

Abstract
We established a method for embryogenic callus induction and highly efficient Agrobacterium-mediated genetic transformation of a table grape cultivar ‘Shine Muscat’ (Vitis labruscana). Embryogenic calli were induced using flower bud filaments from a dormant cane. Agrobacterium strain LBA4404 harboring the binary plasmid pBin19-sgfp, which contains the sgfp and nptII genes, was used to infect embryogenic calli. Infected calli were selected on 1/2 MS medium containing 5% maltose and 2% agar supplemented with 15 mg l−1 kanamycin. Efficiency of transformation of regenerated plants reached nearly 100% as determined by PCR and Southern blot analyses. The developed method will open a new avenue for genome editing of ‘Shine Muscat’ and contribute to the advancement of grape breeding.
Keywords: Agrobacterium, embryogenic callus, genetic transformation, ‘Shine Muscat’ grape
Introduction
A table grape cultivar ‘Shine Muscat’ (Vitis labruscana) was released by the Institute of Fruit Tree and Tea Science, National Agriculture and Food Research Organization in 2006. This cultivar produces large (≥12 g) white seedless berries if streptomycin is sprayed before flowering in combination with two gibberellin applications to flower or fruit clusters (Yamada et al. 2008). The berries have crispy flesh and a muscat flavor, and do not show any cracking. They have thin non-astringent skin and are palatable without peeling, in contrast to berries of ‘Kyoho,’ one of the most important cultivars in Japan. The fruit shelf life is longer than that of ‘Kyoho.’ ‘Shine Muscat’ is moderately tolerant to downy mildew and ripe rot, but is sensitive to anthracnose (Kono et al. 2013; Yamada et al. 2008). The cultivation and management of ‘Shine Muscat’ are relatively easy; its cultivation has rapidly increased to 1195.6 ha (8.7% of grape cultivation area in Japan) in 2016 (MAFF, https://www.e-stat.go.jp), only 9 years after the first sales of young plants in 2007 (Yamada and Sato 2016).
Yet, some ‘Shine Muscat’ traits such as pest and disease resistance, environmental stress tolerance, and accumulation of functional components including resveratrol need to be improved. One method to create new cultivars is genetic transformation. Most of the transformation methods of grape rely on Agrobacterium infection of embryogenic calli (ECs) (Martinelli and Mandolino 1994; Nakajima et al. 2006; Yamamoto et al. 2000). Flower organs, such as anthers (Hirabayashi and Akihama 1982; Nakano et al. 1997), ovaries (Nakano et al. 1997), unfertilized ovules (Nakajima et al. 2000; Notsuka et al. 1992; Srinivasan and Mullins 1980), and filaments (Nakajima and Matsuta 2003), have been used as explants and source materials to induce somatic embryogenesis. Leaves have also been used for this purpose (Matsuta 1992; Matsuta and Hirabayashi 1989; Nakano et al. 1997; Yamamoto et al. 2003), because they are available all year round. Nakano et al. (1997) used leaves, anthers, and ovaries as explants in 11 Vitis vinifera cultivars, 11 V. labruscana cultivars, and a V. rupestris cultivar. In some cultivars, somatic embryogenesis was induced only when ovary or anther explants were used, with ovaries being more efficient. Nakano et al. (1997) also demonstrated the importance of species for the induction of somatic embryos or ECs: they were more effectively induced in V. vinifera than in V. labruscana.
Grapes are transformed mainly by Agrobacterium-mediated transformation of somatic embryos or ECs (Hoshino et al. 1998; Iocco et al. 2001; Kandel et al. 2016; Martinelli and Mandolino 1994; Matsuta et al. 1993; Marchive et al. 2013; Nakajima et al. 2006; Perl et al. 1996; Scorza et al. 1996; Yamamoto et al. 2000, 2003). The success of genetic transformation depends on somatic embryogenesis and on the interaction of Agrobacterium with a particular cultivar.
Somatic embryogenesis and genetic transformation of ‘Shine Muscat’ grape have never been reported. A few reports of genetic transformation of grape via organogenesis have been published, but the number of suitable cultivars remains limited (Dutt et al. 2007; Sabbadini et al. 2019; Xie et al. 2016). Field-grown materials have limited use in the experiments on EC induction because grapes flower only once a year in vineyards. Recurrent cycles of secondary embryogenesis of V. vinifera cv. Thompson Seedless have been reported (Zhou et al. 2014). Here, we established a new procedure for the induction and maintenance of ‘Shine Muscat’ ECs using flower buds from dormant canes. We also report a genetic transformation system mediated by Agrobacterium, which will be applicable to genome editing.
Materials and methods
Plant materials
Dormant canes of ‘Shine Muscat’ were collected in an orchard (Akitsu, Higashihiroshima, Hiroshima, Japan) on January 27, 2014, and kept in a refrigerator at 4°C. The canes were cut into several pieces with 1 or 2 buds per piece on February 17, placed on rock wool, and tap water was poured into the container to about half of the rock-wool height. The canes were incubated at 26°C (16 h light/8 h dark) to induce flower bunches. Flower buds are first stuck to each other, then each bud becomes loose as it develops (here, we designated this status as “slightly apart from each other”). About 1 month after the start of the incubation, flower buds (“slightly apart from each other”) were picked and sterilized for 20 min in sodium hypochlorite solution (1% available chlorine) containing Tween-20 (about 0.2 g l−1). The buds were then rinsed with sterile distilled water, and the filaments were picked aseptically under a stereo microscope.
Culture conditions
Filaments were transferred into 100-ml conical flasks (9 or 10 per flask) with 25 ml of liquid 1/2 MS (Murashige and Skoog 1962) medium containing 1×MS vitamins, 1 µM 2,4-dichlorophenoxyacetic acid (2,4-D), 1 µM N-(1,2,3-thiadiazol-5-yl)-N′-phenylurea, and 30 g l−1 sucrose (1D1T). The medium was adjusted to pH 5.8 before autoclaving. Flasks were agitated continuously at 60 rpm in a gyro-rotary incubator at 26°C in the dark. After 1 month of culture, individual filaments were transferred to solid medium of the same composition as 1D1T with added 0.85% agar (1D1TS) for 2 months. The medium was renewed once a month. Three months after the culture initiation, filaments (some of them formed calli during the culture) were transferred to 1/2 MS medium containing 1×MS vitamins, 1 µM 2,4-D, 50 g l−1 maltose, and 3% agar (1D3A5M) to maintain and propagate the ECs. Maltose was used at 5% because somatic embryogenesis and genetic transformation of ‘Kyoho’ grape, recalcitrant to somatic embryogenesis and genetic transformation, were established using 5% maltose in our previous study (Nakajima et al. 2006). Embryogenic ability was checked by transferring some calli on MS hormone-free medium to examine whether they would regenerate somatic embryos.
Genetic transformation
Agrobacterium tumefaciens strain LBA4404, which harbors two chimeric gene expression cassettes, 35Spro-sgfp-Noster cassette and Nospro-nptII-Noster, in the binary plasmid pBin19-sgfp (Ghorbel et al. 1999), was used for transformation. Agrobacterium was cultured in YEB medium containing 50 mg l−1 kanamycin (Km) and 50 mg l−1 rifampicin with shaking overnight at 140 rpm at 28°C. Agrobacterium was collected by centrifugation and resuspended at a cell density of 1×108 cfu ml−1 in liquid 1/2 MS medium containing 50 g l−1 maltose and 100 µM acetosyringone. The ECs were immersed in the Agrobacterium suspension for 15 min and were then transferred on sterilized filter paper to remove excess Agrobacterium-containing medium. The ECs were co-cultured with Agrobacterium on solid 1/2 MS medium containing 5% maltose, 0.85% agar, and 100 µM acetosyringone for 4 days at 26°C in the dark. After co-cultivation, the ECs (about 2 mm each) were rinsed in 1/2 MS medium containing 5% maltose, 400 mg l−1 cefotaxime and cultured on the 1/2 MS selection medium containing 5% maltose, 15 mg l−1 Km and 200 mg l−1 cefotaxime and 0.85% agar in the dark for 4 weeks. Thereafter, the aggregated embryogenic cells (AEC) (aggregate size, 2 mm) were divided into three groups and were cultured for 1 month on the second selection medium, which consisted of 1/2 MS medium containing 5% maltose, 2% agar, and 0, 15, or 25 mg l−1 Km. The AEC selected on 25 mg l−1 Km were further transferred to the same fresh medium once a month for 2 months. Elongated embryos were transferred to 1/2 MS medium containing 5 µM zeatin, 3% sucrose, 25 mg l−1 Km, and 2% agar to promote shoot elongation under a 16 h light/8 h dark photoperiod. After true leaves appeared, regenerated plants were sub-cultured on 1/2MS medium containing 0.1 µM α-naphthaleneacetic acid, 2.5 µM 6-benzylaminopurine, 0.85% agar, and 3% sucrose (MSNB medium).
Two nodes of each regenerated plant were cut and cultured on MSNB medium in the dark for more than a month (two nodes were used, because not all nodes of axial buds always elongate), and a white elongated shoot in good condition per line was used to detect green fluorescent protein (GFP) fluorescence. GFP expression was visualized under an MZ FL III or M165FC stereo fluorescence microscope (Leica, Germany) with a 480/40 nm excitation filter and 510 nm barrier filter.
DNA analysis
PCR analysis and Southern hybridization were conducted to detect the GFP gene in transgenic plants. Genomic DNA was extracted from leaves with a DNeasy Plant Mini Kit (Qiagen, Germany) for PCR analysis, and with Genomic-tip 20/G (Qiagen) according to Yamamoto et al. (2006) for Southern blotting.
Primers 5′-GAT GTG ATA TCT CCA CTG ACG TAA G-3′, corresponding to the 35S promoter region, and 5′-GTA TAA TTG CGG GAC TCT AAT-3′, corresponding to the Nos terminator region, were used to amplify an about 1-kb fragment of the 35Spro-sgfp-Noster chimeric gene. Primers 5′-GGC TAT TCG GCT ATG ACT GG-3′ and 5′-CAT GTG TCA CGA CGA GAT CC-3′ were used to amplify an about 500-bp fragment of the nptII region. PCR amplification was performed in a total volume of 10 µl containing 5 ng of genomic DNA, 1 µl each of 10 µM primers, and 5 µl of GoTaq DNA polymerase (Promega, USA). Thermal cycler conditions were as follows: 95°C for 5 min; followed by 30 cycles of 1 min at 94°C, 1 min at 55°C, and 1.5 min at 72°C; then 7 min at 72°C.
For Southern hybridization, 5 µg of each genomic DNA sample was digested with EcoRI (Nippon Gene, Japan), separated by electrophoresis on a 1% agarose gel at 30 V for 15 h and blotted onto a nylon membrane (Hybond-N, Amersham, UK). Probes were prepared using a PCR DIG probe synthesis kit (Roche, Germany) and the primers GFP-F 5′-ATG GTG AGC AAG GGC GAG GAG CTG T-3′ and GFP-R 5′-TGA TGC CGT TCT TCT GCT TGT CGG CCA-3′. Hybridization was performed according to the DIG Application Manual (Roche) and signals were detected using a ChemiDoc Touch Imaging system (Bio-Rad, USA).
Results
EC induction and multiplication
Dormant canes were incubated at 26°C (16 h light/8 h dark) in an incubator. After about 2 weeks, buds began to grow. Flower buds were obtained after about a month of incubation. The induced flower bunches with loose flower buds (“slightly apart from each other”) were used as culture materials (Figure 1A). Flower buds at 5 days before anthesis (2.0–2.5 mm in length) were picked and sterilized, and the filaments were picked aseptically under a stereo microscope. Nine or 10 filaments were cultured in one flask containing 1D1T at 26°C in the dark.
Figure 1. Embryogenic callus induction and propagation. (A) Dormant cane with flower bunches after 1-month incubation (bar=10 cm). Inset shows flower buds that were slightly apart from each other and were used for culture (bar=5 mm). Arrows show the buds at optimal stage for culture. (B) Filament culture 1 month after initiation (bar=1 cm). (C) Callus generated from a filament 4 months after culture initiation. Arrows show somatic embryos (bar=5 mm). (D) Part of embryogenic culture was picked and cultivated on 1D3A5M medium. Calli shown were sub-cultured for about 4 months (bar=1 cm). (E) An EC of ‘Shine Muscat’ (macrograph of D) (bar=5 mm).

After 1 month of culture, calli were induced from 87 of 98 filaments (88.8%) (Figure 1B). These calli were transferred to 1D1TS medium to induce ECs. Three months after the transfer, somatic embryos were produced from 44 of 98 filaments (44.9%) (Figure 1C). Calli that included somatic embryos were transferred to 1D3A5M to propagate the ECs (Figure 1D, 1E). Part of each EC was transferred to MS hormone-free medium to check embryogenic ability. Embryogenic ability was considered confirmed if somatic embryos were induced 1 month after the transfer. Embryogenic ability was maintained for about 4 years by transferring ECs to new 1D3A5M medium once every 1.5 months.
Genetic transformation
Immediately after co-culture, GFP spots were observed in about half of the AEC (Figure 2A, B). After 1 month of culture on selection medium containing 15 mg l−1 Km, 45 of 120 (37.5%) AEC had GFP fluorescence; 24 of them had pro-embryos and embryos with GFP fluorescence. These 120 AEC were divided into three groups and incubated in the second selection medium containing 0 (39 AEC), 15 (39 AEC), or 25 mg l−1 Km (42 AEC). After 1 month of culture, embryo length reached about 1 mm (Figure 2C, D). The percentage of AEC with GFP-fluorescent embryos was highest (43.6%) on 15 mg l−1 Km and lowest (33.3%) without Km (Table 1). After 2 months of culture, the percentage of GFP-fluorescent embryos was highest (48.9%) on 25 mg l−1 Km and lowest (36.8%) without Km (Table 1). Afterwards, 7 AEC with GFP embryos selected on 25 mg l−1 Km were sub-cultured to regenerate transgenic plants. Embryos reached approximately 2 mm (Figure 2E, F) and 5 mm or more (Figure 2G) in darkness 2 and 3.5 months, respectively, after Agrobacterium infection. Most embryos showed GFP fluorescence (Figure 2H). In total, 49 plants (7 plants on average per cluster of AEC from 2 to 16 plants; each cluster was designated as no. 1 to no. 7) were regenerated 6 months after Agrobacterium infection. Most of the regenerated plants had normal phenotype (Figure 3A), but several had variegated leaves (Figure 3B). GFP fluorescence was observed in three of seven nodes, with different intensity (Figure 3C, D).
Figure 2. GFP fluorescence of ECs and somatic embryos during Agrobacterium co-culture and selection in darkness. (A, B) GFP spots of the AEC after co-culture (bar=1 mm). (C, D) Embryos were 1 mm in length at 1 month after Agrobacterium infection (bar=1 mm). (E, F) Embryos were approximately 2 mm in length at 2 months after the infection (bar=1 mm). (G, H) Embryos grown were ≥5 mm at 3.5 months after the infection (bar=5 mm).
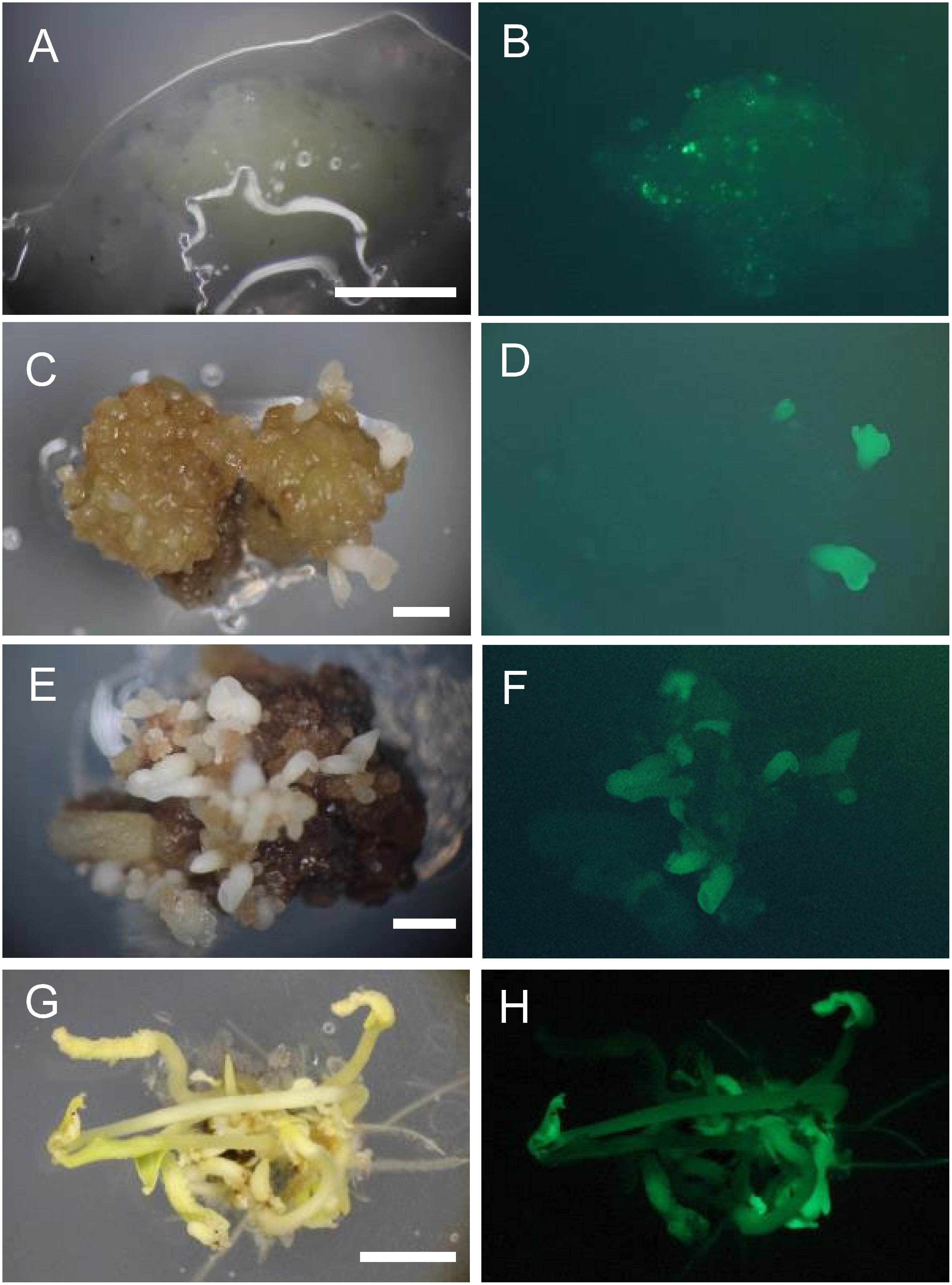
Table 1. Efficiency of transgenic embryo selection at different concentrations of kanamycin.
| Kanamycin concentration (mg l−1) | No. of AEC (A) | No. of AEC with GFP-fluorescent embryos (B)* | Percentage of AEC with GFP-fluorescent embryos (B/A×100) | No. of embryos developed (C)** | No. of GFP-Fluorescent embryos (D)** | Percentage of GFP-fluorescent embryos (D/C×100) |
|---|---|---|---|---|---|---|
| 0 | 39 | 13 | 33.3 | 95 | 35 | 36.8 |
| 15 | 39 | 17 | 43.6 | 166 | 65 | 39.2 |
| 25 | 42 | 17 | 40.5 | 180 | 88 | 48.9 |
AEC: Aggregated embryogenic cells. * After a month of culture. **After 2 months of culture.
Figure 3. Regenerated plants and their GFP fluorescence. (A) A regenerated plant (no. 4-2) with wild-type morphology and without GFP fluorescence. Bar=1 cm. (B) A regenerated plant (no. 3-3) with variegated leaves. Bar=1 cm. (C, D) GFP fluorescence of representative regenerated plants under a stereomicroscope with white incandescent light (C) and with 480-nm-excitation blue light (D). Cont, wild type (non-transformant). Lines 3-3 and 4-6 exhibited weak and strong fluorescence, respectively. Bar=1 cm.

DNA analysis
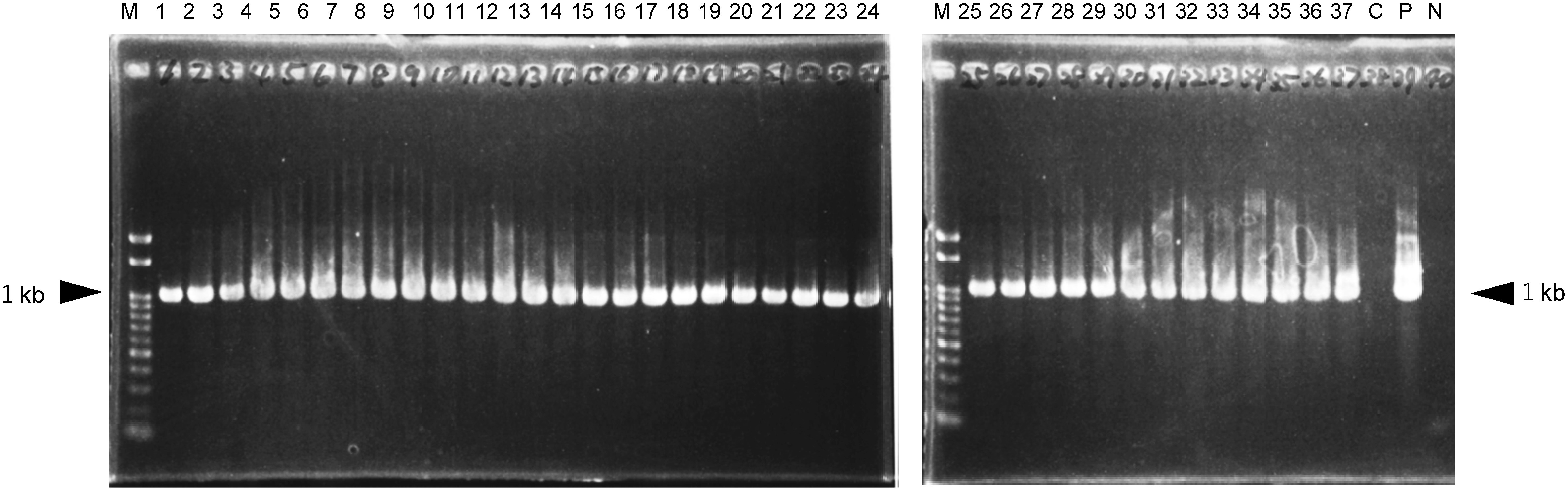
Genomic DNA was extracted from leaves of each regenerated plant. PCR analysis was performed to confirm the integration of sgfp. With primers corresponding to the 35S promoter and Nos terminator, all 49 regenerated plants yielded the expected 1-kb fragment (Figure 4). With nptII primers, 48 of the 49 regenerated plants yielded the expected 0.5-kb fragment (data not shown).
Figure 4. PCR analysis of the 35Spro-sgfp-Noster chimeric gene in regenerated plants. Lanes 1–37, regenerated plants. 1–5, plants regenerated from AEC no. 1; 6–9 from no. 2; 10–13 from no. 3; 14–17 from no. 4; 18–31 from no. 5; 32–35 from no. 6; 36 and 37 from no. 7. C, wild type; P, pBin19-sgfp plasmid; N, no DNA (negative control). M, 100-bp ladder.
In Southern hybridization with the sgfp gene probe, 2 to 8 bands were detected after cutting genomic DNA with EcoRI (Figure 5, Table 2), showing that the regenerated plants were transgenic. The band sizes differed among regenerated plants, even among plants regenerated from the same AEC (nos. 3-2 and 3-3, nos. 4-2 and 4-6), showing that plants were regenerated from different cells. Of 6 transgenic plants analyzed (nos. 1-6, 3-2, 3-3, 4-2, 4-6, 6-6), GFP fluorescence was detected only in nos. 3-3 and 4-6 (Figure 3C, D). One of the reasons for strong GFP fluorescence in no. 4-6 could be a low copy number (2 copies) of the gfp gene. No detection of GFP fluorescence in other plants could be partly ascribed to the relatively high copy number (4–8 copies), which may easily cause gene silencing (Schubert et al. 2004; Tang et al. 2007).
Figure 5. Southern blot analysis of regenerated plants. Genomic DNA was digested with EcoRI and probed with the sgfp fragment. Lane 1, wild type. Lanes 2–8, regenerated plants: nos. 1-6, 3-2, 3-3, 4-2, 4-6, 5-5, and 6-6 (from left to right). M, λ-HindIII digest.

Table 2. Number of DNA fragments detected by Southern blot analysis in Figure 5.
| Lane | DNA sample | No. of bands |
|---|---|---|
| 1 | WT | 0 |
| 2 | no. 1-6 | 4 |
| 3 | no. 3-2 | 8 |
| 4 | no. 3-3 | 2 |
| 5 | no. 4-2 | 6 |
| 6 | no. 4-6 | 3 |
| 7 | no. 5-5 | 2 |
| 8 | no. 6-6 | 3 |
Discussion
We established a procedure for somatic embryogenesis using flower organs from dormant canes of ‘Shine Muscat’ grape and Agrobacterium-mediated genetic transformation using ECs. Genetic transformation can be carried out all year round because ECs can be maintained for 4 years by sub-culturing them on fresh medium every 1.5–2 months. In PCR analysis, the transformation ratio of regenerated plants was close to 100% (all 49 regenerated plants were PCR-positive for the sgfp cassette, and 48 were PCR-positive for the nptII, Figure 5). In Southern blot analysis, positive sgfp signals were detected in all 7 plants analyzed (Figure 6). Our new procedure enables further physiological functional analysis of target genes through over-expression or knockdown experiments.
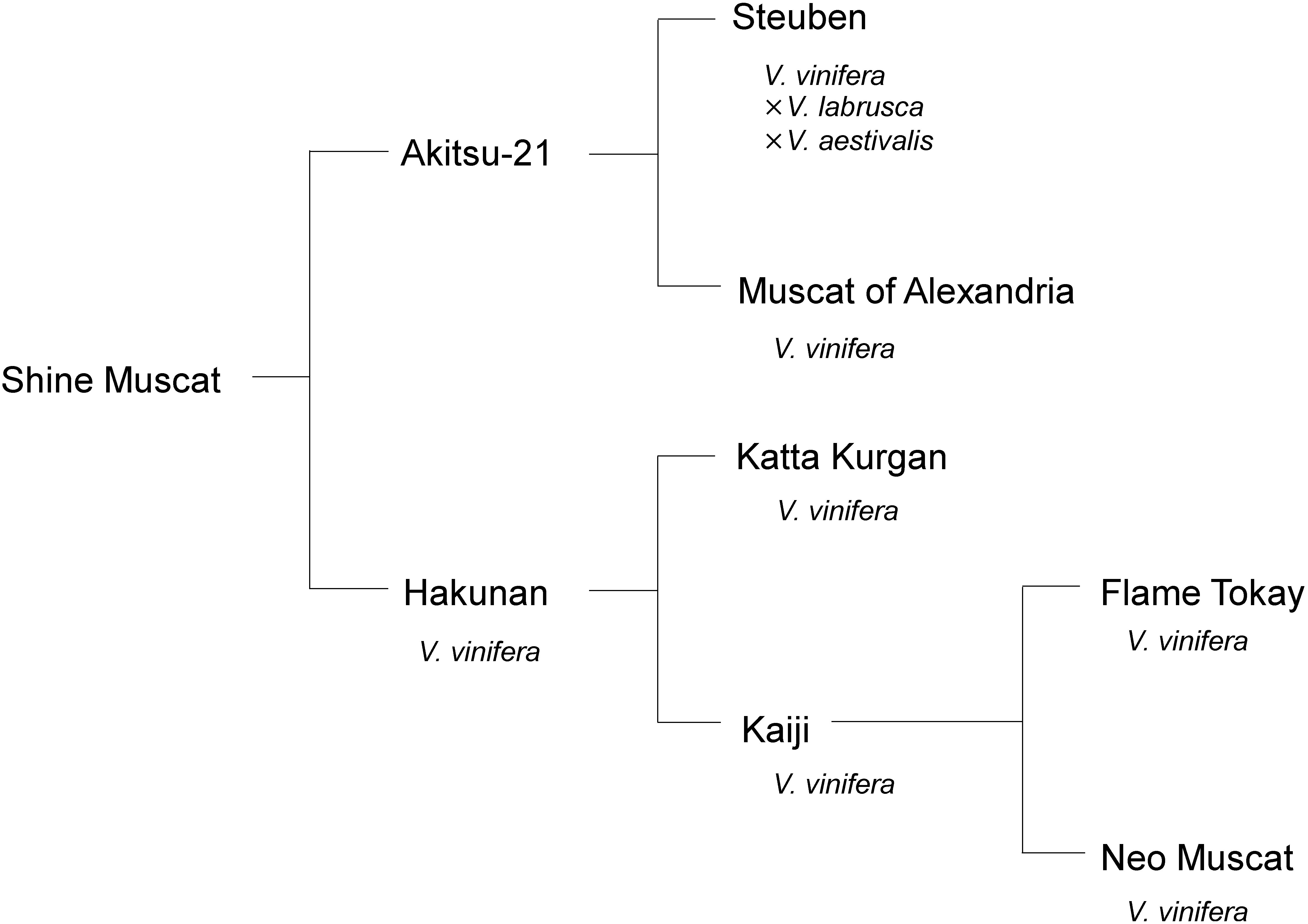
Figure 6. Pedigree of ‘Shine Muscat’ grape. Pedigree was modified from Yamada et al. (2008).
To induce ECs from vineyard-grown ‘Kyoho’ grape, an unfertilized ovule culture needed to be made from flower buds at 19–21 days before anthesis; at this stage, the flower buds are “slightly apart from each other” (see the Plant materials subsection for an explanation of this term) and have translucent yellow anthers (Nakajima et al. 2000). At a later stage, such as 7 days before anthesis, EC induction was not successful (Nakajima et al. 2000). Placing cut dormant canes in an incubator accelerated the development of anther filaments and unfertilized ovules (flower buds from dormant canes bloomed about 1 week after sampling) in comparison with that in the vineyard-grown trees. When the buds were “slightly apart from each other”, they tended to be small. Therefore, we have to pay attention to the developmental status of buds on the dormant cane incubated in a growth chamber because being “slightly apart from each other” could be important for EC induction. Another point to consider is the temperature of dormant cane incubation, which seems to be cultivar-specific: ‘Shine Muscat’ induces flower bunches at 26°C, whereas ‘Kyoho’ flower bunches die at 26°C before attaining the best stage for culture, and incubation at 22°C solves the problem.
Matsuta and Hirabayashi (1989) compared three embryogenic callus induction media (NN, B5, and MS), and concluded that NN medium (without vitamins, myo-inositol or glycine, and supplemented with 1 µM 2,4-D) was optimal. The ingredients and composition of 1/2 MS medium (Matsuta and Hirabayashi 1989) are similar to those of NN medium. Moreover, proliferation of ECs was improved to two- to threefold by using 1×MS vitamins in our previous study (Nakajima et al. 2000). Accordingly, 1/2 MS medium was used instead of NN medium. The use of a high concentration (3%) of agar seems to prolong the embryogenic ability of ECs, especially in V. vinifera cultivars. On 3% agar medium, ECs were compact and very hard. For maintaining ECs, it could be recommended to select compact and hard calli under a stereo microscope. If cultured on 0.85% agar (normal concentration), calli became friable and lost embryogenic ability faster than on 3% agar. The concentration of agar to maintain ECs seemed to depend on the cultivar, especially among V. labruscana cultivars. When many somatic embryos or non-ECs grow on 3% agar medium (1D3A5M) during the maintenance of ECs, the use of lower agar concentrations (e.g., 2%, 1.5%, or 0.85%) helps maintain ECs (Nakajima et al., unpublished observation) and around 0.8% agar (or 0.2% gellan gum) was used in many cases (Das et al. 2002; Hoshino et al. 1998; Perl et al. 1995; Scorza et al. 1996). Vitis vinifera originated in Europe, most likely between the Black and Caspian Seas (Einset and Pratt 1975), and is adapted to dry and moderately warm weather during its growing season (Yamada and Sato 2016). Vitis labruscana was bred in North America in a region with considerable rainfall (Yamada and Sato 2016). In the 19th century, V. vinifera and Vitis species native to North America, including V. labrusca L. (which provided large fruit, cold resistance, and distinctive flavor) were crossed and many new cultivars were developed. L. H. Bailey defined V. labrusca-like cultivars as V. labruscana; this species combines vineyard varieties that show strong V. labrusca likeness and are its derivatives or hybrids (Bailey and Bailey 1930). Therefore, the different origin might affect the optimal maintenance of ECs. The use of 3% or 2% agar medium had a good effect on somatic embryogenesis and plant regeneration, because it avoided vitrification and abnormal somatic embryogenesis, and steadily produced normal somatic embryos, which became normal regenerated plants.
After the establishment of a system for the induction of somatic embryos or ECs, one might consider the next step, Agrobacterium-mediated transformation. ECs were thought to be better than somatic embryos as explants for Agrobacterium infection. Matsuta et al. (1993) used somatic embryo mass, in which embryos showed various stages of development, for Agrobacterium infection. Though most embryos did not grow in the presence of Km, some survived and were white. These embryos did not germinate, suggesting that they were chimeras composed of transgenic and non-transgenic tissues. They were sub-cultured several times, and secondary or tertiary embryos developed and grew to transgenic plants (Matsuta et al. 1993). In our previous study (Nakajima et al. 2006), we used ECs for genetic transformation of ‘Kyoho’ grape, which was recalcitrant to genetic transformation, and obtained transformants. Zhou et al. (2014) also considered a proembryo mass to be a more appropriate tissue for genetic transformation than somatic embryos (transformation through secondary embryogenesis), because it took the least time (about 20 weeks) to develop transgenic somatic embryo lines. In our current study, the high transformation efficiency in a relatively short time could be attributed to the use of ECs for Agrobacterium infection.
Vitis vinifera tends to be relatively easy to transform in comparison with other grape species. Iocco et al. (2001) reported generation of transgenic plants of seven V. vinifera cultivars: ‘Cabernet Sauvignon,’ ‘Shiraz,’ ‘Chardonnay,’ ‘Riesling,’ ‘Sauvignon Blanc,’ ‘Chenin Blanc,’ and ‘Muscat Gordo Blanco’ (synonym of ‘Muscat of Alexandria’). In contrast, V. labruscana has low transformation efficiency or is impossible to transform (Motioike et al. 2002; Nakajima et al. 2006). The genetic background of ‘Shine Muscat’ is about 75% V. vinifera and 25% V. labruscana (Figure 6). ‘Muscat of Alexandria’ (V. vinifera), one of the ancestors of ‘Shine Muscat,’ shows high transformation efficiency (Nakajima et al., unpublished observation), suggesting a reason for the high transformation efficiency of ‘Shine Muscat.’
For the selection of transformants, the optimal Km concentration was 15 mg l−1 for ‘Kyoho’ (Nakajima et al. 2006) and 50 mg l−1 for ‘Neo Muscat’ (V. vinifera) (Yamamoto et al. 2000). Low Km concentrations may be more suitable for V. labruscana than for V. vinifera. Thus, the optimum Km concentration is assumed to be cultivar dependent.
The Agrobacterium strain LBA4404 used in this study allowed us to successfully establish a genetic transformation method in ‘Shine Muscat.’ In our previous report, a supervirulent strain EHA105 (Hood et al. 1993) was used for the transformation of recalcitrant ‘Kyoho’ grape (Nakajima et al. 2006). However, EHA105 tended to overgrow compared to LBA4404 when the antibiotic cefotaxime was used in ‘Kyoho’ transformation. According to Ogawa and Mii (2007), meropenem (at 25 mg l−1) completely suppresses overgrowth of Agrobacterium in the transformation of tobacco, tomato, and rice using LBA4404 and EHA101. Meropenem might also be useful instead of cefotaxime to suppress Agrobacterium overgrowth in grape transformation. Another Agrobacterium strain GV3101 has been used for grape transformation (Mezzetti et al. 2002; Zhao et al. 2017). Zhao et al. (2017) compared the efficiency of transformation of petiole segments of Vitis amurensis with EHA105, GV3101, and LBA4404. GV3101 showed the highest transformation efficiency (29.91%), but it did not differ significantly from that of EHA105 (25.31%). In contrast, the transformation efficiency of LBA4404 was 3.14%. Vitis amurensis produced transformed calli, but generation of transgenic calli was almost negligible when the procedure optimized for V. amurensis and EHA105 was used to transform V. vinifera ‘Muscat Hamburg’ and ‘Centennial Seedless.’ Zhao et al. (2017) concluded that the genetic background is key for successful petiole segment transformation. Thus, the choice of the Agrobacterium strain used to transform grape seems to depend on the cultivar.
Antioxidants may be needed to prevent the deterioration of Agrobacterium-infected explants. Motioike et al. (2002) found that Agrobacterium-infected ‘Niagara’ (V. labruscana) ECs showed browning that increased during the incubation, and most ECs died even in a medium containing an antioxidant (1% polyvinylpolypyrrolidone); in contrast, the browning was less severe in another V. labruscana cultivar, ‘Fredonia,’ and transformed cells could be selected on the same medium. Perl et al. (1996) assumed that necrogenesis after exposure to A. tumefaciens was caused by a hypersensitive response of the grape ECs to the bacterium. They found that a combination of polyvinylpolypyrrolidone and dithiothreitol improved plant viability and inhibited tissue necrosis of the V. vinifera cultivar ‘Superior Seedless’ (Perl et al. 1996). Activated charcoal (AC) was used in grape tissue culture (Oláh 2017). Though AC proved to be effective to reduce browning of calli in transformation experiments, it could also strongly modify the effect of the plant growth regulators added to the medium, probably because of adsorption of the chemicals by AC (Mozsár et al. 1998). Bouquet et al. (2006) proposed the use of 2.5 g l−1 AC for induction of somatic embryos and for co-cultivation on solid medium, while other researchers chose medium without AC. In our experiments, AC has not been used so far.
An increase in the copy number of transgene(s) in a transgenic plant tends to promote gene silencing. Tang et al. (2007) analyzed gfp transgene expression in eastern white pine transformants carrying different numbers of copies of T-DNA insertions. Post-transcriptional gene silencing was mostly observed in transgenic lines with four or more copies of T-DNA, but not in those with one copy. Schubert et al. (2004) reported that GFP transcript levels were high in Arabidopsis plants harboring up to four copies of the GFP gene, whereas five or more transgene loci under the control of the CaMV 35S promoter resulted in silencing. In our study, most of the elongated embryos (3.5 months after infection) showed GFP fluorescence (Figure 2H), demonstrating that Km effectively selected transformed embryos. Then, regenerated plantlets were maintained on medium without Km, implying that the lack of Km might be the reason for the high ratio of silencing of the sgfp gene. Further study will be needed to confirm whether silencing of nptII also occurred; maintaining regenerated plants on Km-containing medium might select transformants with low T-DNA copy number. Another cause of gene silencing might be the use of the 35S promoter. Mishiba et al. (2010) found transgene silencing in gentian in which the introduced 35S promoter region was methylated irrespective of the transgene copy number and integration loci, whereas transgenic tobacco bearing the same T-DNA showed no silencing. In our study, sgfp silencing depended on transgene copy number, although the 35S promoter was used. Therefore, there may be species-specific mechanisms of transgene silencing in plants. In addition, Schubert et al. (2004) reported that the expression of highly similar genes is additive unless transgene transcript levels exceed a gene-specific threshold and consequently trigger gene silencing, and the use of a weaker promoter (e.g., Nos promoter) should reduce variability of transgene expression in populations of transformants. Future studies would be required to examine levels of methylation and to suppress silencing of transgenes in grape.
RNA-guided genome editing using the CRISPR (clustered regularly interspaced short palindromic repeats)/Cas9 (CRISPR-associated protein 9) system has been applied successfully in many plant species, such as rice (Miao et al. 2013), wheat (Shan et al. 2013; Wang et al. 2014), poplar (Fan et al. 2015), tomato (Nonaka et al. 2017), apple (Nishitani et al. 2016), kiwifruit (Wang et al. 2018b), and banana (Naim et al. 2018). CRISPR/Cas9 was developed to induce DNA double-strand breaks at specific sites in the genome. These breaks have a high propensity to induce site-directed mutations through error-prone genome repair via non-homologous end joining. Generating such pinpoint mutations in a specific gene via CRISPR/Cas9 would have advantages over the strategies used in crossbreeding and Targeting Induced Local Lesions In Genomes (TILLING), especially because CRISPR/Cas9 often results in frame-shift mutations that inactivate the target genes. In grape, there have been four reports of genome editing using CRISPR/Cas9. In three of them, genome-edited plants were obtained using transformation with Agrobacterium (Nakajima et al. 2017; Ren et al. 2016; Wang et al. 2018a). Another one was the first report of direct delivery of purified CRISPR/Cas9 ribonucleoproteins to grape protoplasts by PEG-mediated transformation (Malnoy et al. 2016). Indel mutagenesis efficiency of the treated protoplasts was 0.1%. According to Malnoy et al. (2016), protoplast isolation, transfection, and transient gene expression in grape have not been sufficiently explored and most of the available methods have not been updated for two decades. Thus, Malnoy et al. (2016) updated and optimized the method that ensures protoplast viability, yield, and efficient transfection. However, plant regeneration from gene-edited protoplasts has not been achieved. Therefore, Agrobacterium-mediated genetic transformation combined with CRISPR/Cas9-mediated genome editing will be a promising method for targeted modification of genes in ‘Shine Muscat.’ An experiment aimed at changing the color of grape pericarp by combining Agrobacterium-mediated genetic transformation using ECs and a CRISPR/Cas9 vector in ‘Shine Muscat’ grape is now underway.
Acknowledgments
We thank Drs. T. Hirabayashi, S. Kobayashi, N. Matsuta, K. Notsuka, and Y. Nakamura for kind advice on grape culture and genetic transformation, and Drs. A. Sato, A. Azuma, and N. Onoue for plant materials. We also thank Mses. M. Kimura, R. Iwanami, N. Inose, and N. Ito for their technical assistance. A part of this work was supported by the Cabinet Office, Government of Japan, Cross-Ministerial Strategic Innovation Promotion Program (SIP) phase I, “Technologies for creating next-generation agriculture, forestry and fisheries” (funding agency: Bio-oriented Technology Research Advancement Institution, NARO).
Abbreviations
- AC
activated charcoal
- AEC
aggregated embryogenic cells
- 2,4-D
2,4-dichlorophenoxyacetic acid
- EC
embryogenic callus
- GFP
green fluorescent protein
- Km
kanamycin
- Nospro
nopaline synthase promoter
- Noster
nopaline synthase terminator
- nptII
neomycin phosphotransferase II
- sgfp
synthetic GFP (S65T) mutant
- 35Spro
35S promoter
References
- Bailey LH, Bailey EZ (1930) HORTUS, a Concise Dictionary of Gardening, General Horticulture and Cultivated Plants in North America. The Macmillan Company, New York
- Bouquet A, Torregrosa L, Iocco P, Thomas MR (2006) Grapevine (Vitis vinifera L.). Methods Mol Biol 344: 273–285 [DOI] [PubMed] [Google Scholar]
- Das DK, Reddy MK, Upadhyaya KC, Sopory SK (2002) An efficient leaf-disc culture method for the regeneration via somatic embryogenesis and transformation of grape (Vitis vinifera L.). Plant Cell Rep 20: 999–1005 [Google Scholar]
- Dutt M, Li ZT, Dhekney SA, Gray DJ (2007) Transgenic plants from shoot apical meristems of Vitis vinifera L. “Thompson Seedless” via Agrobacterium-mediated transformation. Plant Cell Rep 26: 2101–2110 [DOI] [PubMed] [Google Scholar]
- Einset J, Pratt C (1975) Grapes. In: Janick J, Moore JN (eds) Advances in Fruit Breeding. Purdue University Press, West Lafayette, Indiana, USA, pp 130–153
- Fan D, Liu T, Li C, Jiao B, Li S, Hou Y, Luo K (2015) Efficient CRISPR/Cas9-mediated targeted mutagenesis in Populus in the first generation. Sci Rep 5: 12217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghorbel R, Juárez J, Navarro L, Peña L (1999) Green fluorescent protein as a screenable marker to increase the efficiency of generating transgenic woody fruit plants. Theor Appl Genet 99: 350–358 [Google Scholar]
- Hirabayashi T, Akihama T (1982) In vitro embryogenesis and plant regeneration from the anther-derived callus of Vitis. Proc 5th Intl Cong Plant Tissue and Cell Culture: 547–548
- Hood EE, Gelvin SB, Melchers LS, Hoekema RE (1993) New Agrobacterium helper plasmids for gene transfer to plants. Transgenic Res 2: 208–218 [Google Scholar]
- Hoshino Y, Zhu YM, Nakano M, Takahashi E, Mii M (1998) Production of transgenic grapevine (Vitis vinifera L. cv. Koshusanjaku) plants by co-cultivation of embryogenic calli with Agrobacterium tumefaciens and selecting secondary embryos. Plant Biotechnol 15: 29–33 [Google Scholar]
- Iocco P, Franks T, Thomas MR (2001) Genetic transformation of major wine grape cultivars of Vitis vinifera L. Transgenic Res 10: 105–112 [DOI] [PubMed] [Google Scholar]
- Kandel R, Bergey DR, Dutt M, Sitther V, Li ZT, Grey DJ, Dhekney SA (2016) Evaluation of a grapevine-derived reporter gene system for precision breeding of Vitis. Plant Cell Tissue Organ Cult 124: 599–609 [Google Scholar]
- Kono A, Sato A, Ban Y, Mitani N (2013) Resistance of Vitis germplasm to Elsinoë ampelina (de Bary) Shear evaluated by lesion number and diameter. HortScience 48: 1433–1439 [Google Scholar]
- Malnoy M, Viola R, Jung MH, Koo OJ, Kim S, Kim JS, Velasco R, Kanchiswamy CN (2016) DNA-free genetically edited grapevine and apple protoplast using CRISPR/Cas9 ribonucleoproteins. Front Plant Sci 7: 1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinelli L, Mandolino G (1994) Genetic transformation and regeneration of transgenic plants in grapevine (Vitis rupestris S.). Theor Appl Genet 88: 621–628 [DOI] [PubMed] [Google Scholar]
- Matsuta N (1992) Effect of auxin on somatic embryogenesis from leaf callus in grape (Vitis spp.). Japan J Breed 42: 879–883 [Google Scholar]
- Matsuta N, Hirabayashi T (1989) Embryogenic cell lines from somatic embryos of grape (Vitis vinifera L.). Plant Cell Rep 7: 684–687 [DOI] [PubMed] [Google Scholar]
- Matsuta N, Iketani H, Hayashi T (1993) Transformation in grape and kiwifruit. In: Hayashi T, Omura M, Scott NS (eds) Techniques on Gene Diagnosis and Breeding in Fruit Trees. FTRS/ Japan, pp 184–192
- Marchive C, Léon C, Kappel C, Coutos-Thévenot P, Corio-Costet M, Delrot S, Lauvergeat V (2013) Over-expression of VvWRKY1 in grapevine induces expression of jasmonic acid pathway-related genes and confers higher tolerance to the downy mildew. PLoS One 8: e54185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mezzetti B, Pandolfini T, Navacchi O, Landi L (2002) Genetic transformation of Vitis vinifera via organogenesis. BMC Biotechnol 2: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao J, Guo D, Zhang J, Huang Q, Qin G, Zhang X, Wan J, Gu H, Qu LJ (2013) Targeted mutagenesis in rice using CRISPR-Cas system. Cell Res 23: 1233–1236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishiba K, Yamasaki S, Nakatsuka T, Abe Y, Daimon H, Oda M, Nishihara M (2010) Strict de novo methylation of the 35S enhancer sequence in gentian. PLoS One 5: e9670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motioike SY, Skirvin RM, Norton MA, Otterbacher AG (2002) Development of methods to genetically transform American grape (Vitis×labruscana L. H. Bailey). J Hortic Sci Biotechnol 77: 691–696 [Google Scholar]
- Mozsár J, Vicián O, Süle S (1998) Agrobacterium-mediated genetic transformation of an interspecific grapevine. Vitis 37: 127–130 [Google Scholar]
- Murashige T, Skoog F (1962) A revised medium for rapid growth and bioassays with tobacco tissue cultures. Physiol Plant 15: 473–497 [Google Scholar]
- Naim F, Dugdale B, Kleidon J, Brinin A, Shand K, Waterhouse P, Dale J (2018) Gene editing the phytoene desaturase alleles of Cavendish banana using CRISPR/Cas9. Transgenic Res 27: 451–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima I, Ban Y, Azuma A, Onoue N, Moriguchi T, Yamamoto T, Toki S, Endo M (2017) CRISPR/Cas9-mediated targeted mutagenesis in grape. PLoS One 12: e0177966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima I, Kobayashi S, Nakamura Y (2000) Embryogenic callus induction and plant regeneration from unfertilized ovule of ‘Kyoho’ grape. J Jpn Soc Hortic Sci 69: 186–188 [Google Scholar]
- Nakajima I, Matsuta N (2003) Somatic embryogenesis from filaments of Vitis vinifera L. and Vitis labruscana Bailey. Vitis 42: 53–54 [Google Scholar]
- Nakajima I, Matsuta N, Yamamoto T, Terakami S, Soejima J (2006) Genetic transformation of ‘Kyoho’ grape with a GFP gene. J Jpn Soc Hortic Sci 75: 188–190 [Google Scholar]
- Nakano M, Sakakibara T, Watanabe Y, Mii M (1997) Establishment of embryogenic cultures in several cultivars of Vitis vinifera and V.×labruscana. Vitis 36: 141–145 [Google Scholar]
- Nishitani C, Hirai N, Komori S, Wada M, Okada K, Osakabe K, Yamamoto T, Osakabe Y (2016) Efficient genome editing in apple using a CRISPR/Cas9 system. Sci Rep 6: 31481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonaka S, Arai C, Takayama M, Matsukura C, Ezura H (2017) Efficient increase of γ-aminobutyric acid (GABA) content in tomato fruits by targeted mutagenesis. Sci Rep 7: 7057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notsuka K, Tsuru T, Matsumoto R (1992) Somatic embryo production from unfertilized ovules of grape cultivar, ‘Neo Muscat’. J Jpn Soc Hortic Sci 61: 98–99 (in Japanese) [Google Scholar]
- Ogawa Y, Mii M (2007) Meropenem and moxalactam: Novel β-lactam antibiotics for efficient Agrobacterium-mediated transformation. Plant Sci 172: 564–572 [Google Scholar]
- Oláh R (2017) The use of activated charcoal in grapevine tissue culture. Vitis 56: 161–171 [Google Scholar]
- Perl A, Lotan O, Abu-Abied M, Holland D (1996) Establishment of an Agrobacterium-mediated transformation system for grape (Vitis vinifera L): The role of antioxidants during grape-Agrobacterium interactions. Nat Biotechnol 14: 624–628 [DOI] [PubMed] [Google Scholar]
- Perl A, Saad S, Sahar N, Holland D (1995) Establishment of long-term embryogenic cultures of seedless Vitis vinifera cultivars: A synergistic effect of auxins and the role of abscisic acid. Plant Sci 104: 193–200 [Google Scholar]
- Ren C, Liu X, Zhang Z, Wang Y, Duan W, Li S, Liang Z (2016) CRISPR/Cas9-mediated efficient targeted mutagenesis in Chardonnay (Vitis vinifera L.). Sci Rep 6: 32289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabbadini S, Capriotti L, Molesini B, Pandolfini T, Navacchi O, Limera C, Ricci A, Mezzetti B (2019) Comparison of regeneration capacity and Agrobacterium-mediated cell transformation efficiency of different cultivars and rootstocks of Vitis spp. via organogenesis. Sci Rep 9: 582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert D, Lechtenberg B, Forsbach A, Gils M, Bahadur S, Schmidt R (2004) Silencing in Arabidopsis T-DNA transformants: The predominant role of a gene-specific RNA sensing mechanism versus position effects. Plant Cell 16: 2561–2572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scorza R, Cordts JM, Gray DJ, Gonsalves D, Emershad RL, Ramming DW (1996) Production transgenic ‘Thompson Seedless’ grape (Vitis vinifera L.) plants. J Am Soc Hortic Sci 121: 616–619 [Google Scholar]
- Shan Q, Wang Y, Li J, Zhang Y, Chen K, Liang Z, Zhang K, Liu J, Xi JJ, Qiu JL, et al. (2013) Targeted genome modification of crop plants using a CRISPR-Cas system. Nat Biotechnol 31: 686–688 [DOI] [PubMed] [Google Scholar]
- Srinivasan C, Mullins MG (1980) High-frequency somatic embryo production from unfertilized ovules of grapes. Sci Hortic (Amsterdam) 13: 245–252 [Google Scholar]
- Tang W, Newton RJ, Weidner DA (2007) Genetic transformation and gene silencing mediated by multiple copies of transgene in eastern white pine. J Exp Bot 58: 545–554 [DOI] [PubMed] [Google Scholar]
- Wang X, Tu M, Wang D, Liu J, Li Y, Li Z, Wang Y, Wang X (2018a) CRISPR/Cas9-mediated efficient targeted mutagenesis in grape in the first generation. Plant Biotechnol J 16: 844–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Cheng X, Shan Q, Zhang Y, Liu J, Gao C, Qui JL (2014) Simultaneous editing of three homoeoalleles in hexaploid bread wheat confers heritable resistance to powdery mildew. Nat Biotechnol 32: 947–951 [DOI] [PubMed] [Google Scholar]
- Wang Z, Wang S, Li D, Zhang Q, Li L, Zhong C, Liu Y, Huang H (2018b) Optimized paired-sgRNA/Cas9 cloning and expression cassette triggers high-efficiency multiplex genome editing in kiwifruit. Plant Biotechnol J 16: 1424–1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X, Agüero CB, Wang Y, Walker MA (2016) Genetic transformation of grape varieties and rootstocks via organogenesis. Plant Cell Tissue Organ Cult 126: 541–552 [Google Scholar]
- Yamada M, Sato A (2016) Advances in table grape breeding in Japan. Breed Sci 66: 34–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada M, Yamane H, Sato A, Hirakawa N, Iwanami H, Yoshinaga K, Ozawa T, Mitani N, Shiraishi M, Yoshioka M, et al. (2008) New grape cultivar ‘Shine Muscat.’ Bull Natl Fruit Tree Res Stn 7: 21–38 (in Japanese) [Google Scholar]
- Yamamoto T, Iketani H, Ieki H, Nishizawa Y, Notsuka K, Hibi T, Hayashi T, Matsuta N (2000) Transgenic grapevine plants expressing a rice chitinase with enhanced resistance to fungal pathogens. Plant Cell Rep 19: 639–646 [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Kimura T, Hayashi T, Ban Y (2006) DNA profiling of fresh and processed fruits in pear. Breed Sci 56: 165–171 [Google Scholar]
- Yamamoto T, Nakajima I, Matsuta N (2003) Transgenic grape. In: Jaiwal PK, Singh RP (eds) Plant Genetic Engineering Vol. 6 Improvement of Fruit Crops. Sci Tech Publishing LLC, USA, pp 65–82
- Zhao T, Wang Z, Su L, Sun X, Cheng J, Zhang L, Karungo SK, Han Y, Li S, Xin H (2017) An efficient method for transgenic callus induction from Vitis amurensis petiole. PLoS One 12: e0179730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Dai L, Cheng S, He J, Wang D, Zhang J, Wang Y (2014) A circulatory system useful both for long-term somatic embryogenesis and genetic transformation in Vitis vinifera L. cv. Thompson Seedless. Plant Cell Tissue Organ Cult 118: 157–168 [Google Scholar]