such as HF, this balance can be lost resulting in deleterious Ca2+ handling.
β-Adrenoceptor Signaling
Sympathomimetic stimulation of the β-AR is the primary neurohormonal mechanism which augments the force and frequency of cardiac contraction. In exercise, cardiac output is increased nearly 5-fold within seconds.52 At least two cardio-stimulatory β-ARs are expressed in cardiomyocytes and the sino atrioventricular conducting system of human heart.53−55 Activation of β1-AR and β2-AR causes positive inotropic, chronotropic, dromotropic and lusitropic effects.54−57 Both β1-AR (Ser49Gly, Arg389Gly),58 and β2AR (Arg16Gly, Gln27Glu, and Thr164Ile)59 exist as polymorphisms. Inotropic and lusitropic effects of (−)-noradrenaline through β1-AR and (−)-adrenaline through β2-AR are conserved across all polymorphisms in human right atrium from nonfailing hearts55 despite enhanced β1AR Gsα-protein cyclic AMP–PKA signaling through Arg389-β1AR compared to Gly389-β1AR.58 There is some evidence for a third β-AR, β3-AR, which has been reported to mediate cardio-depressant effects in human ventricle60 and cardiostimulant effects in human atrium,61 but questions remain about its functional significance in human heart.62−64
In the human heart, activation of β1-AR and β2-AR by endogenous agonists (−)-noradrenaline and (−)-adrenaline results in coupling to the Gαs-protein–adenylyl cyclase–cAMP–protein kinase pathway.46,47,55 The intracellular elevation of cAMP activates cAMP-dependent protein kinase (PKA). PKA-catalyzed phosphorylation regulates pivotal proteins responsible for ECC. β1-AR- and β2-AR-mediated lusitropic effects result from phosphorylation of PLN and TnI.46,47,55 Phosphorylation of PLN relieves its inhibition of SERCA2a, increasing the rate of Ca2+ uptake into the SR, accelerating the decline in cytosolic [Ca2+]. Phosphorylation of TnI reduces the affinity of Ca2+ for TnC accelerating myofilament relaxation.55 Together these two mechanisms improve diastolic performance. The inotropic effect is achieved from greater availability of Ca2+. By enhancing SERCA2a activity (via PLN phosphorylation), there is improved diastolic restoration of SR Ca2+, improving its availability for ECC. Furthermore, phosphorylation of LTCCs increases inward Ca2+ influx.17,18,52 A new steady state is achieved from increased cellular Ca2+ and greater amplitude of Ca2+ transients, increasing contraction and reducing time to peak force55 (Figure 5).
Figure 5.

Post-translational phosphorylative signaling. PKA is the main downstream effector of β-AR signaling. CaMKII responds primarily to the local Ca2+ environment. Phosphorylation of RyR2 by PKA and CAMKII increases the Po. Current evidence suggests serine 2808 is the primary target of PKA although evidence also exists for CaMKII. Serine 2814 is a CaMKII phosphor-specific site. PLN is phosphorylated by both PKA and CaMKII, releasing SERCA2a from PLN inhibition, increasing Ca2+ uptake and contractile strength. Both PKA and CaMKII can phosphorylate the LTCC increasing the inward Ca2+ current. PKA-dependent phosphorylation of both TnI and cardiac myosin-binding protein-C (cMyBP-C) exert lusitropic effects, accelerating relaxation.
β1-AR- and β2-AR-mediated inotropic and lusitropic effects in human heart are regulated by chronic administration of β-blockers to patients with coronary artery disease or HF.55,57 Increased inotropic and lusitropic effects through activation of β1-AR and β2-AR were observed following atenolol or metoprolol. However, chronic administration of carvedilol was notable for causing reduced potency of (−)-adrenaline at β2-ARs with smaller reductions for (−)-noradrenaline at β1-ARs.65
The β1-AR can be activated by β-blockers such as (−)-pindolol and (−)-CGP 12177 at higher concentrations (∼2–3 log units) than those required to block β1-AR and β2-ARs.66 (−)-Pindolol and (−)-CGP 12177 cause positive inotropic and lusitropic effects in the human heart which are resistant to the blockade of β1- and β2-ARs by (−)-propranolol at concentrations (200 nM)67,68 that effectively block the inotropic effects of (−)-noradrenaline through β1-AR and (−)-adrenaline through β2-AR.69 These studies indicate β1-AR has two binding sites, one that is sensitive to blockade by (−)-propranolol (activated by (−)-noradrenaline) and another which is relatively insensitive (activated by (−)-pindolol and (−)-CGP 12177). Activation of both sites in the human heart results in β1-AR coupling to the Gsα–cAMP–PKA pathway.68
Redox Signaling
ROS and RNS are highly reactive oxygen-/nitrogen-containing molecules. They are kept in strict balance, as low levels are required for site-specific protein modifications and normal intracellular signaling. ROS are generated by xanthine oxidase (XO) and nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOX) and are byproducts of mitochondrial oxidative phosphorylation.70 The electron transport chain leaks electrons which combine with oxygens forming superoxide anions (O2−•). Superoxide dismutase (SOD) converts O2−• into hydrogen peroxide which may react with transition metals to form hydroxyl radicals (OH·). XO is involved in the purine degradation pathway and produces electrons which transfer to oxygen generating hydrogen peroxide. The enzyme NOX was originally described in neutrophils producing superoxide as a byproduct. In cardiac cells they produce lower levels of ROS. Nitric oxide synthase (NOS) generates nitric oxide (NO·) and RNS. Uncoupled NOS can also produce O2−• which combines with NO· to produce the powerful oxidant peroxynitrate (ONOO·).70 Preventing the uncontrolled production of ROS/RNS are glutathione peroxidase and SOD. Reduced glutathione (GSH) is the major cytosolic ROS scavenger transforming to its oxidized state, GSSG. Glutathione reductase reduces GSSG to GSH and is crucial to maintaining the redox buffer system protecting the cell from ROS-induced damage. NADPH is required for thioredoxin, another ROS scavenger, and helps maintain the ratio of reduced to oxidized glutathione. Oxidative stress occurs when there is excess ROS produced relative to the antioxidant buffer systems. Nitrosative stress results from impaired NO· signaling, increasing RNS. ROS and RNS are tightly coupled within biological systems, working together to modulate signaling, known as the nitro-redox balance. This relationship is complex as both species can modify the same thiols, and there is cross talk between the enzymes that produce them. NO· deficiency can result in increased ROS production. Perturbation of this system can result in impaired contractility and the modification of proteins involved in ECC, a hallmark of HF.71
Heart Failure and Calcium Balance
Failing myocytes display dysfunctional intracellular Ca2+ homeostasis evidenced by fragmentation of the normally synchronized Ca2+ release with spontaneous diastolic Ca2+ leak, associated with contractile dysfunction and arrhythmogenesis (Figure 6). HF studies have consistently shown reduced SERCA2a expression and upregulation of NCX resulting in reduced SR Ca2+ content.22,72 Pogwizd et al. induced HF in rabbits by aortic insufficiency.22 They observed enhanced inward NCX current and a 2-fold increase in NCX mRNA. SERCA2a expression was unchanged but function was reduced by ∼24%. They concluded NCX must compete better with SERCA2a in diastole, opposing restoration of the SR Ca2+ content. This is consistent with findings in human failing hearts which revealed ∼25% reduction in SERCA2a expression and ∼2-fold increase in NCX contributing to systolic dysfunction.72
Figure 6.

Altered calcium handling in HF, a cause of contractile dysfunction and arrhythmogenesis. Increased NCX and reduced SERCA2a expression alters cytosolic Ca2+ cycling reducing SR Ca2+ content, made worse by the increased SR Ca2+ leak. Decreased Ca2+ transients reduce myofilament activation and impair contraction. Enhanced NCX, reduced IKi, and residual β-AR responsiveness contribute to arrhythmogenesis through triggered activity (DADs). Source: Reproduced with permission from ref (22). Copyright 2001 American Heart Association.
Physiologically, CICR controls the release of Ca2+ via the RyR2. The magnitude and frequency of the Ca2+ transient is affected by the Po of RyR2 and SR Ca2+ content, with low levels of Ca2+ release occurring when content is reduced. Under conditions of SR Ca2+ overload the RyR2 is activated independent of cytosolic [Ca2+]. This spontaneous release is known as store-overload-induced Ca2+ release (SOICR) and is one of the proposed mechanisms behind the initiation of DADs leading to arrhythmia.73 Fatal ventricular arrhythmias in nonischemic HF patients result from triggered activity, termed early after depolarizations (EADs) and DADs.74 EADs occur in the late plateau or repolarization phase (phase 3) from decreased outward K+ currents. DADs predominate in the HF population and are oscillations of the transmembrane potential in phase 4 of a preceding AP from Ca2+.9 An initial understanding of the role of Ca2+ came from arrhythmogenic studies of digitalis glycosides. Dogs perfused with an increasing concentration of ouabain produced ventricular ectopic beats. These correlated with DADs, isolated from individual Purkinje fibers, and voltage clamp techniques linked a “transient inward current” in repolarization to a rise of intracellular [Ca2+].75 SOICR occurs when the SR Ca2+ content exceeds a threshold level either in conditions that favor cytosolic and cellular Ca2+ overload or a reduction in the threshold value required to trigger spontaneous release, largely dependent on the properties of the RyR2. However, in HF one would expect less spontaneous Ca2+ release as the SR Ca2+ content is reduced. How can this paradox be resolved?
Stress-Induced Mechanisms Underlying Arrhythmia
Failing myocytes display a greater amount of SR Ca2+ leak for any given SR Ca2+ content. This suggests the threshold content for SOICR to be reduced, with the RyR2 being more sensitive to cytosolic and SR [Ca2+]. However, it may be that RyR2 is dysfunctional, remaining open independent of SR [Ca2+] content. Advanced HF fosters an environment of chronic oxidative stress and elevated sympathetic tone altering the delicate balance of post-translational modifications fine-tuning Ca2+ handling. Although HF demonstrates reduction in β1-AR density53 and reduced β1-AR responses,53,55 there remains residual β-AR responsiveness46 which activates PKA and neighboring components of Ca2+ signaling, elevating local SR Ca2+ content and increasing RyR2 Po. Oxidative stress has been implicated in abnormal RyR2 activity and increased SR Ca2+ leak in many cardiac pathologies including myocardial infarction and HF.39,70 Under normal physiological conditions the RyR2 channel exists in a stable closed, zipped conformation, preventing deleterious Ca2+ leak. The stressed environment of HF can uncouple the delicately balanced post-translational modifications, increasing diastolic RyR2 Po and creating a “leaky” phenotype. The HF environment is one of arrhythmia generation and the dysfunctional RyR2, its catalyst.76
Phosphorylation
Phosphorylation can be performed by several kinases, but pertinent to RyR2 and Ca2+ signaling are CaMKII and PKA. CaMKII responds primarily to the local Ca2+ environment, while PKA provides a systemic effect, being the main downstream effector of β-AR signaling. Present on the cytoplasmic domain of RyR2 are several residues which may undergo phosphorylation. Although it is still under debate, three main functional sites have been identified. Two are PKA-specific sites, serine (S) 2030 and S2808, and one is a CaMKII-specific site, S2814.70
Serine 2808
Controversy exists as to whether S2808 is the primary substrate of PKA, CaMKII, or both, as experiments of phosphospecific antibodies produced mixed results. In 2000, Marx et al. reported PKA hyperphosphorylation was associated with dissociation of FKBP12.6 from the channel complex, increasing RyR2 Ca2+ sensitivity for activation, elevating the Po.77 Their study had access to human failing hearts, which similarly exhibited PKA hyperphosphorylation and reduced FKBP12.6 binding with subsequent increased channel Po. Extensive investigations by Wehrens et al., first utilizing immunoblotting with phopho-epitope-specific antibodies,78,79 and then genetically altered mice in which Ser2808 was replaced by alanine, identified Ser2808 as the functionally important PKA site.80 In RyR2-S2808A mice, β-AR activation was unable to induce PKA phosphorylation of RyR2 and deplete FKBP12.6 from the channel complex. Their mouse models used an ischemic mechanism for deleterious remodeling and induction of HF. Notably, disruption of RyR2 hyperphosphorylation protected against progressive cardiac dysfunction following ischemia. However, this hypothesis is not universally accepted as a number of groups have been unable to confirm the fundamental aspects of this hypothesis. MacDonnell and colleagues, using the same RyR2-S2808A mouse model, were unable to detect PKA-mediated S2808 phosphorylation or an increase in SR Ca2+ leak following activation of β-AR with isoprenaline compared to WT.81 The reasons for these fundamentally different results remain to be understood. While it seems indisputable that S2808 is indeed phosphorylated, its functional effect remains to be agreed upon. This is made more complex by “how much” phosphorylation is required. Under basal conditions, without β-AR stimulation or PKA activation, S2808 has been shown to be constitutively phosphorylated at high levels (>50% of maximum).82 This raises the question of whether the observed PKA phosphorylation in HF is pathological. Indeed some groups have also failed to detect S2808 phosphorylation in HF,83 and another group observed that both minimal and maximal S2808 phosphorylation achieved RyR2 activation, suggesting a “V” shaped curve of dependence.84 Despite the extensive body of work by Marks’ group,77,79,80 it remains a concern that groups using similar models cannot replicate and validate their results. Further studies are required to understand the role of PKA S2808 phosphorylation and its functional effect on RyR2 Po.
Serine 2814
Wehrens et al. demonstrated that CaMKII specifically phosphorylates S2814.79 The implications of this were investigated with knock-in mice, RyR2-2814 (S2814D), mimicking constitutive phosphorylation. Increases in Ca2+ transients were similar between S2814D mice and WT mice with CaMKII activation, suggesting S2814 was the principle site of CaMKII-mediated phosphorylation. This was confirmed by the inhibition of CaMKII with KN-93, preventing phosphorylation in WT and reducing Ca2+ transients, but this had no effect in S2814D mice. The alterations in Ca2+ signaling were associated with increased Ca2+ diastolic leak, ventricular arrhythmias, and the development of HF.85 Uchinoumi et al. sought to uncover the mechanism for this increased Ca2+ leak.86 They used fluorescence resonance energy transfer (FRET) to measure RyR2-bound CaM and binding of fluoroescent-DPc10 (F-DPc10) peptide to prove conformational change. DPc10 is in the RyR2 CPVT mutational hotspot. It is linked to domain unzipping and pathological Ca2+ leak observed in CPVT and HF, which dantrolene can reverse.87 WT mice activated by CaMKII and phophomimetic RyR2-S2814D knock-in mice displayed increased diastolic Ca2+ leak, reduced CaM–RyR2 affinity, and an unzipped (pathological) RyR2 conformational change as evidenced by increased F-DPc10 access. These same observations were revealed in myocytes of rabbits with HF, induced by a nonischemic model, which had elevated CaMKII activity and S2814 phosphorylation. This suggests HF can activate CaMKII to phosphorylate and induce a common pathological (unzipped) conformational change in RyR2, characterized by reduced CaM–RyR2 affinity and increased diastolic Ca2+ leak.86 This conformational change was overcome by dantrolene which binds amino acids 601–620 of RyR2, stabilizing the channel gating in a closed state. The effect of β-AR on CaMKII requires further understanding, but in transgenic mice, with ablation of S2814, spontaneous Ca2+ waves were reduced, and they were protected against catecholaminergic-induced arrhythmias.82 The effect of CaMKII phosphorylation of S2814 has produced more consistent and reproducible observations than S2808, but it remains debated as to which one is more functionally important.
Serine 2030
Serine 2030 has been championed by Xiao et al.88 Using site-phosphor-specific antibodies, they observed S2030, not S2808, was the major PKA site responding to β-AR stimulation. At basal conditions, before β-AR stimulation, S2030 was not phosphorylated, while S2808 was. They also observed increased spontaneous Ca2+ waves suggesting phosphorylation of S2030 was the critical event for stress-induced arrhythmia. In resting conditions, S2030 appears to be unphosphorylated, however we have not defined its role in Ca2+ signaling in healthy cardiomyocytes, let alone HF.26
Multi-Phosphosite Model
Our current models on phosphorylation are simplistic with most evidence supporting activation of RyR2 from PKA phosphorylation of S2808, dissociating FKBP12.6 from the channel complex and CaMKII by phosphorylation of S2814. This model does not consider the effect of S2030, nor does it recognize other undiscovered RyR2 phosphorylation sites.82 They have also been discussed as mutually exclusive events, but it is perfectly conceivable that this is not absolute. Structural models reveal S2808 and S2814 to be co-located on the “phosphorylation hotspot”, a prominent and exposed epitope of the helical domain.82 Phosphorylation maintains a more flexible configuration, favoring an open state conducive with diastolic Ca2+ leak. This provides an interesting question about the current models. Are the effects observed as simple as one kinase phosphorylating one site producing one effect? The sites in the phosphorylation hotspot may indeed have the same end effect, due to their location, but they may also provide redundancy and a graded response dependent on sequential phosphorylation. The integrated effect of S2030 may explain the distinct effects of CaMKII and PKA on the phosphorylation hotspot, providing a “multiphosphosite model” (Figure 7). Of course, there may be other undiscovered sites which may affect these mechanisms. While recognizing these weaknesses in the models, there are also differences in experimental conditions which may explain divergent results. RyR2 function is dependent on SR Ca2+ content and local Ca2+ transients. No one unified method has been utilized to control this, which complicates the interpretation of phosphorylation effects. Second, in intact organelle systems, there are competing intracellular signaling cascades and protective mechanisms reversing phosphorylation to mitigate their effects. β-AR stimulation affects multiple signaling cascades confounding the interpretation of specific β-AR activated PKA events. LTCC can undergo phosphorylation by both CaMKII and PKA, increasing the ICa,L which may activate RyR2, and as previously illustrated, SERCA2a activity is amplified by PLN phosphorylation which increases following β-AR stimulation.
Figure 7.

RyR2 phosphorylation sites. Depiction of the three phosphorylation sites targeted by several kinases on RyR2. S2808 and S2814 are located in the “phosphorylation hotspot”, an exposed protruding epitope on RyR2. There may still be sites to be discovered here (i.e., S2811). S2030 is shown as separate from the hotspot. These sites may work independently or together by differential regulation of PKA and CaMKII. The effect on channel gating may be direct or indirect via interaction with classical Ca2+ activation sites. Reproduced with permission from ref (82). Copyright 2014 Camors et al.
This review focuses on cardiac stress induced by phosphorylation from chronic β-AR stimulation, thiol modifications from oxidative stress, and how they modulate the RyR2 to produce an arrhythmogenic phenotype. We note that there is evidence that RyR2 phosphorylation is modulated by PKG89 and through activation of muscarinic (M2 and M3) receptors.90 In murine ventricular myocytes and canine failing ventricular myocytes, the nonselective muscarinic receptor agonist carbachol increased phosphorylation of S2808 but reduced phosphorylation of S2814. These effects were attributed to to M2-PKG phosphorylation of S2808 and M3-mediated reduction in reactive oxygen species which in turn reduced CaMKII phosphorylation of S2814.90 The net effect was that S2808 phosphorylation increased fractional Ca2+ release from SR and that S2814 dephosphorylation mediated decrease in diastolic Ca2+ leak.90
Redox Balance
Oxidative stress has been observed in the myocardium and plasma of patients with HF. Oxidative stress describes a pathologically mediated redox imbalance, consequential to a diminished antioxidant pool caused by consumption and reduced generation.76 There are several sources of ROS/RNS in HF (mitochondria, NADPH oxidase, xanthine oxidase, and uncoupled NOS), but their relative contributions remain unknown. Kohlhass et al. demonstrated how disturbed Ca2+ and Na+ handling affects mitochondrial ROS generation. Failing myocytes consume increasing amounts of ATP. There is an increase in electron leak from the electron transport chain of mitochondria which either gets oxidized by NADPH or forms O2−•. In order to maintain homeostasis, an increase in NADPH production is required. However, reduced cytosolic Ca2+ transients and increased intracellular [Na+] in HF results in mitochondrial Ca2+ efflux via mitochondrial NCX. Reduced mitochondrial Ca2+ hampers the activation of Kreb cycle dehydrogenases, hampering regeneration of NADPH. Consequently, failing myocytes display pronounced oxidation of NADPH to NADP+ and increased mitochondrial H2O2, worsening oxidative stress.91 As HF progresses, oxidative stress worsens, perpetuating a deleterious cycle. Ca2+ handling is redox-sensitive. In failing myocytes, ROS have been implicated in pathological SR Ca2+ leak from both direct changes to ion channel activity and indirect by amplifying redox-regulated protein kinase activity.70,76
Redox Regulation of RyR2
The RyR2 is highly redox sensitive, due to the presence of 89 cysteine residues (with thiol side chains) per monomer, with 21 of these found “free” of thiol modification.92 ROS/RNS oxidize vulnerable cysteine residue thiol side chains, producing sulfenic, sulfinic, and sulfonic acids. These groups can then react to produce S-nitrosylation, S-glutathionylation, and disulfide bond formation altering the channels Po (Figure 8).70 Generally, the evidence supports activation of RyR2 following cysteine oxidation, but results have been heterogeneous relating to differences in conditions and experimental models used.
Figure 8.

ROS/RNS induced post-translational modifications. Diagram depicts the chemical relationships between different ROS and RNS and how they impact post-translational modifications. GSNO: S-nitrosoglutathione. GSSG: oxidized glutathione.
Initial models with rat cardiomyocytes revealed concentrations of 1 μM H2O2 had no effect on cytosolic Ca2+, while 100 μM H2O2 caused a marked and sustained increase in cytosolic Ca2+.93 Guinea pig models showed a biphasic response with an initial increase followed by suppression of cellular Ca2+ at 5 min to 1 mM H2O2.94 Jones et al. explored this relationship, observing that oxidation caused a concentration and time-dependent effect on the threshold for SOICR.95 Following an initial reduction in threshold for SOICR, prolonged exposure increased the threshold, represented by an abrupt loss in SOICR events. They exposed HEK293 cells expressing RyR2 to supraphysiological amounts of H2O2 (0.01–1 mM). Molecular models suggest thiol groups present in the channel pore and transmembrane domains have different threshold levels for oxidation, with less reactive thiols inducing a delayed inhibition of RyR2, explaining this biphasic response.95 Importantly, the supraphysiological H2O2 levels in these experimental models are unlikely to represent the lower levels of pathological oxidative stress induced by HF. This would account for the decrease in release threshold observed in HF models, representing milder oxidizing conditions. In canine models of tachycardia-paced-mediated HF, single RyR2 channel activity was significantly higher than control. They observed a rightward shift in the ratio of reduced and oxidized glutathione which correlated with a reduction in free thiols on RyR2, consistent with redox imbalance. The increased Po resulted from an increase in sensitivity of RyR2 to SR luminal Ca2+, linking redox modifications to reduced threshold for SOICR to occur. This effect was partially reversed in the presence of reducing agents, while oxidation of controls reproduced the HF phenotype and luminal Ca2+ sensitivity.96 This incomplete reversal could be explained by permanent oxidative modifications or synergistic post-translational effects like phosphorylation. Lastly, RyR2 exists in a basal oxidized state, the degree of which is unknown. Using an artificial bilayer model, Hanna et al. noticed that RyR2 requires mild oxidizing conditions to mimic the healthy in situ state.97 They observed that in reduced conditions, the Po of RyR2 did not increase with rising luminal [Ca2+], as would be expected in healthy myocardium, but was corrected in mildly oxidizing states. Compared to lipid bilayer models, the junctional microdomain of intact organelles may create an enclosed microenvironment, where an oxidizing redox potential may be maintained by NADPH oxidase NOS2 and not reflect the reduced global environment of the cytoplasm. Unfortunately, it has proven challenging to characterize the sites of modifications. In RyR1, cysteine 3635 is found in the CaM binding site and can undergo functionally significant S-nitrosylation. Despite RyR1 and RyR2 displaying ∼70% homology, the corresponding hyper-reactive cysteines found in RyR2 (cysteine 3602) have revealed inconsistent results with Ca2+ handling. While it did not affect CaM regulation of RyR2, it did suppress SOICR and was an important determinant of the activation and termination of Ca2+ release.98
Taken altogether, redox modifications by ROS consistently cause RyR2 Ca2+ mishandling which are time- and concentration-dependent.70 The cysteine residues that confer significant functional change have yet to be defined. HF induces chronic increases in ROS which may account for the observed increased RyR2 activity. These ex vivo models, using isolated myocytes, have inherent limitations. Exogenous models cause a global increase in ROS; however, free radicals are highly active diffusing only nanometers. This suggests endogenous systems compartmentalize, creating microenvironments where ROS have local effects on closely apposed targets. Furthermore, these models do not fully reflect the dynamic redox environment present in intact organelles, nor do they measure the integrative effect of competing intracellular signaling cascades and modulating proteins that regulate RyR2 activity which may also undergo redox modulation (Figure 9).
Figure 9.

Direct and indirect effects of redox dependent post translation modifications of cardiac RyR2.ROS/RNS induce direct modifications by S-glutathionylation, S-nitrosylation, and disulfide bond formation on RyR2 (solid arrows). Indirect effects are communicated by the activation of PKA and CaMKII and dissociation of the regulatory ligand CaM (dashed arrows). The principal effect is increased RyR2 Po causing pathological SR diastolic Ca2+ leak. Reproduced with permission from ref (70). Copyright 2018 Nikolaienko et al.
Redox Regulation of the RyR2 Junctional Complex
The altered association of accessory proteins within the RyR2 junctional complex may contribute and underpin redox-mediated changes in HF. The binding affinity of CaM to RyR2 is reduced when oxidized by H2O2, increasing diastolic Ca2+ leak. Oda et al. performed an elegant study of permeabilized rat ventricular myocytes.39 H2O2 (50 μM) reduced the content of free thiols on RyR2, decreased CaM–RyR2 binding but did not alter FKBP12.6–RyR2 association. They confirmed increased DPc10 association indicating domain unzipping. When evaluating the relative contributions, 60% was from H2O2 changes on RyR2, and 40% was from effects on CaM, suggesting a combined mechanism for RyR2 domain unzipping. The conformational change was reversed with dantrolene and restored CaM–RyR2 binding. The study controlled for the effects of CaMKII with a direct inhibitor, KN-93. Redox reactions activate CaMKII in a fashion similar to phosphorylation, by oxidation of methionine residues 281/282 found in the regulatory domain, preserving kinase activity. While models of ROS and CaMKII produce phenotypically similar effects of RyR2 activation, the contribution of redox-activated CaMKII is still not understood. H2O2-activated CaMKII can contribute to arrhythmogenesis which is reversed by KN-93 inhibition. Rabbit ventricular myocytes had greater potential for EADs from impairment of late Na+ current inactivation reducing repolarization reserve.99 Finally, PKA is activated independent of cAMP by oxidation. Cysteines found within the regulatory subunits of PKA are oxidized leading to their dissociation.70 In a study examining RyR2 dissociation from FKBP12.6, oxidation was not shown to directly effect RyR2-FKBP12.6 binding; instead, its effect was indirect via phosphorylation.77 In SR vesicles, H2O2 alone or in combination with CaMKII had no effect on FKBP12.6 dissociation from RyR2. However, RyR2 oxidation with PKA caused almost complete FKBP12.6 dissociation. It is important to realize that concurrent hyper-phosphorylation is present in studies examining RyR2 redox modifications and effects were not always evaluated.
These studies highlight the indirect effects of redox modifications on the RyR2 activity. Lipid bilayer experiments using purified RyR2 do not mimic the dynamic and competing signaling cascades present in cellular systems, nor do they recreate the microenvironment of the couplon. Key Ca2+ and Na+ channels that are sensitive to redox regulation contribute to the local ionic environment affecting RyR2 Po. Direct ROS effects on RyR2 are most likely a local effect, a consequence of low diffusion capacity, while protein kinases provide a mechanism by which ROS can translate their effect across the cell. These inherent limitations may contribute to the disappointing results obtained when translating findings to an intact organelle system.
Redox Regulation of SERCA2a and NCX
SERCA2a contains 25 cysteine residues, although only 1 or 2 have shown functional importance. By exposing permeabilized myocytes to NADH, Zima et al. found SR Ca2+ was reduced by >50% with associated reduction in SERCA2a activity. This was attenuated by the addition of superoxide dismutase, a reducing agent.100 The mechanism is unclear, but oxidation denatures the SERCA2a ATP-binding site, reducing its activity.101 This inhibitory effect is offset, indirectly, by the phosphorylation of PLN by redox-activated PKA and CaMKII leading to stimulation of SERCA2a activity. The predominant effect of ROS/RNS on SERCA2a is unknown, but likely to be negative, and may depend on the time and concentration of exposure and the predominant kinase that mediate indirect ROS/RNS effects.76 NCX contains cysteine residues in functional significant domains. H2O2 and O2−• have been shown to activate NCX but results have been inconsistent, relating to concentration and source.102 The α1 subunit of the LTCC contains over 10 cysteines which can be the subject of redox modifications. Like SERCA2a, direct oxidation appears to inhibit inward ICa,L, while CaMKII and PKA increase it.76,102
S-Nitrosylation of RyR2
NO· exerts its effects via two mechanisms. First, NO· binds guanylate cyclase stimulating cGMP production which allosterically activates phosphodiesterase-2 which decreases the levels of cAMP reducing PKA activity. Second, NO· may transfer to a nucleophilic thiol group, a process called S-nitrosylation. For this to occur, thiol groups must first be oxidized. The cystine residues involved in NO-mediated RyR2 modulation are not well understood. In skeletal muscle RyR1, S-nitrosylation of Cys3635 activates the channel, and the effect was abolished with the expression of a C3635A mutant.103 The corresponding RyR2 Cys3602 was not required for activation.102 The effect of S-nitrosylation on gating properties is controversial and likely relates to experimental conditions: using endogenous or exogenous NO·, the concentration of NO·, and cytosolic environment. Purified canine RyR2 incorporated into proteoliposomes revealed increased Ca2+ transients with SIN-1, a NO·/ONOO−• donor. As the number of nitrosylated thiol groups increased, progressive activation was noted. The use of dithiothreitol, a reducing agent, reversed the number of NO groups and activation.92 Studies using an endogenous source have utilized NOS1, found in the SR near RyR2, which induces S-nitrosylation. RyR2 of NOS1–/– mice had reduced nitrosylation, but there was also a measurable increase in ROS and concomitant thiol oxidation. Cardiomyocytes displayed reduced SR Ca2+ content and marked diastolic Ca2+ leak, generating proarrhythmic Ca2+ waves.104 The isolated cardiomyocytes were paced at 4 Hz and displayed a HF phenotype with reduced contractility. Wang et al. challenged this view, observing hyponitrosylation and reduced SR Ca2+ content with NOS1 deficiency.105 The RyR2 Po was reduced as was the frequency of SR Ca2+ sparks. Here cardiomyocytes were placed in lipid bilayers and observed under basal conditions. The difference in physiological state (paced vs quiescence) and the additional oxidative effects recorded in the first experiment may explain the discrepancy in results. Elevated ROS are a hallmark of HF, and in normal physiology, the balance in activity of nitric oxide and ROS is tightly coupled, coined the nitroso-redox balance. Cutler et al. set out to clarify this relationship using paced whole-heart studies of Langendorff perfused guinea pigs.106 Reduced NOS1 activity was associated with significant nitroso-redox imbalance. There was reduced S-nitrosylation and increased oxidation of RyR2 with a decrease in RyR2 thiol content. Isolated myocytes exhibited an increase in spontaneous Ca2+ release which, unlike control, were enough to mediate triggered arrhythmic beats. The nitroso-redox balance is tightly controlled in healthy hearts. Physiological levels of S-nitrosylation may be required for normal RyR2 functioning, reducing RyR2 Po. In an environment of oxidative stress, S-nitrosylation may be protective, preventing oxidation of reactive thiols and overactivation.70 However, the experiments by Cutler et al. revealed further insights. First, the arrhythmic events only occurred under conditions of elevated intracellular Ca2+ and were not appreciated under normal intracellular Ca2+ conditions.106 Second, the arrhythmic phenotype was compounded by the addition of H2O2, producing an increase in sustained ventricular arrhythmias of ∼150%, compared to NOS1 inhibition or H2O2 alone. Initially, greater nitroso-redox imbalance was thought to underlie this, but no significant difference in RyR2 oxidative state was appreciated. Instead, there was an increase in peak LTCC current which can further increase intracellular Ca2+. Together this suggests that nitro-redox imbalance may not, on its own, account for arrhythmogenesis but increase the potential, and it is the integrated effects of multiple pathways that together, result in arrhythmogenesis.
Combining the Models
In its simplest form, HF creates a stressed environment promoting pathological diastolic Ca2+ leak and reducing SR lumen [Ca2+] and systolic Ca2+ release, causing reduced contractility. The increased cytosolic [Ca2+] promotes NCX, which HF upregulates, to extrude Ca2+ causing a net sarcolemmal depolarization and DAD generation (Figure 10). One of the fundamental challenges scientists face is translating how an experimental model, usually in its purest form (single cell), performs within the complex dynamics of competing cascade signaling of an intact organism. Unfortunately, many models fail to mimic the combined effects of these post-translational modifications in human cardiomyocytes with HF.
Figure 10.
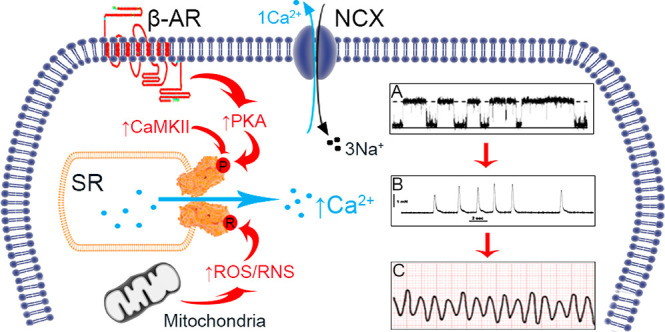
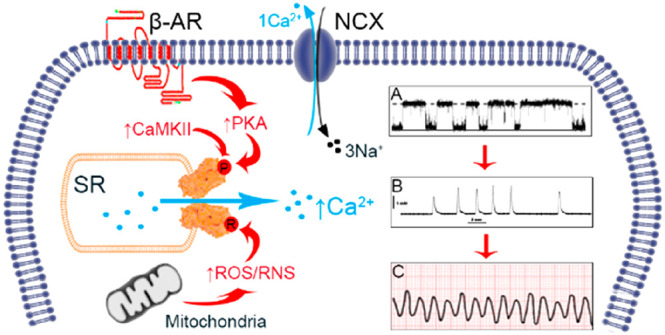
Stress-induced mechanisms of arrythmia. In HF, there is chronic β-AR activity and increased ROS/RNS generation. Vulnerable residues on RyR2 are susceptible to phosphorylation and redox modifications, which increase RyR2 Po (diagram A). In diastole there is pathological SR Ca2+ leak which can activate neighboring NCX, extruding Ca2+ causing sarcolemma depolarization; the generation of DADs and spontaneous contractions (diagram B). Ultimately this may degenerate into ventricular arrythmia (diagram C) and SCD.
HF leads to a progressive destabilization of RyR2 function. Initial phospho-modulating effects predominate, partially driven by β-AR activity, with late thiol oxidative effects in advanced HF. RyR2 exhibits a progressive loss in ability to become refractory to luminal SR [Ca2+], and the potential for spontaneous release occurs at lower luminal Ca2+ content.107 The progression of HF parallels the worsening potential for diastolic Ca2+ leak, Ca2+ waves, and DADs. Walweel et al. investigated these combined effects in an elegant study using healthy and failing human hearts.108 S2808 and S2814 were hyperphosphorylated implicating both CaMKII and PKA pathways, and increased thiol modification was associated with reduced FBP12.6 affinity for RyR2 which, as discussed, may increase channel activity. Two further observations were made. First, phosphorylation and thiol oxidation were synergistic, significantly increasing the Po. Second, within individual hearts there was heterogeneity in RyR2 activity. While normal hearts displayed 95% low-activity RyR2, failing hearts displayed 30%. Approximately 20% of RyR2 were not regulated by intracellular Ca2+; consequently, in diastolic conditions they would be near maximally activated.108 These differences may result from compartmentalization, leading to pockets of cells with higher degrees of post-translational modifications and chronic activation of regulatory proteins PKA and CaMKII. Graded sequential modifications may provide a unifying mechanism for gradual deterioration in contractile function and worsening arrhythmogenic potential.
Another issue scientists face is translating the observed single-cell events to the coordinated and sustained activity of myocytes at a ventricular level. Cutler et al. observed that the arrhythmic events produced by NOS1 inhibition only occurred with increased intracellular Ca2+.106 We now appreciate the negative cardiac remodeling of HF may predispose to abnormal electro-mechanical coupling and electrophysiological dysfunction due to changes in spatial temporal relationships. In rodents, HF disrupted the T tubule architecture, separating it from the SR luminal surface where RyR2 locates.19,109 This was associated with a temporal delay in ECC and, in a large number of CRUs, prevented RyR2 activation via CICR.110 The ultrastructure of the T tubule interface is important as it contains the extracellular space within the T tubule and both the cytosolic and junctional SR subspaces which exert local effects on the constituent ion channels.23 RyR2 molecules that are uncoupled from sarcolemmal Ca2+ channels may amplify pathological Ca2+ release. By having a large population of available CRUs after phase 2 of an AP, a mechanism is provided whereby spontaneous Ca2+ release can produce a Ca2+ wave.110,111 Cardiac resynchronization has been shown to restore T tubule architecture and reduce the burden of arrythmia in HF.112
Heart Failure Medications
Inotropes
A 1986 review article on novel positive inotropic agents predicted the future of therapy for systolic HF.113 They altered cardiac performance by targeting a common principle, altering cardiomyocyte handling of Ca2+. After three decades, only digoxin remains in HF guidelines as a long-term agent, although it is a weak recommendation from evidence suggesting reduction in hospitalizations.114 Digoxin, a cardiac glycoside, inhibits Na+/K+ ATPase increasing cytosolic Na+. This influences the NCX driving force, thermodynamically favoring cytosolic Ca2+ accumulation improving.115 In patients presenting in cardiogenic shock, with impaired end organ perfusion, conventional inotropic agents are recommended.114 Catecholamines are direct β-AR agonists exploiting the Gαs-protein–adenylyl cyclase–cAMP–protein kinase pathway. Milrinone, a phosphodiesterase-3 inhibitor, prevents cAMP hydrolysis increasing PKA activity. Because β-AR density is reduced in HF and many patients are prescribed long-term β-blockers, they were assumed to be more effective, but trials have been neutral with increased arrhythmic burden. Their unifying mechanism is the exploitation of Ca2+ handling, which is a dual-edged sword. Although improving contractile dynamics, catecholamines put further stress on failing cardiomyocytes with detrimental alterations to myocardial energetics and arrhythmic potential by mechanisms discussed. Clinical trials are at best neutral and at worst negative from increased arrhythmic death opposing their use in long-term management.116 Novel agents are now being developed. Omecamtiv mecarbil is a first-in-class direct myosin activator targeting the myocardial machinery independent of Ca2+.117 Alternatively, we are discovering additional properties of current GDMT. The β2-AR-selective (13-fold selective for β2-AR vs β1-AR) inverse antagonist/β-arrestin-biased agonist carvedilol65,118,119 has proven positive effects over other β-blockers for the treatment of HF. Unlike other β-blockers, carvedilol displays an additional antioxidant effect.120
Carvedilol
The additional ROS-scavenging properties of carvedilol may explain its beneficial effect on HF and the prevention of ventricular arrhythmias over other β-blockers. In a canine model of pacing-induced HF, cells incubated with carvedilol had reduced ROS levels and restoration of defective interdomain interactions, promoting a stable zipped state, improved Ca2+ transients, and cell shortening.120 They also exposed SR from normal hearts to SIN-1 leading to reduction of reactive thiols and defective interdomain interaction (unzipping) of the RyR2 with increased SR Ca2+ leakage. Once again, carvedilol restored normal interdomain interaction preventing defective RyR2 Ca2+ handling, thus improving myocyte function. This antioxidant effect occurred at low concentrations (30 nmol/L). Nonetheless, chronic administration of carvedilol was associated with reduced S2808 phosphorylation suggesting β-AR modulation, but the predominant effect was attributed to its antioxidant ability. A similar dose of metoprolol, eliciting an equivalent β-blocking effect, had no beneficial effect on cardiomyocyte function. To explain this, DPc10 was administered, abolishing carvedilols beneficial effects. As DPc10 induces domain unzipping without affecting PKA and FKBP12.6 phosphorylation, this added further weight that the primary action was via reducing oxidative stress. However, the poor translation of clinical effect with antioxidants suggest another mechanism may explain its positive effects in clinical trials. Zhou et al. compared 14 different β-blockers.121 They found that only carvedilol suppressed SOICR by reducing the duration and increasing the frequency of RyR2 channel opening. SOICR can generate DADs, and this attribute may contribute to its favorable antiarrhythmic effects, independent of its antioxidant and β-AR antagonistic effects. However, supraphysiological concentrations (0.3–1 μM) were required to suppress SOICR, while the concentration required for β-blockade is only ∼1 nM. These studies used HEK293 cells, not human failing cardiomyocytes. It is therefore important to systematically assess the efficacy of carvedilol and establish whether the pleotropic effects (β-AR antagonism, ROS scavenging, and direct RyR2 modulation) act alone or in concert, protecting against the generation of malignant arrhythmias in human failing cardiomyocytes. Notably, the pharmacological properties of carvedilol also extend to β-arrestin-biased agonism through occupation of the orthosteric ligand-binding pocket of β2-ARs118,119 to cause G-protein-independent β2-AR phosphorylation, internalization, EGFR transactivation, and ERK1/2 phosphorylation. In mouse cardiomyocytes, despite carvedilol exhibiting β-arrestin-biased agonism through the β2-AR, it does not increase contractility, unlike the selective β2-AR pepducin ICL1-9 which does through a Ca2+-independent mechanism.119 Gαi-protein stabilizes β1-AR orthosteric bound carvedilol in a conformation that favors β-arrestin 1/2 signaling.122 Interestingly, carvedilol does not recruit Gαi-protein to β2-ARs, nor do other tested β-blockers (acebutolol, alprenolol, propranolol, metoprolol, and carazolol) recruit Gαi-protein to β1-AR.122 It is important to understand the significance of carvedilol-induced biased agonism in human heart and whether it confers beneficial outcomes in disease settings of HF and arrhythmogenesis.
Future Medicines Directly Targeting the Ryanodine Receptor
Chronic oxidative stress and phosphorylation have been identified to impart deleterious post-translational modifications of cardiac RyR2. They converge to produce excessive spontaneous diastolic SR Ca2+ leak facilitating arrhythmogenesis via DADs. Intuitively, medicines that target RyR2 to selectively inhibit diastolic SR Ca2+ leak could provide benefit to patients with HF at risk of ventricular arrhythmias.
Some drugs have already been shown to reduce diastolic RyR2 Ca2+ leak. Notably, dantrolene reduces diastolic Ca2+ leak in failing canine,123 rabbit,124 and human heart125 but not corresponding species (canine, rabbit, and human) nonfailing heart.124,125 Despite these promising results, the implementation of dantrolene into clinical practice is limited by adverse effects including hepatotoxicity. More recently, it was shown that another hydantoin compound, phenytoin, inhibits diastolic Ca2+ leak from human failing heart but not nonfailing heart.125 Phenytoin (100 μM) caused 50% reduction in Po of RyR2 channels from ventricle of HF patients with IHD but had no effect on RyR2 channels from nonfailing hearts under diastolic (cytoplasmic Ca2+ 100 nM) conditions.125 In complementary experiments in sheep ventricle, it was shown that both phenytoin and dantrolene reduce mean RyR2 channel open time, increase RyR2 closed time, but have no effect on RyR2 Po at systolic Ca2+ concentrations (Ca2+ 100 μM).125 It is important that therapies do not depress systolic function, especially in the compromised failing heart. Interestingly, phenytoin and dantrolene do not compete for the same binding site on RyR2.125 Furthermore, inhibition by dantrolene but not phenytoin was dependent on the presence of calmodulin.125 These studies suggest phenytoin and dantrolene can stabilize the remodeled RyR2 by restoring the “zipped” configuration of the N-terminal and central domains to maintain the closed state of RyR2 during diastole. At least two separate sites for phenytoin and dantrolene may be targeted with the same effect. Dantrolene binds to domain 601–620 of RyR2,123,126 but the specific phenytoin-binding site is not yet known.125 Phenytoin is currently indicated for epilepsy and status epilepticus. It is estimated the concentrations of phenytoin required to stabilize RyR2 are approximately 1/2 log unit less than those used clinically.125 The repurposing of phenytoin may provide an opportunity to cautiously test RyR2 inhibition in HF patients. Interestingly, phenytoin also has class IB antiarrhythmic properties but has been replaced for clinical use by other medicines with lower toxicity profiles.127 The development of high-throughput screens may expedite the discovery of high-affinity (nM), selective RyR2 diastolic Ca2+ inhibitors128 for testing in the human failing heart.
Gene Therapy
The novel mechanism of gene transfer is being studied by the Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease (CUPID) investigators.129,130 Adeno-associated viruses (AAV) are utilized as vectors to deliver and transfer known genes of interest to cardiomyocytes. SERCA2a is perhaps the first of many to be tested in the clinical realm. In animals, SERCA2a gene transfer resulted in reverse left ventricular remodeling, improved survival, and normalized Ca2+ handling.131 The antiarrhythmic properties may originate from a reduction in Ca2+ alternans which protects from re-entrant excitation by regional AP duration gradients.132 The CUPID 1 trial performed a single intracoronary infusion of recombinant AAV serotype 1 in patients with advanced HF. At 12 months, there were no safety concerns, and gene therapy reduced HF event rates.130 Unfortunately, the phase 2b CUPID 2 trial of 250 HF patients revealed no evidence of improved outcomes.129 This was despite the entry requirements and treatment algorithms being similar. The mechanism of delivery optimizes vector uptake. Intracoronary delivery is practical, while other forms, such as myocardial injection, would be invasive and negatively affect the risk/benefit ratio. Perhaps counterintuitive, an increased ratio of empty capsid particles to AAV viral capsid particles may increase gene transfer as empty capsid particles may act to neutralize antibodies.133 Is affecting one gene enough, especially in the context of advanced HF, whereby, irrespective of the success in gene therapy delivery, it will remain insufficient to alter the trajectory of disease? Importantly, the two CUPID trials have shown SERCA2a gene therapy and its delivery to be safe. Gene therapy remains a promising field for development, and other proteins could be targeted in the future to reduce SCD in patients with HF.
Conclusions
HF induces a state of increased sympathetic tone and redox imbalance. The pathognomonic feature of HFrEF, impaired contractility, and the generation of fatal arrhythmias share a common mechanism, deleterious intracellular Ca2+ signaling. Experimental models researching redox modifications and phosphorylation have thrown light on how post-translational modifications affect Ca2+ and Na+ cycling proteins within the T tubule microdomain and terminal cisternae of the SR. Much interest has surrounded the RyR2 macromolecular complex. Increased phosphorylation and oxidation are associated with increased diastolic activity and pathological RyR2 Ca2+ handling which is recognized as a key component of the arrhythmic phenotype in HF. Currently, the causative molecular changes remain controversial and undefined. Current experimental models have been limited in dissecting the individual and synergistic contributions of RyR2 stress-induced modifications and how single-cell studies translate to ventricular arrythmias in the clinical realm. We are beginning to appreciate the complex interplay of competing cascade signaling and the importance of spatial temporal relationship, within microdomains, which compromise increased sympathetic tone, redox imbalance, and negative remodeling of HF. It follows that antiarrhythmic treatment will likely require the modification of multiple targets. First, the reversal of HF and promotion of positive remodeling is paramount to reduce both oxidative and phosphorylative stress. Second, by defining the molecular sites involved in arrhythmia genesis on RyR2 and other Na+ and Ca2+ cyclic proteins associated within the calcium release units, novel medication targets can be developed to work synergistically and reduce the rate of sudden cardiac death in heart failure.
Glossary
List of Abbreviations
- AAV
Adeno-associated viruses
- AP
Action potential
- Ca2+
Calcium
- CRU
Ca2+ release units
- CaM
Calmodulin
- CaMKII
Ca2+/calmodulin-dependent protein kinase II
- CASQ2
Calsequestrin 2
- CICR
Calcium-induced calcium release
- CPVT
Catecholaminergic polymorphic ventricular tachycardia
- DAD
Delayed after depolarization
- ECC
Excitation–contraction coupling
- Em
Membrane potential
- F-DPc10
Fluoroescent-DPc10
- FRET
Fluorescence resonance energy transfer
- GDMT
Guideline-directed medical therapy
- GSH
Reduced glutathione
- GSNO
S-Nitrosoglutathione
- GSSG
Oxidized glutathione
- HF
Heart failure
- HFrEF
Heart failure with reduced ejection fraction
- ICa,L
Inward Ca2+ current (LTCC)
- ICD
Intracardiac defibrillators
- IHD
Ischemeic heart disease
- INa
Inward sodium current
- K+
Potassium
- FKBP12.6
12.6 kDa FK506-binding protein
- LTCC
L-Type calcium channels
- LVF
Left ventricular function
- Na+
Sodium
- Nav
Voltage-gated Na+
- NCX
Na+/Ca2+ exchanger
- NO·
Nitric oxide
- NOS
Nitric oxide synthase
- O2·
Superoxide anions
- OH·
Hydroxyl radicals
- ONOO·
Peroxynitrate
- PKA
Protein kinase A
- PLN
Phospholamban
- Po
Open probability
- ROS/RNS
Reactive oxygen species/reactive nitrogen species
- RyR2
Ryanodine receptor
- SCD
Sudden cardiac death
- SERCA2a
Sarcoplasmic reticulum Ca2+-ATPase
- SLN
Sarcolipin
- SOD
Superoxide dismutase
- SOICR
Store-overload-induced Ca2+ release
- SR
Sarcoplasmic reticulum
- Tm
Tropomyosin
- TnC
Troponin C
- TnI
Troponin I
- T tubules
Transverse tubules
- WT
Wild-type
- β-AR
β-Adrenoceptor
The authors declare no competing financial interest.
References
- Ambrosy A. P.; Fonarow G. C.; Butler J.; Chioncel O.; Greene S. J.; Vaduganathan M.; Nodari S.; Lam C. S. P.; Sato N.; Shah A. N.; Gheorghiade M. (2014) The global health and economic burden of hospitalizations for heart failure: lessons learned from hospitalized heart failure registries. J. Am. Coll. Cardiol. 63 (12), 1123–1133. 10.1016/j.jacc.2013.11.053. [DOI] [PubMed] [Google Scholar]
- Sahle B. W.; Owen A. J.; Mutowo M. P.; Krum H.; Reid C. M. (2016) Prevalence of heart failure in Australia: a systematic review. BMC Cardiovasc. Disord. 16, 32. 10.1186/s12872-016-0208-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newgard C. B.; Sharpless N. E. (2013) Coming of age: molecular drivers of aging and therapeutic opportunities. J. Clin. Invest. 123 (3), 946–50. 10.1172/JCI68833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho K. K.; Anderson K. M.; Kannel W. B.; Grossman W.; Levy D. (1993) Survival after the onset of congestive heart failure in Framingham Heart Study subjects. Circulation 88 (1), 107–15. 10.1161/01.CIR.88.1.107. [DOI] [PubMed] [Google Scholar]
- Crespo-Leiro M. G.; Anker S. D.; Maggioni A. P.; Coats A. J.; Filippatos G.; Ruschitzka F.; Ferrari R.; Piepoli M. F.; Delgado Jimenez J. F.; Metra M.; et al. (2016) European Society of Cardiology Heart Failure Long-Term Registry (ESC-HF-LT): 1-year follow-up outcomes and differences across regions. Eur. J. Heart Failure 18 (6), 613–625. 10.1002/ejhf.566. [DOI] [PubMed] [Google Scholar]
- Kober L.; Thune J. J.; Nielsen J. C.; Haarbo J.; Videbaek L.; Korup E.; Jensen G.; Hildebrandt P.; Steffensen F. H.; Bruun N. E.; et al. (2016) Defibrillator Implantation in Patients with Nonischemic Systolic Heart Failure. N. Engl. J. Med. 375 (13), 1221–1230. 10.1056/NEJMoa1608029. [DOI] [PubMed] [Google Scholar]
- Elming M. B.; Nielsen J. C.; Haarbo J.; Videbaek L.; Korup E.; Signorovitch J.; Olesen L. L.; Hildebrandt P.; Steffensen F. H.; Bruun N. E.; Eiskjaer H.; Brandes A.; Thogersen A. M.; Gustafsson F.; Egstrup K.; Videbaek R.; Hassager C.; Svendsen J. H.; Hofsten D. E.; Torp-Pedersen C.; Pehrson S.; Kober L.; Thune J. J. (2017) Age and Outcomes of Primary Prevention Implantable Cardioverter-Defibrillators in Patients With Nonischemic Systolic Heart Failure. Circulation 136 (19), 1772–1780. 10.1161/CIRCULATIONAHA.117.028829. [DOI] [PubMed] [Google Scholar]
- Nerbonne J. M.; Kass R. S. (2005) Molecular physiology of cardiac repolarization. Physiol. Rev. 85 (4), 1205–53. 10.1152/physrev.00002.2005. [DOI] [PubMed] [Google Scholar]
- Bers D. M. (2001) Excitation-Contraction Coupling and Cardiac Contractile Force, 2nd ed., Kluwer Academic Publishers. [Google Scholar]
- Solaro R. J.; Rarick H. M. (1998) Troponin and tropomyosin: proteins that switch on and tune in the activity of cardiac myofilaments. Circ. Res. 83 (5), 471–80. 10.1161/01.RES.83.5.471. [DOI] [PubMed] [Google Scholar]
- Fozzard H. A. (2002) Cardiac sodium and calcium channels: a history of excitatory currents. Cardiovasc. Res. 55 (1), 1–8. 10.1016/S0008-6363(02)00407-8. [DOI] [PubMed] [Google Scholar]
- Tristani-Firouzi M.; Jensen J. L.; Donaldson M. R.; Sansone V.; Meola G.; Hahn A.; Bendahhou S.; Kwiecinski H.; Fidzianska A.; Plaster N.; Fu Y. H.; Ptacek L. J.; Tawil R. (2002) Functional and clinical characterization of KCNJ2 mutations associated with LQT7 (Andersen syndrome). J. Clin. Invest. 110 (3), 381–8. 10.1172/JCI15183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponce-Balbuena D.; Guerrero-Serna G.; Valdivia C. R.; Caballero R.; Diez-Guerra F. J.; Jimenez-Vazquez E. N.; Ramirez R. J.; Monteiro da Rocha A.; Herron T. J.; Campbell K. F.; Willis B. C.; Alvarado F. J.; Zarzoso M.; Kaur K.; Perez-Hernandez M.; Matamoros M.; Valdivia H. H.; Delpon E.; Jalife J. (2018) Cardiac Kir2.1 and NaV1.5 Channels Traffic Together to the Sarcolemma to Control Excitability. Circ. Res. 122 (11), 1501–1516. 10.1161/CIRCRESAHA.117.311872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page E.; Surdyk-Droske M. (1979) Distribution, surface density, and membrane area of diadic junctional contacts between plasma membrane and terminal cisterns in mammalian ventricle. Circ. Res. 45 (2), 260–7. 10.1161/01.RES.45.2.260. [DOI] [PubMed] [Google Scholar]
- Bailey F. R., Kelly D. E., Wood R. L., and Enders A. C. (1984) Bailey’s Textbook of Microscopic Anatomy, Williams & Wilkins, Baltimore, MD. [Google Scholar]
- Santulli G.; Lewis D.; des Georges A.; Marks A. R.; Frank J. (2018) Ryanodine Receptor Structure and Function in Health and Disease. Subcell. Biochem. 87, 329–352. 10.1007/978-981-10-7757-9_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bers D. M. (2002) Cardiac excitation-contraction coupling. Nature 415 (6868), 198–205. 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- Eisner D.; Bode E.; Venetucci L.; Trafford A. (2013) Calcium flux balance in the heart. J. Mol. Cell. Cardiol. 58, 110–7. 10.1016/j.yjmcc.2012.11.017. [DOI] [PubMed] [Google Scholar]
- Lyon A. R.; MacLeod K. T.; Zhang Y.; Garcia E.; Kanda G. K.; Lab M. J.; Korchev Y. E.; Harding S. E.; Gorelik J. (2009) Loss of T tubules and other changes to surface topography in ventricular myocytes from failing human and rat heart. Proc. Natl. Acad. Sci. U. S. A. 106 (16), 6854–9. 10.1073/pnas.0809777106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H.; Lederer W. J.; Cannell M. B. (1993) Calcium sparks: elementary events underlying excitation-contraction coupling in heart muscle. Science 262 (5134), 740–4. 10.1126/science.8235594. [DOI] [PubMed] [Google Scholar]
- Landstrom A. P.; Dobrev D.; Wehrens X. H. T. (2017) Calcium Signaling and Cardiac Arrhythmias. Circ. Res. 120 (12), 1969–1993. 10.1161/CIRCRESAHA.117.310083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pogwizd S. M.; Schlotthauer K.; Li L.; Yuan W.; Bers D. M. (2001) Arrhythmogenesis and contractile dysfunction in heart failure: Roles of sodium-calcium exchange, inward rectifier potassium current, and residual beta-adrenergic responsiveness. Circ. Res. 88 (11), 1159–67. 10.1161/hh1101.091193. [DOI] [PubMed] [Google Scholar]
- Radwanski P. B.; Johnson C. N.; Gyorke S.; Veeraraghavan R. (2018) Cardiac Arrhythmias as Manifestations of Nanopathies: An Emerging View. Front. Physiol. 9, 1228. 10.3389/fphys.2018.01228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner G. (2017) The structural basis of ryanodine receptor ion channel function. J. Gen. Physiol. 149 (12), 1065–1089. 10.1085/jgp.201711878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng W.; Shen H.; Wu J.; Guo W.; Pan X.; Wang R.; Chen S.R.; Yan N. (2016) Structural basis for the gating mechanism of the type 2 ryanodine receptor RyR2. Science 354 (6310), aah5324. 10.1126/science.aah5324. [DOI] [PubMed] [Google Scholar]
- Denniss A.; Dulhunty A. F.; Beard N. A. (2018) Ryanodine receptor Ca(2+) release channel post-translational modification: Central player in cardiac and skeletal muscle disease. Int. J. Biochem. Cell Biol. 101, 49–53. 10.1016/j.biocel.2018.05.004. [DOI] [PubMed] [Google Scholar]
- Laver D. R. (2018) Regulation of the RyR channel gating by Ca(2+) and Mg(2). Biophys. Rev. 10 (4), 1087–1095. 10.1007/s12551-018-0433-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balog E. M.; Fruen B. R.; Shomer N. H.; Louis C. F. (2001) Divergent effects of the malignant hyperthermia-susceptible Arg(615)-->Cys mutation on the Ca(2+) and Mg(2+) dependence of the RyR1. Biophys. J. 81 (4), 2050–8. 10.1016/S0006-3495(01)75854-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezprozvanny I.; Watras J.; Ehrlich B. E. (1991) Bell-shaped calcium-response curves of Ins(1,4,5)P3- and calcium-gated channels from endoplasmic reticulum of cerebellum. Nature 351 (6329), 751–4. 10.1038/351751a0. [DOI] [PubMed] [Google Scholar]
- des Georges A.; Clarke O. B.; Zalk R.; Yuan Q.; Condon K. J.; Grassucci R. A.; Hendrickson W. A.; Marks A. R.; Frank J. (2016) Structural Basis for Gating and Activation of RyR1. Cell 167 (1), 145–157. 10.1016/j.cell.2016.08.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitsapesan R.; Williams A. J. (1994) Regulation of the gating of the sheep cardiac sarcoplasmic reticulum Ca(2+)-release channel by luminal Ca2+. J. Membr. Biol. 137 (3), 215–226. 10.1007/BF00232590. [DOI] [PubMed] [Google Scholar]
- Laver D. R. (2007) Ca2+ stores regulate ryanodine receptor Ca2+ release channels via luminal and cytosolic Ca2+ sites. Biophys. J. 92 (10), 3541–55. 10.1529/biophysj.106.099028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner G.; Henderson J. S. (1987) Rapid calcium release from cardiac sarcoplasmic reticulum vesicles is dependent on Ca2+ and is modulated by Mg2+, adenine nucleotide, and calmodulin. J. Biol. Chem. 262 (7), 3065–3073. [PubMed] [Google Scholar]
- Xin H. B.; Senbonmatsu T.; Cheng D. S.; Wang Y. X.; Copello J. A.; Ji G. J.; Collier M. L.; Deng K. Y.; Jeyakumar L. H.; Magnuson M. A.; Inagami T.; Kotlikoff M. I.; Fleischer S. (2002) Oestrogen protects FKBP12.6 null mice from cardiac hypertrophy. Nature 416 (6878), 334–8. 10.1038/416334a. [DOI] [PubMed] [Google Scholar]
- Brillantes A. B.; Ondrias K.; Scott A.; Kobrinsky E.; Ondriasova E.; Moschella M. C.; Jayaraman T.; Landers M.; Ehrlich B. E.; Marks A. R. (1994) Stabilization of calcium release channel (ryanodine receptor) function by FK506-binding protein. Cell 77 (4), 513–23. 10.1016/0092-8674(94)90214-3. [DOI] [PubMed] [Google Scholar]
- Yamaguchi N.; Xu L.; Pasek D. A.; Evans K. E.; Meissner G. (2003) Molecular basis of calmodulin binding to cardiac muscle Ca(2+) release channel (ryanodine receptor). J. Biol. Chem. 278 (26), 23480–6. 10.1074/jbc.M301125200. [DOI] [PubMed] [Google Scholar]
- Xu L.; Meissner G. (2004) Mechanism of calmodulin inhibition of cardiac sarcoplasmic reticulum Ca2+ release channel (ryanodine receptor). Biophys. J. 86 (2), 797–804. 10.1016/S0006-3495(04)74155-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sondergaard M. T.; Liu Y.; Larsen K. T.; Nani A.; Tian X.; Holt C.; Wang R.; Wimmer R.; Van Petegem F.; Fill M.; Chen S. R.; Overgaard M. T. (2017) The Arrhythmogenic Calmodulin p.Phe142Leu Mutation Impairs C-domain Ca2+ Binding but Not Calmodulin-dependent Inhibition of the Cardiac Ryanodine Receptor. J. Biol. Chem. 292 (4), 1385–1395. 10.1074/jbc.M116.766253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda T.; Yang Y.; Uchinoumi H.; Thomas D. D.; Chen-Izu Y.; Kato T.; Yamamoto T.; Yano M.; Cornea R. L.; Bers D. M. (2015) Oxidation of ryanodine receptor (RyR) and calmodulin enhance Ca release and pathologically alter, RyR structure and calmodulin affinity. J. Mol. Cell. Cardiol. 85, 240–8. 10.1016/j.yjmcc.2015.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postma A. V.; Denjoy I.; Hoorntje T. M.; Lupoglazoff J. M.; Da Costa A.; Sebillon P.; Mannens M. M.; Wilde A. A.; Guicheney P. (2002) Absence of calsequestrin 2 causes severe forms of catecholaminergic polymorphic ventricular tachycardia. Circ. Res. 91 (8), e21–e26. 10.1161/01.RES.0000038886.18992.6B. [DOI] [PubMed] [Google Scholar]
- Bers D. M. (2008) Calcium cycling and signaling in cardiac myocytes. Annu. Rev. Physiol. 70, 23–49. 10.1146/annurev.physiol.70.113006.100455. [DOI] [PubMed] [Google Scholar]
- DeMaria C. D.; Soong T. W.; Alseikhan B. A.; Alvania R. S.; Yue D. T. (2001) Calmodulin bifurcates the local Ca2+ signal that modulates P/Q-type Ca2+ channels. Nature 411 (6836), 484–9. 10.1038/35078091. [DOI] [PubMed] [Google Scholar]
- Schultz J. E. J.; Glascock B. J.; Witt S. A.; Nieman M. L.; Nattamai K. J.; Liu L. H.; Lorenz J. N.; Shull G. E.; Kimball T. R.; Periasamy M. (2004) Accelerated onset of heart failure in mice during pressure overload with chronically decreased SERCA2 calcium pump activity. Am. J. Physiol Heart Circ Physiol 286 (3), H1146–H1153. 10.1152/ajpheart.00720.2003. [DOI] [PubMed] [Google Scholar]
- Neubauer S.; Horn M.; Naumann A.; Tian R.; Hu K.; Laser M.; Friedrich J.; Gaudron P.; Schnackerz K.; Ingwall J. S.; et al. (1995) Impairment of energy metabolism in intact residual myocardium of rat hearts with chronic myocardial infarction. J. Clin. Invest. 95 (3), 1092–100. 10.1172/JCI117756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Periasamy M.; Bhupathy P.; Babu G. J. (2007) Regulation of sarcoplasmic reticulum Ca2+ ATPase pump expression and its relevance to cardiac muscle physiology and pathology. Cardiovasc. Res. 77 (2), 265–73. 10.1093/cvr/cvm056. [DOI] [PubMed] [Google Scholar]
- Kaumann A.; Bartel S.; Molenaar P.; Sanders L.; Burrell K.; Vetter D.; Hempel P.; Karczewski P.; Krause E. G. (1999) Activation of beta2-adrenergic receptors hastens relaxation and mediates phosphorylation of phospholamban, troponin I, and C-protein in ventricular myocardium from patients with terminal heart failure. Circulation 99 (1), 65–72. 10.1161/01.CIR.99.1.65. [DOI] [PubMed] [Google Scholar]
- Molenaar P.; Bartel S.; Cochrane A.; Vetter D.; Jalali H.; Pohlner P.; Burrell K.; Karczewski P.; Krause E. G.; Kaumann A. (2000) Both beta(2)- and beta(1)-adrenergic receptors mediate hastened relaxation and phosphorylation of phospholamban and troponin I in ventricular myocardium of fallot infants, consistent with selective coupling of beta(2)-adrenergic receptors to G(s)-protein. Circulation 102 (15), 1814–1821. 10.1161/01.CIR.102.15.1814. [DOI] [PubMed] [Google Scholar]
- Kranias E. G.; Hajjar R. J. (2012) Modulation of cardiac contractility by the phospholamban/SERCA2a regulatome. Circ. Res. 110 (12), 1646–60. 10.1161/CIRCRESAHA.111.259754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayasinghe I. D.; Cannell M. B.; Soeller C. (2009) Organization of ryanodine receptors, transverse tubules, and sodium-calcium exchanger in rat myocytes. Biophys. J. 97 (10), 2664–73. 10.1016/j.bpj.2009.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W.; Wasserstrom J. A.; Shiferaw Y. (2009) Role of coupled gating between cardiac ryanodine receptors in the genesis of triggered arrhythmias. Am. J. Physiol Heart Circ Physiol 297 (1), H171–80. 10.1152/ajpheart.00098.2009. [DOI] [PubMed] [Google Scholar]
- Schlotthauer K.; Bers D. M. (2000) Sarcoplasmic reticulum Ca(2+) release causes myocyte depolarization. Underlying mechanism and threshold for triggered action potentials. Circ. Res. 87 (9), 774–80. 10.1161/01.RES.87.9.774. [DOI] [PubMed] [Google Scholar]
- El-Armouche A.; Eschenhagen T. (2009) Beta-adrenergic stimulation and myocardial function in the failing heart. Heart Failure Rev. 14 (4), 225–41. 10.1007/s10741-008-9132-8. [DOI] [PubMed] [Google Scholar]
- Bristow M. R.; Ginsburg R.; Umans V.; Fowler M.; Minobe W.; Rasmussen R.; Zera P.; Menlove R.; Shah P.; Jamieson S.; et al. (1986) Beta 1- and beta 2-adrenergic-receptor subpopulations in nonfailing and failing human ventricular myocardium: coupling of both receptor subtypes to muscle contraction and selective beta 1-receptor down-regulation in heart failure. Circ. Res. 59 (3), 297–309. 10.1161/01.RES.59.3.297. [DOI] [PubMed] [Google Scholar]
- Buxton B. F.; Jones C. R.; Molenaar P.; Summers R. J. (1987) Characterization and autoradiographic localization of beta-adrenoceptor subtypes in human cardiac tissues. Br. J. Pharmacol. 92 (2), 299–310. 10.1111/j.1476-5381.1987.tb11324.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molenaar P.; Chen L.; Semmler A. B.; Parsonage W. A.; Kaumann A. J. (2007) Human heart beta-adrenoceptors: beta1-adrenoceptor diversification through ’affinity states’ and polymorphism. Clin. Exp. Pharmacol. Physiol. 34 (10), 1020–8. 10.1111/j.1440-1681.2007.04730.x. [DOI] [PubMed] [Google Scholar]
- Molenaar P.; Savarimuthu S. M.; Sarsero D.; Chen L.; Semmler A. B.; Carle A.; Yang I.; Bartel S.; Vetter D.; Beyerdorfer I.; Krause E. G.; Kaumann A. J. (2007) (−)-Adrenaline elicits positive inotropic, lusitropic, and biochemical effects through beta2 -adrenoceptors in human atrial myocardium from nonfailing and failing hearts, consistent with Gs coupling but not with Gi coupling. Naunyn-Schmiedeberg's Arch. Pharmacol. 375 (1), 11–28. 10.1007/s00210-007-0138-x. [DOI] [PubMed] [Google Scholar]
- Kaumann A. J.; Hall J. A.; Murray K. J.; Wells F. C.; Brown M. J. (1989) A comparison of the effects of adrenaline and noradrenaline on human heart: the role of beta 1- and beta 2-adrenoceptors in the stimulation of adenylate cyclase and contractile force. Eur. Heart J. 10, 29–37. 10.1093/eurheartj/10.suppl_B.29. [DOI] [PubMed] [Google Scholar]
- Mason D. A.; Moore J. D.; Green S. A.; Liggett S. B. (1999) A gain-of-function polymorphism in a G-protein coupling domain of the human beta1-adrenergic receptor. J. Biol. Chem. 274 (18), 12670–4. 10.1074/jbc.274.18.12670. [DOI] [PubMed] [Google Scholar]
- Green S. A.; Cole G.; Jacinto M.; Innis M.; Liggett S. B. (1993) A polymorphism of the human beta 2-adrenergic receptor within the fourth transmembrane domain alters ligand binding and functional properties of the receptor. J. Biol. Chem. 268 (31), 23116–23121. [PubMed] [Google Scholar]
- Gauthier C.; Tavernier G.; Charpentier F.; Langin D.; Le Marec H. (1996) Functional beta3-adrenoceptor in the human heart. J. Clin. Invest. 98 (2), 556–62. 10.1172/JCI118823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skeberdis V. A.; Gendvilieneη V.; Zablockaiteη D.η; Treinys R.; Macχianskieneη R.; Bogdelis A.; Jurevicχius J.; Fischmeister R. (2008) beta3-adrenergic receptor activation increases human atrial tissue contractility and stimulates the L-type Ca2+ current. J. Clin. Invest. 118 (9), 3219–3227. 10.1172/JCI32519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo W.; Michel M. C.; Lee X. W.; Kaumann A. J.; Molenaar P. (2017) The beta3 -adrenoceptor agonist mirabegron increases human atrial force through beta1 -adrenoceptors: an indirect mechanism?. Br. J. Pharmacol. 174 (16), 2706–2715. 10.1111/bph.13897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molenaar P.; Sarsero D.; Kaumann A. J. (1997) Proposal for the interaction of non-conventional partial agonists and catecholamines with the 'putative beta 4-adrenoceptor' in mammalian heart. Clin. Exp. Pharmacol. Physiol. 24 (9-10), 647–656. 10.1111/j.1440-1681.1997.tb02107.x. [DOI] [PubMed] [Google Scholar]
- Christ T.; Molenaar P.; Klenowski P. M.; Ravens U.; Kaumann A. J. (2011) Human atrial beta(1L)-adrenoceptor but not beta(3)-adrenoceptor activation increases force and Ca(2+) current at physiological temperature. Br. J. Pharmacol. 162 (4), 823–839. 10.1111/j.1476-5381.2010.00996.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molenaar P.; Chen L.; Parsonage W. A. (2006) Cardiac implications for the use of beta2-adrenoceptor agonists for the management of muscle wasting. Br. J. Pharmacol. 147 (6), 583–6. 10.1038/sj.bjp.0706670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaumann A. J.; Molenaar P. (2008) The low-affinity site of the beta1-adrenoceptor and its relevance to cardiovascular pharmacology. Pharmacol. Ther. 118 (3), 303–36. 10.1016/j.pharmthera.2008.03.009. [DOI] [PubMed] [Google Scholar]
- Joseph S. S.; Lynham J. A.; Molenaar P.; Grace A. A.; Colledge W. H.; Kaumann A. J. (2003) Intrinsic sympathomimetic activity of (−)-pindolol mediated through a (−)-propranolol-resistant site of the beta(1)-adrenoceptor in human atrium and recombinant receptors. Naunyn-Schmiedeberg's Arch. Pharmacol. 368 (6), 496–503. 10.1007/s00210-003-0835-z. [DOI] [PubMed] [Google Scholar]
- Sarsero D.; Russell F. D.; Lynham J. A.; Rabnott G.; Yang I.; Fong K. M.; Li L.; Kaumann A. J.; Molenaar P. (2003) -)-CGP 12177 increases contractile force and hastens relaxation of human myocardial preparations through a propranolol-resistant state of the beta 1-adrenoceptor. Naunyn-Schmiedeberg's Arch. Pharmacol. 367 (1), 10–21. 10.1007/s00210-002-0652-9. [DOI] [PubMed] [Google Scholar]
- Gille E.; Lemoine H.; Ehle B.; Kaumann A. J. (1985) The affinity of (−)-propranolol for beta 1- and beta 2-adrenoceptors of human heart. Differential antagonism of the positive inotropic effects and adenylate cyclase stimulation by (−)-noradrenaline and (−)-adrenaline. Naunyn-Schmiedeberg's Arch. Pharmacol. 331 (1), 60–70. 10.1007/BF00498852. [DOI] [PubMed] [Google Scholar]
- Nikolaienko R.; Bovo E.; Zima A. V. (2018) Redox Dependent Modifications of Ryanodine Receptor: Basic Mechanisms and Implications in Heart Diseases. Front. Physiol. 9, 1775. 10.3389/fphys.2018.01775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutsui H.; Kinugawa S.; Matsushima S. (2011) Oxidative stress and heart failure. Am. J. Physiol Heart Circ Physiol 301 (6), H2181–90. 10.1152/ajpheart.00554.2011. [DOI] [PubMed] [Google Scholar]
- Hasenfuss G.; Schillinger W.; Lehnart S. E.; Preuss M.; Pieske B.; Maier L. S.; Prestle J.; Minami K.; Just H. (1999) Relationship between Na+-Ca2+-exchanger protein levels and diastolic function of failing human myocardium. Circulation 99 (5), 641–8. 10.1161/01.CIR.99.5.641. [DOI] [PubMed] [Google Scholar]
- Volders P. G.; Vos M. A.; Szabo B.; Sipido K. R.; de Groot S. H.; Gorgels A. P.; Wellens H. J.; Lazzara R. (2000) Progress in the understanding of cardiac early afterdepolarizations and torsades de pointes: time to revise current concepts. Cardiovasc. Res. 46 (3), 376–92. 10.1016/S0008-6363(00)00022-5. [DOI] [PubMed] [Google Scholar]
- Weiss J. N.; Garfinkel A.; Karagueuzian H. S.; Chen P. S.; Qu Z. (2010) Early afterdepolarizations and cardiac arrhythmias. Heart Rhythm 7 (12), 1891–9. 10.1016/j.hrthm.2010.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen M. R.; Gelband H.; Merker C.; Hoffman B. F. (1973) Mechanisms of digitalis toxicity. Effects of ouabain on phase four of canine Purkinje fiber transmembrane potentials. Circulation 47 (4), 681–9. 10.1161/01.CIR.47.4.681. [DOI] [PubMed] [Google Scholar]
- Sag C. M.; Wagner S.; Maier L. S. (2013) Role of oxidants on calcium and sodium movement in healthy and diseased cardiac myocytes. Free Radical Biol. Med. 63, 338–49. 10.1016/j.freeradbiomed.2013.05.035. [DOI] [PubMed] [Google Scholar]
- Marx S. O.; Reiken S.; Hisamatsu Y.; Jayaraman T.; Burkhoff D.; Rosemblit N.; Marks A. R. (2000) PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell 101 (4), 365–76. 10.1016/S0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- Wehrens X. H.; Lehnart S. E.; Reiken S. R.; Deng S. X.; Vest J. A.; Cervantes D.; Coromilas J.; Landry D. W.; Marks A. R. (2004) Protection from cardiac arrhythmia through ryanodine receptor-stabilizing protein calstabin2. Science 304 (5668), 292–6. 10.1126/science.1094301. [DOI] [PubMed] [Google Scholar]
- Wehrens X. H.; Lehnart S. E.; Reiken S. R.; Marks A. R. (2004) Ca2+/calmodulin-dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circ. Res. 94 (6), e61–e70. 10.1161/01.RES.0000125626.33738.E2. [DOI] [PubMed] [Google Scholar]
- Wehrens X. H.; Lehnart S. E.; Reiken S.; Vest J. A.; Wronska A.; Marks A. R. (2006) Ryanodine receptor/calcium release channel PKA phosphorylation: a critical mediator of heart failure progression. Proc. Natl. Acad. Sci. U. S. A. 103 (3), 511–8. 10.1073/pnas.0510113103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonnell S. M.; Garcia-Rivas G.; Scherman J. A.; Kubo H.; Chen X.; Valdivia H.; Houser S. R. (2008) Adrenergic regulation of cardiac contractility does not involve phosphorylation of the cardiac ryanodine receptor at serine 2808. Circ. Res. 102 (8), e65–e72. 10.1161/CIRCRESAHA.108.174722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camors E.; Valdivia H. H. (2014) CaMKII regulation of cardiac ryanodine receptors and inositol triphosphate receptors. Front. Pharmacol. 5, 101. 10.3389/fphar.2014.00101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao B.; Jiang M. T.; Zhao M.; Yang D.; Sutherland C.; Lai F. A.; Walsh M. P.; Warltier D. C.; Cheng H.; Chen S. R. (2005) Characterization of a novel PKA phosphorylation site, serine-2030, reveals no PKA hyperphosphorylation of the cardiac ryanodine receptor in canine heart failure. Circ. Res. 96 (8), 847–55. 10.1161/01.RES.0000163276.26083.e8. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Makarewich C. A.; Kubo H.; Wang W.; Duran J. M.; Li Y.; Berretta R. M.; Koch W. J.; Chen X.; Gao E.; Valdivia H. H.; Houser S. R. (2012) Hyperphosphorylation of the cardiac ryanodine receptor at serine 2808 is not involved in cardiac dysfunction after myocardial infarction. Circ. Res. 110 (6), 831–40. 10.1161/CIRCRESAHA.111.255158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier L. S.; Zhang T.; Chen L.; DeSantiago J.; Brown J. H.; Bers D. M. (2003) Transgenic CaMKIIdeltaC overexpression uniquely alters cardiac myocyte Ca2+ handling: reduced SR Ca2+ load and activated SR Ca2+ release. Circ. Res. 92 (8), 904–11. 10.1161/01.RES.0000069685.20258.F1. [DOI] [PubMed] [Google Scholar]
- Uchinoumi H.; Yang Y.; Oda T.; Li N.; Alsina K. M.; Puglisi J. L.; Chen-Izu Y.; Cornea R. L.; Wehrens X. H. T.; Bers D. M. (2016) CaMKII-dependent phosphorylation of RyR2 promotes targetable pathological RyR2 conformational shift. J. Mol. Cell. Cardiol. 98, 62–72. 10.1016/j.yjmcc.2016.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda T.; Yang Y.; Nitu F. R.; Svensson B.; Lu X.; Fruen B. R.; Cornea R. L.; Bers D. M. (2013) In cardiomyocytes, binding of unzipping peptide activates ryanodine receptor 2 and reciprocally inhibits calmodulin binding. Circ. Res. 112 (3), 487–97. 10.1161/CIRCRESAHA.111.300290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao B.; Zhong G.; Obayashi M.; Yang D.; Chen K.; Walsh M. P.; Shimoni Y.; Cheng H.; Ter Keurs H.; Chen S. R. (2006) Ser-2030, but not Ser-2808, is the major phosphorylation site in cardiac ryanodine receptors responding to protein kinase A activation upon beta-adrenergic stimulation in normal and failing hearts. Biochem. J. 396 (1), 7–16. 10.1042/BJ20060116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takasago T.; Imagawa T.; Furukawa K.; Ogurusu T.; Shigekawa M. (1991) Regulation of the cardiac ryanodine receptor by protein kinase-dependent phosphorylation. J. Biochem. 109 (1), 163–70. 10.1093/oxfordjournals.jbchem.a123339. [DOI] [PubMed] [Google Scholar]
- Ho H. T.; Belevych A. E.; Liu B.; Bonilla I. M.; Radwanski P. B.; Kubasov I. V.; Valdivia H. H.; Schober K.; Carnes C. A.; Gyorke S. (2016) Muscarinic Stimulation Facilitates Sarcoplasmic Reticulum Ca Release by Modulating Ryanodine Receptor 2 Phosphorylation Through Protein Kinase G and Ca/Calmodulin-Dependent Protein Kinase II. Hypertension 68 (5), 1171–1178. 10.1161/HYPERTENSIONAHA.116.07666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlhaas M.; Liu T.; Knopp A.; Zeller T.; Ong M. F.; Bohm M.; O’Rourke B.; Maack C. (2010) Elevated cytosolic Na+ increases mitochondrial formation of reactive oxygen species in failing cardiac myocytes. Circulation 121 (14), 1606–13. 10.1161/CIRCULATIONAHA.109.914911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L.; Eu J. P.; Meissner G.; Stamler J. S. (1998) Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science 279 (5348), 234–7. 10.1126/science.279.5348.234. [DOI] [PubMed] [Google Scholar]
- Suzuki Y. J.; Cleemann L.; Abernethy D. R.; Morad M. (1998) Glutathione is a cofactor for H2O2-mediated stimulation of Ca2+-induced Ca2+ release in cardiac myocytes. Free Radical Biol. Med. 24 (2), 318–25. 10.1016/S0891-5849(97)00227-X. [DOI] [PubMed] [Google Scholar]
- Goldhaber J. I.; Liu E. (1994) Excitation-contraction coupling in single guinea-pig ventricular myocytes exposed to hydrogen peroxide. J. Physiol. 477, 135–47. 10.1113/jphysiol.1994.sp020178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waddell H. M. M.; Zhang J. Z.; Hoeksema K. J.; McLachlan J. J.; McLay J. C.; Jones P. P. (2016) Oxidation of RyR2 Has a Biphasic Effect on the Threshold for Store Overload-Induced Calcium Release. Biophys. J. 110 (11), 2386–2396. 10.1016/j.bpj.2016.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terentyev D.; Gyorke I.; Belevych A. E.; Terentyeva R.; Sridhar A.; Nishijima Y.; Carcache de Blanco E.; Khanna S.; Sen C. K.; Cardounel A. J.; Carnes C. A.; Gyorke S. (2008) Redox modification of ryanodine receptors contributes to sarcoplasmic reticulum Ca2+ leak in chronic heart failure. Circ. Res. 103 (12), 1466–1472. 10.1161/CIRCRESAHA.108.184457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna A. D.; Lam A.; Thekkedam C.; Gallant E. M.; Beard N. A.; Dulhunty A. F. (2016) Correction: Cardiac ryanodine receptor activation by a high Ca2+ store load is reversed in a reducing cytoplasmic redox environment. J. Cell Sci. 129 (22), 4317. 10.1242/jcs.198705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi T.; Xiao Z.; Guo W.; Tang Y.; Hiess F.; Xiao J.; Wang Y.; Zhang J. Z.; Zhang L.; Wang R.; Jones P. P.; Chen S. R. (2015) Role of Cys(3)(6)(0)(2) in the function and regulation of the cardiac ryanodine receptor. Biochem. J. 467 (1), 177–90. 10.1042/BJ20141263. [DOI] [PubMed] [Google Scholar]
- Xie L. H.; Chen F.; Karagueuzian H. S.; Weiss J. N. (2009) Oxidative-stress-induced afterdepolarizations and calmodulin kinase II signaling. Circ. Res. 104 (1), 79–86. 10.1161/CIRCRESAHA.108.183475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zima A. V.; Copello J. A.; Blatter L. A. (2004) Effects of cytosolic NADH/NAD(+) levels on sarcoplasmic reticulum Ca(2+) release in permeabilized rat ventricular myocytes. J. Physiol. 555, 727–741. 10.1113/jphysiol.2003.055848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K. Y.; Zweier J. L.; Becker L. C. (1997) Hydroxyl radical inhibits sarcoplasmic reticulum Ca(2+)-ATPase function by direct attack on the ATP binding site. Circ. Res. 80 (1), 76–81. 10.1161/01.RES.80.1.76. [DOI] [PubMed] [Google Scholar]
- Zima A. V.; Blatter L. A. (2006) Redox regulation of cardiac calcium channels and transporters. Cardiovasc. Res. 71 (2), 310–21. 10.1016/j.cardiores.2006.02.019. [DOI] [PubMed] [Google Scholar]
- Sun J.; Xin C.; Eu J. P.; Stamler J. S.; Meissner G. (2001) Cysteine-3635 is responsible for skeletal muscle ryanodine receptor modulation by NO. Proc. Natl. Acad. Sci. U. S. A. 98 (20), 11158–62. 10.1073/pnas.201289098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez D. R.; Beigi F.; Treuer A. V.; Hare J. M. (2007) Deficient ryanodine receptor S-nitrosylation increases sarcoplasmic reticulum calcium leak and arrhythmogenesis in cardiomyocytes. Proc. Natl. Acad. Sci. U. S. A. 104 (51), 20612–7. 10.1073/pnas.0706796104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H. L.; Viatchenko-Karpinski S.; Sun J. H.; Gyorke I.; Benkusky N. A.; Kohr M. J.; Valdivia H. H.; Murphy E.; Gyorke S.; Ziolo M. T. (2010) Regulation of myocyte contraction via neuronal nitric oxide synthase: role of ryanodine receptor S-nitrosylation. J. Physiol. 588 (15), 2905–2917. 10.1113/jphysiol.2010.192617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutler M. J.; Plummer B. N.; Wan X.; Sun Q. A.; Hess D.; Liu H.; Deschenes I.; Rosenbaum D. S.; Stamler J. S.; Laurita K. R. (2012) Aberrant S-nitrosylation mediates calcium-triggered ventricular arrhythmia in the intact heart. Proc. Natl. Acad. Sci. U. S. A. 109 (44), 18186–91. 10.1073/pnas.1210565109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belevych A. E.; Terentyev D.; Terentyeva R.; Nishijima Y.; Sridhar A.; Hamlin R. L.; Carnes C. A.; Gyorke S. (2011) The relationship between arrhythmogenesis and impaired contractility in heart failure: role of altered ryanodine receptor function. Cardiovasc. Res. 90 (3), 493–502. 10.1093/cvr/cvr025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walweel K.; Molenaar P.; Imtiaz M. S.; Denniss A.; Dos Remedios C.; van Helden D. F.; Dulhunty A. F.; Laver D. R.; Beard N. A. (2017) Ryanodine receptor modification and regulation by intracellular Ca(2+) and Mg(2+) in healthy and failing human hearts. J. Mol. Cell. Cardiol. 104, 53–62. 10.1016/j.yjmcc.2017.01.016. [DOI] [PubMed] [Google Scholar]
- Brette F.; Salle L.; Orchard C. H. (2004) Differential modulation of L-type Ca2+ current by SR Ca2+ release at the T-tubules and surface membrane of rat ventricular myocytes. Circ. Res. 95 (1), e1–e7. 10.1161/01.RES.0000135547.53927.F6. [DOI] [PubMed] [Google Scholar]
- Shiferaw Y.; Aistrup G. L.; Wasserstrom J. A. (2012) Intracellular Ca2+ waves, afterdepolarizations, and triggered arrhythmias. Cardiovasc. Res. 95 (3), 265–8. 10.1093/cvr/cvs155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aistrup G. L.; Balke C. W.; Wasserstrom J. A. (2011) Arrhythmia triggers in heart failure: the smoking gun of [Ca2+]i dysregulation. Heart Rhythm 8 (11), 1804–8. 10.1016/j.hrthm.2011.06.012. [DOI] [PubMed] [Google Scholar]
- Sachse F. B.; Torres N. S.; Savio-Galimberti E.; Aiba T.; Kass D. A.; Tomaselli G. F.; Bridge J. H. (2012) Subcellular structures and function of myocytes impaired during heart failure are restored by cardiac resynchronization therapy. Circ. Res. 110 (4), 588–97. 10.1161/CIRCRESAHA.111.257428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colucci W. S.; Wright R. F.; Braunwald E. (1986) New positive inotropic agents in the treatment of congestive heart failure. Mechanisms of action and recent clinical developments. 1. N. Engl. J. Med. 314 (5), 290–9. 10.1056/NEJM198601303140506. [DOI] [PubMed] [Google Scholar]
- Ponikowski P.; Voors A. A.; Anker S. D.; Bueno H.; Cleland J. G. F.; Coats A. J. S.; Falk V.; Gonzalez-Juanatey J. R.; Harjola V. P.; Jankowska E. A.; et al. (2016) 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC)Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 37 (27), 2129–2200. 10.1093/eurheartj/ehw128. [DOI] [PubMed] [Google Scholar]
- Hasenfuss G.; Teerlink J. R. (2011) Cardiac inotropes: current agents and future directions. Eur. Heart J. 32 (15), 1838–45. 10.1093/eurheartj/ehr026. [DOI] [PubMed] [Google Scholar]
- Elkayam U.; Tasissa G.; Binanay C.; Stevenson L. W.; Gheorghiade M.; Warnica J. W.; Young J. B.; Rayburn B. K.; Rogers J. G.; DeMarco T.; Leier C. V. (2007) Use and impact of inotropes and vasodilator therapy in hospitalized patients with severe heart failure. Am. Heart J. 153 (1), 98–104. 10.1016/j.ahj.2006.09.005. [DOI] [PubMed] [Google Scholar]
- Malik F. I.; Hartman J. J.; Elias K. A.; Morgan B. P.; Rodriguez H.; Brejc K.; Anderson R. L.; Sueoka S. H.; Lee K. H.; Finer J. T.; Sakowicz R.; Baliga R.; Cox D. R.; Garard M.; Godinez G.; Kawas R.; Kraynack E.; Lenzi D.; Lu P. P.; Muci A.; Niu C.; Qian X.; Pierce D. W.; Pokrovskii M.; Suehiro I.; Sylvester S.; Tochimoto T.; Valdez C.; Wang W.; Katori T.; Kass D. A.; Shen Y. T.; Vatner S. F.; Morgans D. J. (2011) Cardiac myosin activation: a potential therapeutic approach for systolic heart failure. Science 331 (6023), 1439–43. 10.1126/science.1200113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisler J. W.; DeWire S. M.; Whalen E. J.; Violin J. D.; Drake M. T.; Ahn S.; Shenoy S. K.; Lefkowitz R. J. (2007) A unique mechanism of beta-blocker action: carvedilol stimulates beta-arrestin signaling. Proc. Natl. Acad. Sci. U. S. A. 104 (42), 16657–62. 10.1073/pnas.0707936104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr R. 3rd; Schilling J.; Song J.; Carter R. L.; Du Y.; Yoo S. M.; Traynham C. J.; Koch W. J.; Cheung J. Y.; Tilley D. G.; Benovic J. L. (2016) beta-arrestin-biased signaling through the beta2-adrenergic receptor promotes cardiomyocyte contraction. Proc. Natl. Acad. Sci. U. S. A. 113 (28), E4107–16. 10.1073/pnas.1606267113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki M.; Yano M.; Oda T.; Tateishi H.; Kobayashi S.; Yamamoto T.; Ikeda Y.; Ohkusa T.; Ikemoto N.; Matsuzaki M. (2007) Scavenging free radicals by low-dose carvedilol prevents redox-dependent Ca2+ leak via stabilization of ryanodine receptor in heart failure. J. Am. Coll. Cardiol. 49 (16), 1722–32. 10.1016/j.jacc.2007.01.064. [DOI] [PubMed] [Google Scholar]
- Zhou Q.; Xiao J.; Jiang D.; Wang R.; Vembaiyan K.; Wang A.; Smith C. D.; Xie C.; Chen W.; Zhang J.; Tian X.; Jones P. P.; Zhong X.; Guo A.; Chen H.; Zhang L.; Zhu W.; Yang D.; Li X.; Chen J.; Gillis A. M.; Duff H. J.; Cheng H.; Feldman A. M.; Song L. S.; Fill M.; Back T. G.; Chen S. R. (2011) Carvedilol and its new analogs suppress arrhythmogenic store overload-induced Ca2+ release. Nat. Med. 17 (8), 1003–9. 10.1038/nm.2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Hanada K.; Staus D. P.; Makara M. A.; Dahal G. R.; Chen Q.; Ahles A.; Engelhardt S.; Rockman H. A. (2017) Galphai is required for carvedilol-induced beta1 adrenergic receptor beta-arrestin biased signaling. Nat. Commun. 8 (1), 1706. 10.1038/s41467-017-01855-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi S.; Yano M.; Suetomi T.; Ono M.; Tateishi H.; Mochizuki M.; Xu X.; Uchinoumi H.; Okuda S.; Yamamoto T.; Koseki N.; Kyushiki H.; Ikemoto N.; Matsuzaki M. (2009) Dantrolene, a therapeutic agent for malignant hyperthermia, markedly improves the function of failing cardiomyocytes by stabilizing interdomain interactions within the ryanodine receptor. J. Am. Coll. Cardiol. 53 (21), 1993–2005. 10.1016/j.jacc.2009.01.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell J. T.; Domeier T. L.; Blatter L. A. (2012) Dantrolene prevents arrhythmogenic Ca2+ release in heart failure. Am. J. Physiol Heart Circ Physiol 302 (4), H953–63. 10.1152/ajpheart.00936.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashna A.; van Helden D. F.; Dos Remedios C.; Molenaar P.; Laver D. R. (2020) Phenytoin Reduces Activity of Cardiac Ryanodine Receptor 2; A Potential Mechanism for Its Cardioprotective Action. Mol. Pharmacol. 97 (4), 250–258. 10.1124/mol.119.117721. [DOI] [PubMed] [Google Scholar]
- Paul-Pletzer K.; Yamamoto T.; Ikemoto N.; Jimenez L. S.; Morimoto H.; Williams P. G.; Ma J.; Parness J. (2005) Probing a putative dantrolene-binding site on the cardiac ryanodine receptor. Biochem. J. 387, 905–909. 10.1042/BJ20041336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L. W.; Subbiah R. N.; Kilborn M. J.; Dunn R. F. (2013) Phenytoin: an old but effective antiarrhythmic agent for the suppression of ventricular tachycardia. Med. J. Aust. 199 (3), 209–11. 10.5694/mja13.10224. [DOI] [PubMed] [Google Scholar]
- Rebbeck R. T.; Essawy M. M.; Nitu F. R.; Grant B. D.; Gillispie G. D.; Thomas D. D.; Bers D. M.; Cornea R. L. (2017) High-Throughput Screens to Discover Small-Molecule Modulators of Ryanodine Receptor Calcium Release Channels. SLAS Discov 22 (2), 176–186. 10.1177/1087057116674312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg B.; Butler J.; Felker G. M.; Ponikowski P.; Voors A. A.; Desai A. S.; Barnard D.; Bouchard A.; Jaski B.; Lyon A. R.; Pogoda J. M.; Rudy J. J.; Zsebo K. M. (2016) Calcium upregulation by percutaneous administration of gene therapy in patients with cardiac disease (CUPID 2): a randomised, multinational, double-blind, placebo-controlled, phase 2b trial. Lancet 387 (10024), 1178–86. 10.1016/S0140-6736(16)00082-9. [DOI] [PubMed] [Google Scholar]
- Jessup M.; Greenberg B.; Mancini D.; Cappola T.; Pauly D. F.; Jaski B.; Yaroshinsky A.; Zsebo K. M.; Dittrich H.; Hajjar R. J. (2011) Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease (CUPID): a phase 2 trial of intracoronary gene therapy of sarcoplasmic reticulum Ca2+-ATPase in patients with advanced heart failure. Circulation 124 (3), 304–313. 10.1161/CIRCULATIONAHA.111.022889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawase Y.; Ly H. Q.; Prunier F.; Lebeche D.; Shi Y.; Jin H.; Hadri L.; Yoneyama R.; Hoshino K.; Takewa Y.; Sakata S.; Peluso R.; Zsebo K.; Gwathmey J. K.; Tardif J. C.; Tanguay J. F.; Hajjar R. J. (2008) Reversal of cardiac dysfunction after long-term expression of SERCA2a by gene transfer in a pre-clinical model of heart failure. J. Am. Coll. Cardiol. 51 (11), 1112–9. 10.1016/j.jacc.2007.12.014. [DOI] [PubMed] [Google Scholar]
- Cutler M. J.; Wan X.; Plummer B. N.; Liu H.; Deschenes I.; Laurita K. R.; Hajjar R. J.; Rosenbaum D. S. (2012) Targeted sarcoplasmic reticulum Ca2+ ATPase 2a gene delivery to restore electrical stability in the failing heart. Circulation 126 (17), 2095–104. 10.1161/CIRCULATIONAHA.111.071480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mingozzi F.; Anguela X. M.; Pavani G.; Chen Y.; Davidson R. J.; Hui D. J.; Yazicioglu M.; Elkouby L.; Hinderer C. J.; Faella A.; Howard C.; Tai A.; Podsakoff G. M.; Zhou S.; Basner-Tschakarjan E.; Wright J. F.; High K. A. (2013) Overcoming preexisting humoral immunity to AAV using capsid decoys. Sci. Transl. Med. 5 (194), 194ra92. 10.1126/scitranslmed.3005795. [DOI] [PMC free article] [PubMed] [Google Scholar]