

SUMMARY
Sensory inputs activate sparse neuronal ensembles in the dentate gyrus of the hippocampus, but how eligibility of individual neurons to recruitment is determined remains elusive. We identify thousands of largely bistable (CpG methylated or unmethylated) regions within neuronal gene bodies, established during mouse dentate gyrus development. Reducing DNA methylation and the proportion of the methylated epialleles at bistable regions compromises novel context-induced neuronal activation. Conversely, increasing methylation and the frequency of the methylated epialleles at bistable regions enhances intrinsic excitability. Single-nucleus profiling reveals enrichment of specific epialleles related to a subset of primarily exonic, bistable regions in activated neurons. Genes displaying both differential methylation and expression in activated neurons define a network of proteins regulating neuronal excitability and structural plasticity. We propose a model in which bistable regions create neuron heterogeneity and constellations of exonic methylation, which may contribute to cell-specific gene expression, excitability, and eligibility to a coding ensemble.
Graphical Abstract
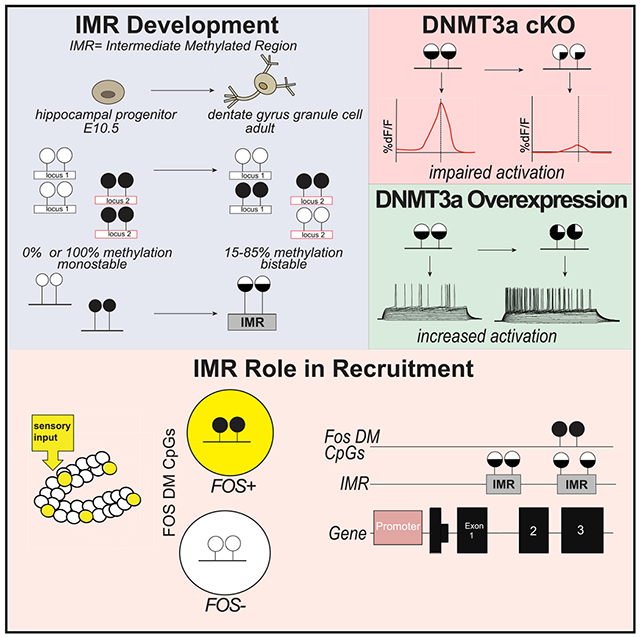
In Brief
Odell et al. show regions within neuronal genes with bistable DNA methylation states that are associated with gene expression, excitability, and activation in the dentate gyrus of the hippocampus. These data suggest that the methylation state of bistable regions dictates, via modulating gene expression, neuron eligibility to a coding ensemble.
INTRODUCTION
Neurocomputational and experimental models predict that environmental inputs are encoded by small and largely distinct ensembles of hippocampal neurons (Rolls and Treves, 2011). Sparsely distributed coding is thought to be an efficient way to produce high representational capacity with low interference. Sparse coding is particularly prominent in the dentate gyrus (DG) of the hippocampus because of the unusually low firing rates of DG granule cells (DGCs) and their higher number, relative to their number of entorhinal cortex input and CA3 (cornu ammonis 3) output neurons (Leutgeb et al., 2007). The first step in coding is the recruitment of a subpopulation of neurons to an ensemble. Neurotransmitters, paracrine signals, and hormones can prime neurons to increase their chance of recruitment (Abraham and Tate, 1997). For example, synaptically released acetylcholine, by inhibiting the KV7/M current via increased axonal Ca2+, lowers the action potential, enhancing intrinsic excitability and synaptic potential-spike coupling in DGCs (Martinello et al., 2015). Yet the state of the neuron receiving these modulatory inputs is also essential (Picciotto et al., 2012), because neurons with high intrinsic excitability are preferentially recruited during context exposure (Park et al., 2016). Excitability is increased by a host of factors, from changes in the level and function of voltage-gated sodium and potassium channels to transcription factors such as activated CREB (Han et al., 2007). However, it is not known how these, or as-yet unidentified excitability-related molecules, are regulated to produce a small and ever-changing (Cai et al., 2016) population of neurons eligible for coding at a given time. Epigenetics (referring to modifications to the genome that do not involve a change in the nucleotide sequence), because of its cell-to-cell variability, may explain why one cell is recruited to a coding ensemble, whereas a neighboring cell is not.
RESULTS
Emergence of Intermediate Methylated Regions in Neuronal Gene Bodies during Hippocampal Development
We found evidence for the developmental emergence of epigenetic heterogeneity in otherwise morphologically homogeneous and genetically identical dorsal DGCs (dDGCs) in male C57BL/6 male mice (Sharma et al., 2016). DGCs or their progenitors were microdissected from the granule-cell layer of hippocampal slices at embryonic day (E) 10.5, postnatal day (P) 6, or 10–12 weeks of age, followed by profiling cytosine methylation at CpG and non-CpG sites by an enhanced version of reduced representation bisulfite sequencing (RRBS) (Akalin et al., 2012a) (Figure 1A). Loss and gain of methylation during the transition of P6 to adult DGCs produced 170,198 differentially methylated (DM) CpG sites (q < 0.01, RRBS ≥ 10× coverage, average change 22.23%). In adult DGCs, most DM CpGs were not fully methylated or unmethylated but rather were in a state of intermediate methylation (IM) (i.e., were methylated across the entire 0% to 100% range) (Figure 1C, pink). Many of these sites were already IM at P6 (Figure 1C, blue), indicating that the IM process began earlier. Indeed, comparing adult DGCs with their earlier, E10.5, hippocampal progenitors (HPs) still yielded DM sites (282,155 with an average change of 26.80%), which were IM in adult DGCs (Figure 1B, pink). These sites exhibited more genome-typical bimodal methylation distribution in HPs (Figure 1B, blue). IM in DGCs was unexpected, because CpG sites in morphologically homogeneous and genetically identical cells are typically uniformly methylated or unmethylated in all cells (i.e., most CpG sites are in the 0%–10% and 90%–100% methylation range), resulting in bimodal methylation distribution, as was the case at CpGs outside of the developmental IM CpGs (Figures 1D and 1E).
Figure 1. Emergence of Genomic Regions with IM during Hippocampal Development.
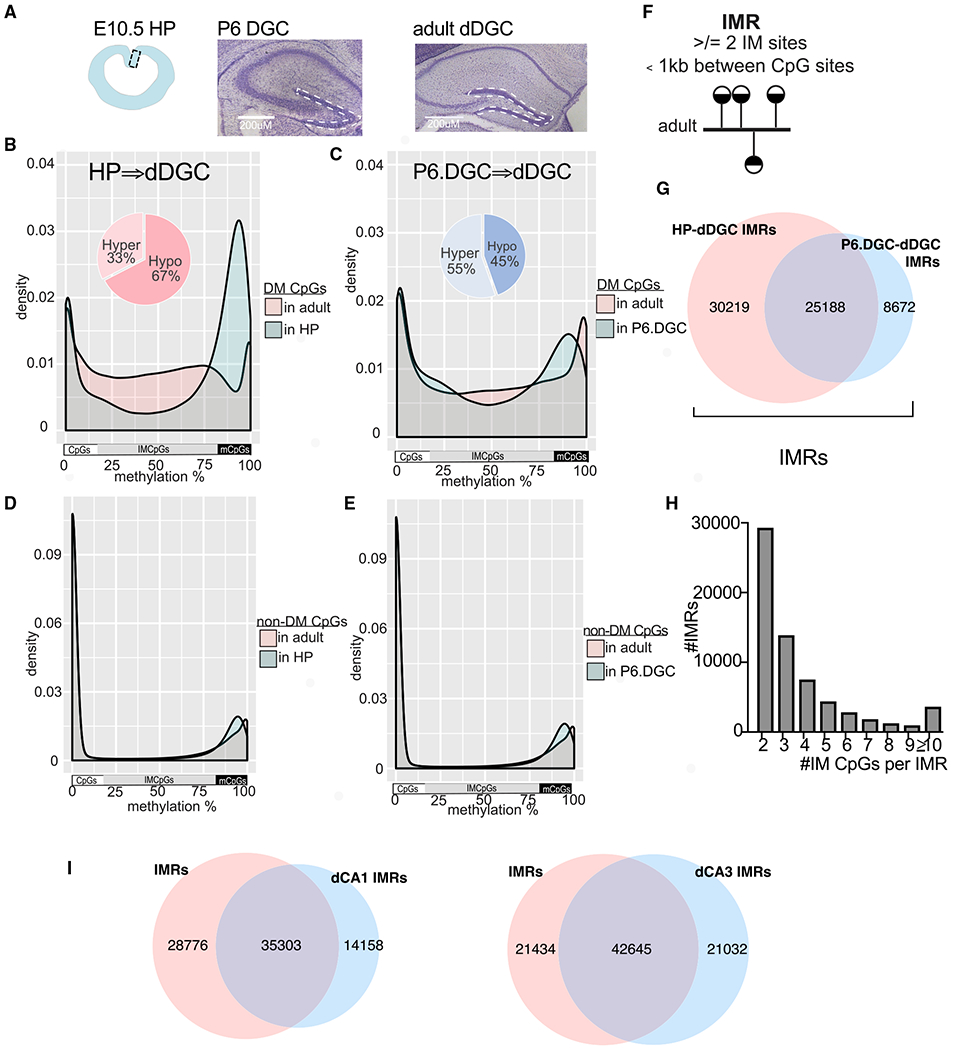
(A) Cells used in DNA methylation profiling. HPs, P6.DGCs, and adult DGCs were microdissected from E10.5, P6, and adult brains, respectively (scale bars, 200 μm).
(B and C) Distribution of methylation levels at clustered (≥2) and DM CpGs between E10.5 HPs and adult DGCs (B) and between P6.DGCs and adult DGCs (C). Methylation at E10.5 is bimodal and later becomes IM.
(D and E) Methylation at non-DM CpGs between HPs and adult DGCs (D) and P6.DGCs and adult DGCs (E) is bimodal.
(F) IMR definition: regions with ≥2 clustered IM CpGs in adult DGCs.
(G) IMRs include HP-dDGC IMRs and P6.DGC-dDGC IMRs.
(H) Distribution of IMRs according to their number of IM CpGs.
(I) Overlap of IMRs and CA1/CA3 IMRs in adult neurons.
See also Figure S1.
Around 80% of the IM CpGs, produced during the P6.DGC-to-DGC and HP-to-DGC transitions, clustered to IM regions (IMRs) of 2 or more IM CpGs, with an average size of 266 and 300 bp, respectively (Figures 1F–1H). The large overlap of P6.DGC-DGC and HP-DGC IMRs indicated selective DNA methylation/demethylation dynamics at the same CpGs across neuronal development, but the unique set of P6.DGC-DGC IMRs indicated additional methylation changes during DGC maturation. Because of the presence of IM at these regions in adult DGCs, all further analyses were performed on the combined set of IMRs (HP-DGC and P6.DGC-DGC) (Figure 1G; see Tables S1A and S1B for links to custom tracks in the Integrative Genomics Viewer [IGV]). Overall, we identified 64,079 autosomal IMRs that on average contained 4 IM CpGs.
The IM CpG profile of adult DGCs pooled from three males (displayed in Figure 1) was highly similar to that of six individual males (r2 = 0.89–0.90) (Figures S1A and S1B). This indicates that developmental IMRs do not result from interindividual variation in methylation, as reported at transposon-derived sequences (Kazachenka et al., 2018). Rather, the IM pattern in adult DGCs arises stochastically during development. IMRs are not related to genomic imprinting; they were not exclusively methylated around 50% (Figures 1B and 1C, pink) and did not map to classical imprinting control regions or non-canonical imprinted genes (Bonthuis et al., 2015; Xie et al., 2012). IMRs are also different from so-called partially methylated domains (PMDs), which are large regions spanning hundreds of kilobases, covering almost 40% of the genome of some cells (Gaidatzis et al., 2014). Finally, IM did not result from differential methylation between developmentally and adult-born DGCs, because immature (<6 weeks old) neurons make up less than 10% of the total population size (Cameron and McKay, 2001), which is below the relative proportion of most methylated or unmethylated IMR CpGs in the DGC population.
IMRs were not limited to DGCs in the hippocampus. Comparing the methylome of young, E17.5 CA neurons and adult CA1 or CA3 cells microdissected from pyramidal layers yielded 49,461 and 63,677 autosomal regions, respectively, exhibiting bimodal distribution in E17.5 CA cells but exhibiting IM in adult neurons (Figures S1C and S1D). DGC, CA1, and CA3 IMRs showed extensive overlaps, consistent with their common origin and shared neurotransmitter phenotype (Figure 1I).
Analysis of individual sequence reads from DGCs (Figure 2A) showed that two adjacent IMR CpGs more frequently occupy the same epigenetic state than occupy the opposite states (positive correlation with Fisher’s test, adjusted p < 0.05) (Figure 2B). Extending this analysis to multiple (>2) CpGs showed that neighboring CpGs tend to be uniformly methylated/unmethylated (Figure 2C). Methylation profiling also showed the emergence of IM cytosines in CpA, CpT, and CpC dinucleotide contexts during DG maturation, but IM CpHs were not enriched at IMRs.
Figure 2. IMRs Are Largely Bistable Regions Enriched in Neuronal Gene Bodies.
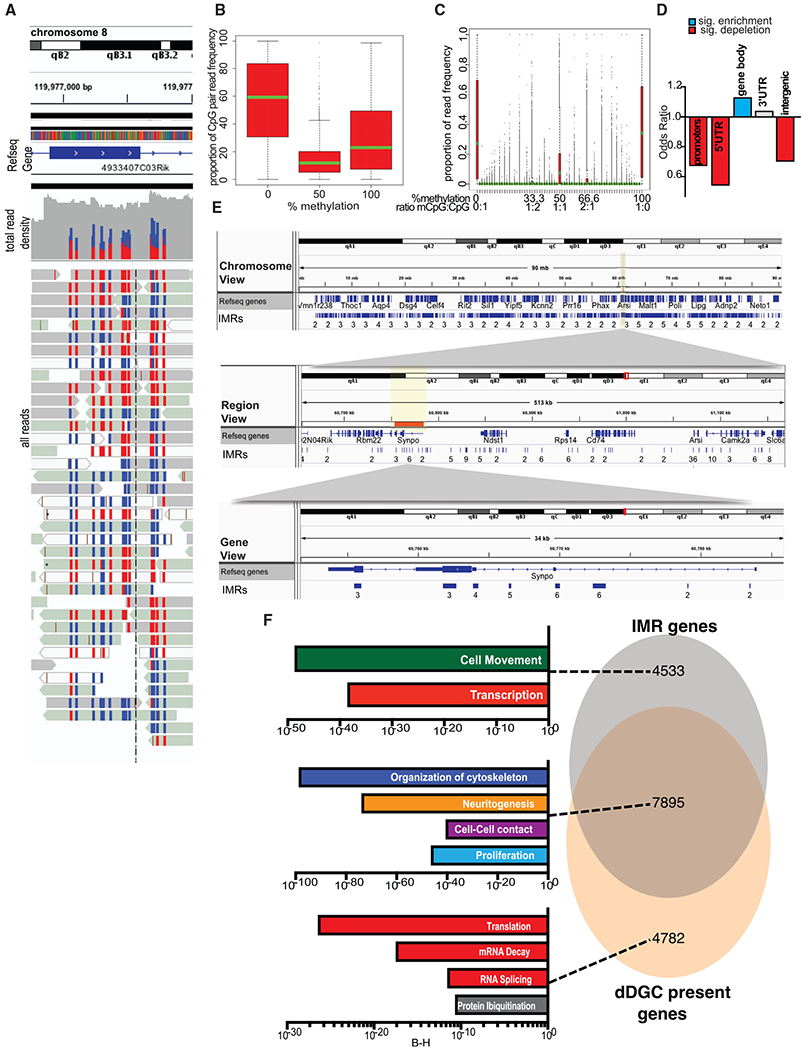
(A) Snapshot of individual whole-genome bisulfite sequencing (WGBS) reads at an IMR. CpGs are either methylated (red) or unmethylated (blue). RRBS produced a similar result.
(B) Two adjacent IM CpGs in adult DGCs tend to occupy the same methylation state. Means, SDs; 5/95 percentiles are indicated.
(C) Neighboring IMR CpGs in individual reads (~4.37/IMR).
(D) IMR genomic feature enrichment.
(E) Representative IGV browser view of Synpo1 with custom IMR tracks showing multiple IMRs spanning exons and introns. Numbers under regions indicate the number of CpGs within an IMR.
(F) DGC-expressed IMR gene enrichment (IPA, Benjamini-Hochberg corrected p values). Gene expression is from single PROX1+ DGC nuclei, separated from other dorsal hippocampal neuronal and non-neuronal nuclei by FACS.
Because some of the methylation signal in bisulfite sequencing (BS) can originate from 5-hydroxymethylated cytosine (5hmC), an intermediary between 5-methylcytosine (5mC) and 5-cytosine (5C) with potentially unique regulatory functions (Kozlenkov et al., 2018), 5mC-selective oxidative BS was performed. However, 63.87% of IMRs were not hydroxymethylated, and those that were (36.13%) contained only 1.56 5-hydroxymethylated CpG (5hmCpG) per IMR. Furthermore, intermediate hydroxymethylated CpGs were not enriched at IMRs (Figure S1E); therefore, hydroxymethylation at IMRs likely reflects ongoing demethylation across the genome (Skvortsova et al., 2017). Finally, IMRs that contain hydroxymethylation retained their IM profile even after the removal of 5hmC sites (Figure S1F).
IMRs were slightly enriched in CpG shelves (odds ratio [OR] = 1.15, p = 2.36E–9) but depleted in CpG islands (OR = 0.66, p < 2.20E–16). Interestingly, IMRs were enriched in genic regions but depleted in intergenic areas. Within genic regions, IMRs were depleted in promoters and 5′ UTRs but enriched in gene bodies, exons, introns, and last exon/3′ UTRs (Figure 2D). The IGV view of representative IMRs in the gene body of Synpo, encoding the actin-associated protein synaptopodin (Kremerskothen et al., 2005; Mundel et al., 1997), is displayed in Figure 2E.
Consistent with their high number and preferential gene-body association, IMRs were found in more than 12,000 genes mapped in Ingenuity Pathway Analysis (IPA), ~2/3rd of which were expressed in adult DGCs (Figure 2F; see Tables S1C and S2 for IMR- and DGC-expressed genes). Expressed genes containing IMRs showed the most significant enrichment in four main functions: (1) cytoskeleton organization, (2) neurite morphogenesis, (3) cell-to-cell interactions/contact, and (4) proliferation (Figure 2F, IPA). The remaining ~1/3rd of IMR genes, not expressed/detected in our assay, were related to functions in cell movement and transcription. These IMR genes may have been expressed earlier (e.g., cell movement) or expressed below the detection level in adult DGCs (e.g., transcription factors). DGC-expressed genes with no IMRs were associated with more universal cellular functions, such as RNA splicing, mRNA decay, translation, and protein ubiquitination.
Reducing DNA Methylation and the Proportion of the Methylated IMR Epialleles Impair Neuronal Activation
Given their DNA methylation-based heterogeneity, association with gene bodies, and functional enrichments, we hypothesized that IMRs, via their combinatorial methylation states, individualize the structure and connectivity of DGCs and that the resulting cellular heterogeneity is essential to support specific neuronal functions and behaviors. Because methylation bistability suggested sensitivity to the dosage of DNA methyltransferases (DNMTs), we tested whether IMR methylation can be reduced en masse by the downregulation of neuronal Dnmt3a. Although Dnmt3a has been implicated in the ensuing process of memory formation (Levenson et al., 2006), its role in neuronal recruitment is unknown. Partial (~75%) and conditional deletion of Dnmt3a (conditional knockout [cKO]) in nestin-creERT2 mice (Chen et al., 2009) by a single dose of tamoxifen (TAM) at E13.5 (Sharma et al., 2016), a time point selected to interfere with the emergence of IMRs (Figure 3A), resulted in the hypomethylation of a substantial number of IM CpG sites (18,825, q < 0.01) (Figure 3B). CpGs that were not affected in cKO DGCs exhibited bimodal distribution (Figure 3C), indicating that hypomethylation in cKO cells was largely limited to IM CpGs. Hypomethylated CpGs clustered to 4,194 DM regions (DMRs, ≥2 CpGs) that highly overlapped with the identified IMRs (85.96%, OR = 5.52, p < 2.20E–16) (Figure 3D; see Table S1B for the IGV track). Furthermore, essentially all genes associated with cKO-DMRs (98.09%) were also IMR genes (Figure 3E). However, in line with the partial nature of the cKO, not all IMR genes were affected, and only some IMRs within genes with multiple IMRs were hypomethylated. The partial and conditional deletion of Dnmt3a had no effect on the size of the DG and the extent of adult neurogenesis, indicating no apparent developmental effect of cKO on the number of adult DGCs (Figures S2A and S2B).
Figure 3. Emergence of IMRs during Development Depends on DNMT3A and Is Essential for Proper Neuronal Activation.
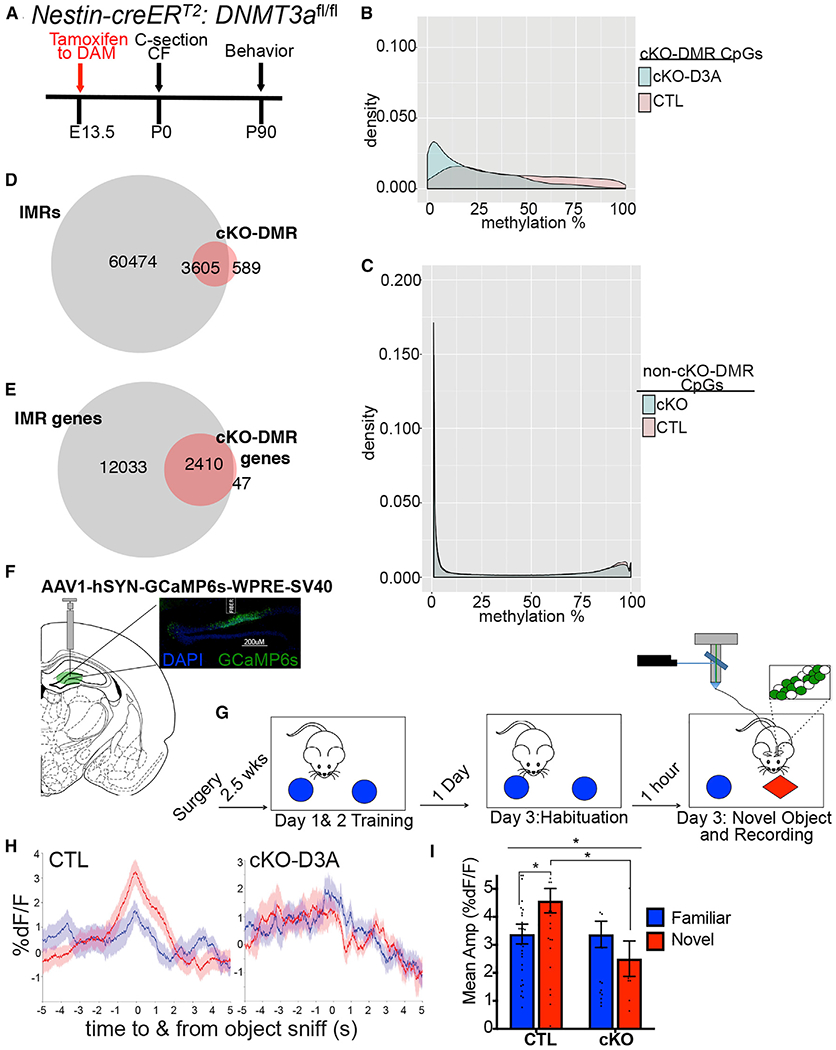
(A) Timeline for generating Dnmt3a cKO mice by TAM in Nestin-creERT2-Dnmt3af/f mice. cKO (cre+/TAM); control (TAM); CF, crossfostering.
(B) Distribution of methylation levels of CpG sites at cKO-DMRs (≥2 CpGs).
(C) Distribution of methylation levels at CpG sites with no significant methylation change in cKO neurons.
(D) cKO-DMR and cKO-IMR overlap.
(E) Essentially all cKO-DMR-containing genes are IMR genes.
(F) Schematic of AAV1-hSYN-GCaMP6s injection and fiber implantation into dDG to record Ca2+ transients in the DG in real time. Inset: image of GCaMP6s expression and fiber implantation in the dDG.
(G) Timeline of the novel object recognition test, with recording of neuronal activity in the dDG by fiber photometry.
(H) Averaged traces of the percentage of fluorescence changes in the DG during interaction with the novel object (red) or habituated object (blue), normalized to baseline (%ΔF/F) for cKO and control mice. Data are mean ± SEM.
(I) Quantification of average amplitude ΔF/F during object sniff of novel and familiar objects in control and cKO mice. Two-way ANOVA genotype × environment, F(1,88) = 4.009, p = 0.048; genotype, F(1,88) = 4.073, p = 0.047; control versus cKO in novel environment, adjusted *p = 0.0134; n = 5 cKO animals, 5 control animals. Significant difference in activity during novel versus familiar object exploration of control mice is also displayed (adjusted *p = 0.032). Novel versus familiar of cKO, p = 0.320. Data are mean ± SEM.
We next tested whether the partial hypomethylation of IMRs was sufficient to interfere with coding in the hippocampus. We assessed neuronal activation, elicited by a novel object in a familiar environment, by monitoring population Ca2+ signal, a proxy for neuronal activity in the dorsal DG (dDG) by fiber photometry (Gunaydin et al., 2014) (Figure 3F). Novelty is known to enhance extracellular glutamate levels in the hippocampus (Cohen et al., 2013) and to increase firing rates and frequencies of Ca2+ events of DGCs (Barbosa et al., 2013; Cohen et al., 2013). Although control (TAM) mice displayed a robust increase in the Ca2+ signal at the novel, but not familiar, object in the test session, no apparent increase in signal was detected at the novel, and familiar, object in cKO (cre+/TAM) mice, suggesting a failure of the dDG to detect/recognize novel objects (Figures 3G–3I). However, novel object exploration and preference of cKO mice was not impaired and was similar to that of controls (Figure S2C), consistent with previous studies reporting spared novel object recognition after permanent hippocampal lesions because of compensatory changes in the perirhinal cortex (Good et al., 2007; Winters et al., 2004). Collectively, these data indicate that expression of DNMT3A in developing neurons is essential for the emergence of IMRs and novelty-induced activation of DGCs.
Increasing DNA Methylation and the Proportion of the Methylated IMR Epialleles Lead to Neuronal Hyperexcitability
Failure of Dnmt3a cKO DGCs to respond to a novel environment prompted us to test whether the opposite, i.e., increasing DNA methylation by the DNMT3A overexpression (OE) in adult dDG, increases the proportion of the methylated IMR epialleles and generates neurons with higher-than-average excitability. Again, we exploited the increased sensitivity of IMR CpGs to methylate them while avoiding the methylation of non-IMR CpGs (e.g., promoters and CpG islands) that are typically protected against de novo methylation (Ginno et al., 2012). Adult mice were injected with either Dnmt3a-expressing AAV-DJ-SYN-DNMT3A-GFP virus or control AAV-DJ-SYN-GFP virus bilaterally in the dDG, titrated to achieve a sparse (10%–30%) expression pattern (Figure 4A). Twenty days later, both Dnmt3a and control virus-injected animals exhibited GFP expression in DGCs. Although limited to a fraction of cells, OE resulted in population-level hypermethylation at a large number of CpG sites (43,586, q < 0.01). These CpGs, like IMR CpGs, displayed mostly IM in control cells and were hypermethylated in OE neurons (Figure 4B). CpGs that were unaffected in OE cells exhibited bimodal distribution (Figure 4C), indicating that hypermethylation in OE cells was largely limited to IM CpGs. Hypermethylated CpGs clustered to 13,745 DMRs (≥2 CpGs) that substantially overlapped with the previously identified IMRs (67.38%, OR = 2.85, p < 2.20E–16) (Figure 4D; see Table S1B for the IGV track). Furthermore, most genes associated with OE-DMRs (92.01%) were also IMR genes (Figure 4E). Similar to hypomethylation by cKO, hypermethylation by OE was partial, because not all IMR genes were affected, and only some IMRs in genes with multiple IMRs were hypermethylated.
Figure 4. DNMT3A OE in DGCs Increases Methylation at a Subset of IMRs and Enhances Intrinsic Excitability.
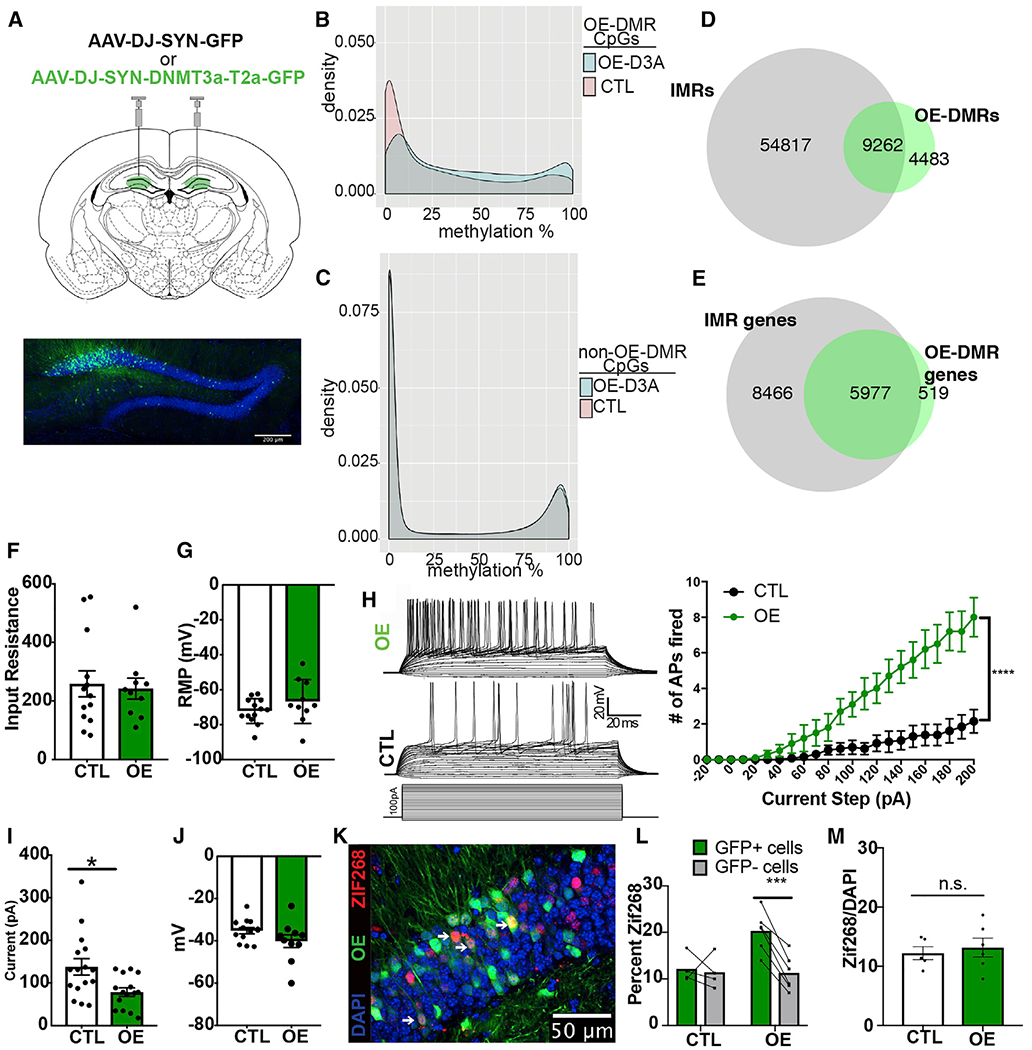
(A) Injection of DNMT3A OE of AAV-DJ-SYN-DNMT3A-T2a-GFP and control AAV-DJ-SYN-GFP into the dDG bilaterally (scale bar, 200 μm).
(B) Distribution of methylation levels of CpG sites at OE-DMRs.
(C) Distribution of methylation levels at CpG sites with no significant methylation change in OE neurons.
(D) OE-DMR and OE-IMR overlap.
(E) OE-DMR-containing genes are IMR genes.
(F) Input resistance of OE and control GFP+ DGCs in slices (t = 0.2783, p = 0.7835; n = OE, 10 cells/6 animals; control, 13 cells/7 animals). Data are mean ± SEM.
(G) Resting potential of OE and control GFP+ cells (t = 1.316, p = 0.2023). Data are mean ± SEM.
(H) Evoked action potentials in OE and control cells across a depolarizing current injection curve while cells are held at −70 mV. Representative traces from an OE and a control cell are at the left top and middle; current step protocol is at the left bottom. Graphical representation of the data is on the right. Two-way ANOVA genotype × pA, F(22,462) = 15.10, p < 0.0001; genotype, F(1,21) = 14.85, ***p = 0.0009; n = OE, 10 cells/6 animals, control, 15 cells/8 animals. Data are mean ± SEM.
(I) Rheobase in OE compared with control cells (Welch corrected, t = 2.754, *p = 0.0114). Data are mean ± SEM.
(J) Membrane potential at the occurrence of the first spike during current injection in OE and control neurons (t = 1.573, p = 0.1306). Data are mean ± SEM.
(K) Representative immunohistochemistry image of dDG of mice after 15 min of novel environment exploration. White arrows indicate OE and ZIF268 colabeled cells (scale bar, 50 μm).
(L) ZIF268+ positivity counts in GFP+ versus GFP− neurons. Two-way ANOVA GFP3×experimental group, F(1,8) = 10.95, p = 0.0107; FOS+ versus FOS− in OE and control groups, adjusted ***p = 0.0009 and 0.9182, respectively; n = 6 OE, 4 control, 2–4 sections per animal.
(M) Total recruitment in control and OE DGCs after novel environment exploration (t = 0.426 p = 0.6477, n = 5, 6). Data are mean ± SEM.
Next, we tested whether the partial hypermethylation of IMRs in dDGCs was sufficient to increase their excitability. Because OE and control cells were labeled by GFP, instead of population activity recording, we performed whole-cell patch-clamp slice electrophysiology. The input resistance levels of both OE GFP+ and control GFP+ neurons were similarly low (Figure 4F), consistent with the 100–300 MΩ range reported for fully matured DGCs (Staley et al., 1992). Although the resting membrane potential (RMP) did not differ (Figure 4G), we found a significant increase in the number of evoked action potentials in OE compared with control cells across increasing steps of depolarizing current injection (Figure 4H). Furthermore, we found a decrease in the rheobase (minimum current required to elicit an action potential) in OE neurons using a current injection ramp (Figure 4I), whereas the membrane potential at the occurrence of the first spike did not differ between groups (Figure 4J). This indicates that OE neurons are more excitable in response to depolarization than control neurons. However, synaptic transmission was unaltered in OE neurons, because the frequency and amplitude of spontaneous excitatory postsynaptic currents (EPSCs) and inhibitory postsynaptic currents (IPSCs) were similar to those of control neurons (Figures S2D and S2E). These data show that DNMT3A OE in DGCs enhances their propensity to generate action potentials in response to depolarizing current and presumably to excitatory synaptic inputs (i.e., increased intrinsic excitability).
Because excitability is thought to be a main factor driving neuron recruitment for an ensemble (Lisman et al., 2018), we asked whether OE cells have a higher-than-random chance to be included into a neuronal ensemble. OE and control mice exposed to a novel environment for 15 and 60 min were later sacrificed to visualize and calculate the fraction of DGCs positive for the immediate-early gene (IEG) product ZIF268 in the infected (GFP+) and non-infected (GFP−) populations of DGCs (Figure 4K). OE-GFP+ neurons (i.e., with increased excitability) were more likely to be recruited (ZIF268+) during novel environment exploration than their non-infected GFP− neighbors, whereas GFP+ and GFP− neurons in control mice had equal probability for recruitment (Figure 4L). There was no difference in the overall number of ZIF268+ DGCs relative to all DGCs (DAPI+), suggesting that OE does not increase the recruited population size in the DG (Figure 4M), presumably because strong γ-aminobutyric acid (-GABAergic) feedback inhibition limits the activated population to DGCs with the highest excitability (Temprana et al., 2015).
Single-Cell Methylomes of Activated Cells Differ from the Rest of DGCs
Because hypermethylation of IMRs in OE neurons was accompanied by increased neuronal excitability, we predicted that the methylated state of some IMRs is enriched in neurons recruited to a coding ensemble. DGCs, activated by 15 min of exposure to a novel environment, were isolated 60 min later for single-nucleus RRBS (snRRBS). DGC nuclei (PROX+), expressing the IEG protein FOS (FOS+), a proxy for neuronal activity, were separated from PROX1+/FOS− cells by fluorescence-activated cell sorting (FACS) from dissected dorsal hippocampi (Jaeger et al., 2018; Lacar et al., 2016) (Figure 5A). Exposure to a novel environment, relative to the home cage, increased the number of FOS+ neurons, but, consistent with sparse encoding in the DG, they represented ~2.3% of DGCs (Figure 5B). The 264/288 FOS+ and 233/288 FOS− single-nucleus methylomes (sn-methylomes) that passed quality control identified 1,272,335 CpGs, which were also present in the bulk DGC methylome (≥3 cells in both FOS+ and FOS− populations, average 46.30 cells/CpG). Aggregate CpG methylation in FAC-sorted PROX1+ cells, as determined by snRRBS, was highly concordant with methylation by bulk RRBS (r2 = 0.8918). Direct comparison of methylation levels in FOS+ and FOS− cells identified 14,881 FOS-DM CpG sites (1.17% of all detected CpGs, p < 0.05, Bernard’s exact test) (see Tables S1B and S3A for links to custom tracks in IGV). Half of FOS-DM CpGs (49.4%) were preferentially methylated and the other half (50.6%) were preferentially unmethylated in FOS+ versus FOS− cells.
Figure 5. sn-Methylomes of Activated DGCs Reveal DM CpGs that Exhibit IM, Map to Developmental IMRs, and Are Associated with Specific Neuronal Gene Functions.
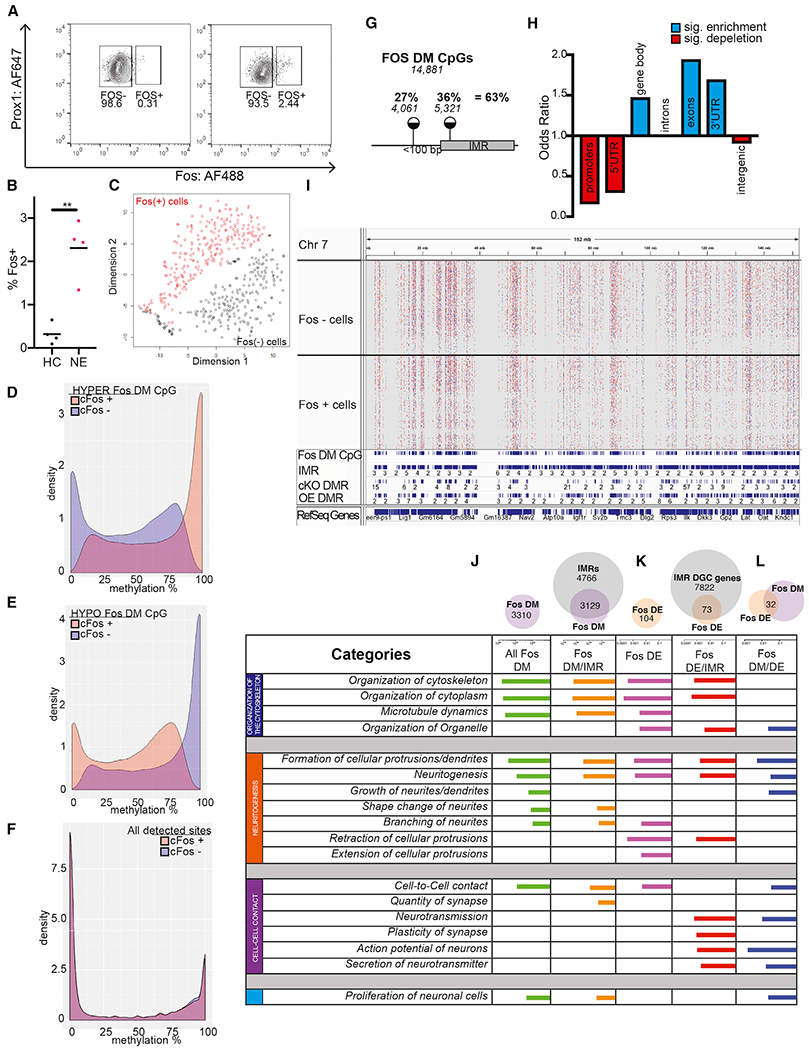
(A) Representative FACS plots showing the proportion of FOS+ nuclei in the population of PROX+ DGCs in a home cage and after novel environment exposure.
(B) Proportion of FOS+ nuclei is increased by novel environment exposure. Four independent experiments (Welch corrected, t = 5.504, **p = 0.0066, n = 2–4 mice per group/experiment).
(C) t-SNE plot of FOS-DM CpG sites shows the separation of FOS+ and FOS− nuclei.
(D and E) DM CpGs between FOS+ and FOS− cells, identified by snRRBS, exhibit IM across the entire 0% to 100% range. About half of FOS-DM CpGs are hypermethylated in FOS+ (D), whereas the other half are hypomethylated in FOS+ (E).
(F) Bimodal distribution of methylation levels at non-DM (~1.2 million) CpGs in FOS− and FOS+ DGCs.
(G) Around 1/3rd of DM CpGs between FOS+ and FOS− cells mapped to IMRs, whereas an additional 1/4th were near IMRs.
(H) Enrichment of IMRs in genomic features.
(I) Distribution of DMCpGs within a representative 150 Mb region of chromosome 7 (Chr7) in 233 FOS− cells (upper tracks) and 264 FOS+ cells (lower tracks). Red, hypermethylated, and blue, hypomethylated, in FOS+ versus FOS− cells. The IGV track of aggregate sn-methylomes is also displayed below the single-cell tracks. FOS-DM CpGs tend to accumulate at regions occupied by IMRs, cKO-DMRs, and OE-IMRs.
(J–L) sn-Methylomes and transcriptomes of activated DGCs reveal common functional gene ontologies. Ingenuity molecular and cellular functions associated with DM (J), DE (K), and both DM and DE (L) genes between FOS+ and FOS− cells (IPA). Top ten functions are displayed based on BH-corrected p values.
See also Figures S3 and S4.
Methylation data from single FOS+ and FOS− cell nuclei, pooled for unsupervised cluster analysis by t-distributed stochastic neighbor embedding (t-SNE), showed no separation of the two populations, presumably because of sparse CpG coverage and the low number of FOS-DM CpGs relative to all detected CpGs. A similar analysis with FOS-DM CpGs assigned individual FOS+ and FOS− cells into separate but individually homogeneous clusters (Figure 5C), indicating no apparent methylation-based subpopulations.
Remarkably, FOS-DM CpGs, similar to IMR CpGs, were IM (Figures 5D and 5E). Although hypermethylated DM sites exhibited a wide range of IM distribution in FOS− cells, their methylation was in a higher IM range or fully methylated state (23.1% with >90% methylation) in the small population of FOS+ cells (Figure 5D, blue or orange areas, respectively). Hypomethylated DM CpGs were also IM in FOS− cells but contained a fraction of sites (27.2%) in the fully methylated state, which were in the IM range in FOS+ cells (Figure 5E, blue and orange, respectively). The preferential inclusion/exclusion of methylated epialleles from FOS+ cells may indicate their compatibility or incompatibility with recruitment. CpGs with unaltered methylation, similar to non-IMR CpGs, exhibited bimodal methylation distribution, demonstrating that they were either fully methylated or unmethylated across all cells, whether FOS+ and FOS− (Figure 5F). These data indicate that recruited/activated cells differ from the rest of DGCs at DM CpGs in the relative proportions of the methylated and unmethylated epialleles.
Because IM was a unique feature of both developmental IMR and DM CpGs, we asked whether they colocalize in the genome. We found that 36% of FOS-DM CpGs mapped to IMRs, a significant enrichment over chance (p = 0.01291), whereas another 27% mapped less than 100 bp from IMRs, indicating the concentration of FOS-DM sites (63% of all) at, and around, developmental IMRs (Figure 5G; see Table S3A for FOS-DM CpGs). Furthermore, FOS-DM CpGs colocalized with both cKO-DMRs (OR = 2.586, p < 2.2E–16) and OE-DMRs (OR = 4.808, p < 2.2E–16). A representative genome view of colocalization of DM CpGs in 233 FOS− and 264 FOS+ cells with developmental IMRs, cKO-DMRs, and OE-DMRs is displayed in Figure 5I. These data show that FOS-DM CpGs specify regions that become epigenetically bistable during development and whose methylation tends to be sensitive to DNMT3A dosage. Typically, only one DM CpG fell within the boundaries of an IMR (1.21 DM CpG/IMR), likely because of the inherent sparsity of coverage by snRRBS and thus the low number of reads over CpGs, relative to bulk RRBS. When two DM CpGs mapped to an IMR, they were typically uniformly methylated or unmethylated in FOS+ versus FOS− cells, much like neighboring CpGs in single IMR reads. These data indicated that the methylation state of DM CpGs may be used as a proxy to predict the preferred methylation state of their associated IMRs in FOS+ cells.
Further analysis showed that FOS-DM CpGs (and related FOS/IMRs), similar to developmental IMRs, were depleted in promoters, 5′ UTRs, and intergenic regions and were enriched in gene bodies (Figure 5H; Table S3B). FOS-DM CpGs were enriched in exons and 3′ UTRs, but not in introns (unlike developmental IMRs), suggesting that FOS CpG-IMRs correspond to a subset of IMRs with high evolutionary sequence conservation. Finally, association of FOS-DM CpGs with developmental IMRs was higher at the gene level (94.5% of the 3,323 FOS-DM genes harbored IMRs), indicating the convergence of DM CpGs with a subset of IMRs on the same genes, but not necessarily at the same location (Figure 5J, top). This indicates the presence of additional IM areas within these genes that were not captured through developmental methylation changes among the three selected developmental time points (Figure 1A).
Gene ontology by IPA showed the enrichment of DM CpG-containing genes in four main functional categories (Figure 5J; Table S3C), which were essentially the same as those we identified containing IMR-expressed genes (Figure 2F): (1) cytoskeleton organization, (2) neurite morphogenesis, (3) cell-to-cell interactions, and (4) proliferation. Lack of enrichment of FOS-DM genes in RNA or protein expression or modification and metabolic processes was notable, suggesting enrichment in neuron-specific functions.
Next, we asked whether FOS-DM genes are differentially expressed (DE) between FOS+ and FOS− cells. After filtering, we analyzed 4,319 PROX+/FOS− and 1,392 PROX+/Fos+ DGC nuclei by single-nucleus RNA sequencing (snRNA-seq) using the 10× Genomics platform. Although 10× snRNA-seq generates sparse gene expression profiles, especially from nuclei (median number of genes = 307 genes/cell), we identified 13,382 expressed genes (Table S2). Consistent with the preselection of DGCs for snRNA-seq by FACS, t-SNE analysis (Butler et al., 2018) assigned most cells to one large cluster. However, within this cluster, FOS+ and FOS− cells segregated from each other, indicating gene expression differences between the two populations (Figure S3A). Furthermore, FOS+ and FOS− cells could be subdivided to three groups each: FOS+1–FOS+3 and FOS−1–FOS−3 (Figures S3B and S3C). The largest population of FOS+ nuclei, FOS+1, was enriched in genes related to protein synthesis and turnover, and the smaller FOS+2 and FOS+3 populations were prominently enriched in the CREB pathway and synaptogenesis genes (Figure S3D), functions consistent with an activated neuronal state. In contrast, the three FOS− subclusters shared the biological functions of synaptogenesis and calcium signaling (Figure S3E).
Direct comparison of FOS+ and FOS− transcriptomes identified 104 FOS-DE genes (adjusted p < 0.05), with 62.5% upregulated and 37.5% downregulated (Table S3D). Most FOS-DE genes (72%) contained IMRs (OR = 1.35) (Figure 5K), and FOS-DE genes were enriched in functional categories similar to those of FOS-DM/IMR genes (Figure 5K), suggesting a methylated epiallele × gene expression relationship. Indeed, 32 genes (31%) were both DE and DM (OR = 83.35, p < 2.2E–16) (Figure 5L; Table S3E).
Remarkably, 75% of DE × DM genes were part of a single interactive network (Figure S4A). This network included several voltage-gated channels, regulators of actin/cytoskeleton, and proteins related to cell-to-cell contact that all can be linked to intrinsic excitability and cell/neurite morphogenesis. Overall, the network of DE × DM genes defined interacting proteins regulating neuronal excitability and structural plasticity.
Upregulation and downregulation in expression FOS+ cells were often associated with increased (Trpc5 and Cadps) and decreased (Kcnq2) methylated epiallele frequency, respectively (Figure S4B), but the methylation state at FOS-DM CpGs was not predictive of expression, presumably because methylation can influence gene expression by numerous mechanisms. Furthermore, although epiallelic frequencies in FOS+ cells were only moderately different from those in FOS− cells, their combinatorial and cumulative effects may change the gene network and neuronal functions.
DISCUSSION
We identified thousands of small genomic regions, IMRs, that exhibit stochastic CpG methylation-based variability within neuronal gene bodies, predominantly bistability among cells and alleles. Similar DNA methylation variability was observed at promoters and CpG island shores in stem cells and cancer cells (Jenkinson et al., 2017; Landan et al., 2012; Shipony et al., 2014). The theoretical model of Jenkinson et al. (2017) posits that methylation bistability is produced when fully methylated and unmethylated epialleles have low potential energy relative to epialleles with a mix of methylated and unmethylated CpGs. They also hypothesized that de novo methylation and demethylation allow random methylation patterns to be modified with high probability to assume a lower potential energy state. Our data show that methylation bistability in hippocampal neurons is established during development by partial methylation and demethylation of unmethylated and methylated regions, respectively. We hypothesize that local activity of DNMT3A, presumably in conjunction with TET enzymes, in individual cells determines whether epiallele frequencies are shifted toward the methylated or unmethylated epiallele. However, maintenance and modification of epigenetic bistability also includes chromatin factors (Jenkinson et al., 2017).
Although we recognized IMRs through developmental changes in methylation between multiple time points, IMRs are likely present in adult hippocampal neurons that were not captured in our study. snRRBS profiling of FOS+ and FOS− cells, an approach that relies not on developmental changes but rather on existing epigenetic heterogeneity and neuronal activation in the adult DGC population, revealed DM CpGs that were mostly within or close to developmental IMRs. Furthermore, FOS-DM CpGs colocalized with both cKO-DMRs and OE-DMRs. Our data show the presence of IM and methylation bistability within neuronal gene bodies in adult hippocampal neurons by two independent approaches and connect developmentally produced IM sites and regions within gene bodies to neuronal recruitment.
Bistability in regional CpG methylation in bulk methylomes has been recognized in human cells and tissue; however, this primarily resulted from sequence-dependent allele-specific methylation (Onuchic et al., 2018). Sequence-independent (and non-parental) variability in methylation was reported in embryonic stem cells and progenitors, suggesting their transient nature and role in early developmental processes (Singer et al., 2014; Stadler et al., 2011). In contrast, the sequence-independent bistable methylation at IMRs described here is present in mature DGCs, implying a function in the adult hippocampus such as encoding of experiences.
Although our snRRBS data suggest that the preexisting methylation state of a subset of IMRs dictates neuron eligibility to recruitment, we cannot exclude the possibility that de novo methylation (and demethylation) during the short 60-min period between recruitment and IEG expression plays a role. However, because IMR CpG sites that were preferentially methylated in FOS+ neurons were hypermethylated above chance (OR = 2.065, p = 0.0018) in OE neurons (which are even more excitable), the preexisting methylation state of IMRs likely contributes to excitability and the neuron’s eligibility to recruitment. These data suggest a relationship between the methylation state of IM CpGs/regions within gene bodies and the propensity of the cell to be recruited to a coding ensemble.
Because IMRs were depleted in promoter regions, they may not directly regulate overall transcription. Instead, the exonic and 3′ UTR enrichment of FOS-IMRs suggests more complex DNA-methylation-dependent regulation that may include splicing, alternative 3′ length, and consecutive changes in mRNA trafficking, subcellular compartmentalization, and local translation (Lev Maor et al., 2015; Maunakea et al., 2013; Shukla et al., 2011). Technical limitations do not currently allow mRNA isoform-specific profiling at single-cell levels. Furthermore, the presence of multiple DM CpGs/IMRs, with occasionally opposing methylations states, within a gene complicates establishing a direct relationship between epialleles and expression. Epigenetic editing (Liu et al., 2016) could help in understanding how individual epialleles affect spicing and isoform-specific gene expression.
Although most of this work was conducted in DGCs, we detected similar IMRs in other glutamatergic neurons in the hippocampus. This indicates that establishment of IMRs during neuronal development and subsequent maintenance in adulthood are a general phenomenon in principal neurons of the hippocampus. Therefore, we propose a model in which bistable regions create neuron heterogeneity and constellations of exonic methylation, which may contribute to cell-specific gene expression, excitability, and eligibility to a coding ensemble.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information or requests for resources should be directed to the Lead Contact, Miklos Toth (mtoth@med.cornell.edu).
Materials Availability
This study did not generate new unique reagents
Data and Code Availability
The data reported in this study can be found in GEO under accession number GEO: GSE72700 and GEO: GSE150551. Most analyses were done using publicly available tools as listed in STAR Methods, with custom code available in the Supplemental Information.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal experiments were performed in accordance with the Weill Cornell Medical College Institutional Animal Care and Use Committee guidelines. All mice were group-housed two to five per cage, with a 12 h light/dark cycle and with lights on at 6:00 A.M. Food and water were available ad libitum. Wild-type C57BL/6 males were purchased from The Jackson Laboratory. Conditional Dnmt3a knock-out mice were generated as previously described (Sharma et al., 2016). C57BL/6 mice carrying floxed Dnmt3a alleles (Dnmt3af/f (Kaneda et al., 2004)), provided by Riken BioResource Center, were crossed with mice heterozygous for the tamoxifen (TAM)-inducible nestin-cre-ERT2 transgene (Chen et al., 2009), kindly provided by Luis Parada (University of Texas Southwestern Medical Center, Dallas, TX). To induce Cre-mediated Dnmt3a knockout at E13.5, pregnant cre-negative homozygous females were bred with cre-heterozygous males and were injected with 150-μl TAM solution (1mg), prepared by dissolving TAM (Sigma-Aldrich, St. Louis, MO) in ethanol and then in sunflower oil (Sigma-Aldrich, St. Louis, MO) (9:1) solution at 6.7 mg/ml. Because gestational TAM injection interferes with females’ maternal care behavior and labor, newborn pups were delivered via C-section on E20 and transferred into the nest of an early post-partum adoptive dam. Adult males (between 10 and 20 weeks of age) were used for all sequencing, novel environment exploration, and slice recordings. Adult males and females used for Fiber Photometry. IMRs from males and females largely overlapped, so just male data are shown.
METHOD DETAILS
Intracranial virus injection and optical fiber implantation
Intracranial fiber implantation into the dDG for fiber photometry. Male and female animals were anesthetized with 2% isoflurane and placed onto a stereotaxic frame to perform craniotomy. First, we delivered 275 nL viral vector AAV1.Syn.GCaMP6s at a rate of 75 nL per minute by a 10 μL Hamilton 700 series syringe with a 33 gauge blunt needle attached to a microsyringe pump and controller at the following coordinates: −1.94 mm anterior-posterior, −1.0 mm medial-lateral, and −1.85 mm dorsal-ventral. The needle remained at the injection site for 7 minutes post injection and was withdrawn slowly. A 400 μm diameter optical fiber was then implanted using the following coordinates: −1.94 mm anterior-posterior, −1.0 mm medial-lateral, and −1.83 mm dorsal-ventral. Optical fibers were secured with Metabond.
Intracranial virus injection to overexpress DNMT3A in the dDG. C57BL/6 males for virus mediated DNMT3A overexpression were purchased from Taconic Biosciences. Males were injected bilaterally, 500 nL/side with a rate of 50 nL/min into the dDG with either DNMT3A overexpressing virus, AAV-DJ-Syn-Dnmt3a-T2a-GFP (1.2E+07 IU./mL), or control virus, AAV-DJ-Syn-GFP (6.81E+08 IU/ml). Viruses were prepared by the Stanford Vector Core.
Behavior
All tests were conducted using mice between 10 and 20 weeks of age. During all behavioral tests, the investigators who performed the tests were blind to the genotype and the treatment of the animals. Behavioral tests were conducted during the light on phase, between 1000 hours and 1600 hours. All mice were habituated to the behavior room for at least 1 h prior to testing.
Novel Object Recognition
This behavioral test was performed in conjunction with fiber photometry to assess the activation of the dDG during familiar and novel object exploration. The test was conducted 2-2.5 weeks after the implantation of fibers and performed as previously described (Huynh et al., 2015). Different shaped and colored lego blocks (approximately 1 × 1 × 2 in) were used for the familiar and novel objects. Two days of training to familiar objects were performed in a 10-minute session, where male and female mice were exposed to the arena with the same two identical objects. On day 3, mice were again exposed to the same two identical objects in the arena for 10 min. One hr later, mice were reintroduced to the arena in which a novel object replaced one of the familiar objects on the nonpreferred position (measured in previous session). Interaction with the objects for frequency, time, and duration was scored for 5 min from video recordings.
Novel Environment Exploration. Exposure to a novel environment was used to ‘mark,’ by the expression of the early immediate protein FOS or zif268, the small population of ‘encoding’ DG neurons for cellular and molecular studies. Mice were individually housed in dark boxes at least 2 hours prior to exposure to minimize background FOS/zif268 expression. Animals were placed one at a time into a large novel arena (24 x45 in) with many huts, tubes, and objects scattered throughout. Animals were allowed to freely explore for 15-minutes. Animals were then placed back in the dark for one hour to allow for maximum FOS or zif268 expression prior to transcardial perfusion or hippocampal dissection for FACs.
Fiber Photometry
Fiber photometry was performed as previously described (Gunaydin et al., 2014) in the novel object recognition task as described above. To compare calcium signals across animals, fluorescence recordings were normalized to ΔF/F by taking the median signal across the recording period, subtracting it from each data point and then dividing by the median signal.
Tissue preparation and immunostaining
Animals were sacrificed by transcardial perfusion with 4% paraformaldehyde (wt/vol), followed by one day post fix in 4% paraformaldehyde and at least 24 h in 30% sucrose cryoprotectant. Brains were flash frozen and were embedded in OCT followed by sectioning (40 μm) in a cryostat. For DCX cell counts, sections were incubated in 50% methanol for 30 min. Tissue was blocked in 1% BSA and incubated at 22–24°C for 48 h with a goat polyclonal antibody to DCX (1:300, Santa Cruz, C18). Tissue was then incubated in secondary biotinylated rabbit antibody to goat antibody (1:250, Sigma, B-7024) for 2 h, followed by 1 h in ABC and DAB reagents for 2-0 min. Sections were counterstained with Nissl. DCX cell density in the dentate gyrus was measured using 4 sample sections (2 dorsal, 2 ventral) by using the Stereoinvestigator software (MBF Bioscience). For granule cell layer (GCL) volume measurement, immunohistochemistry was performed by incubating the slices in DAPI (1:5000 Thermo Fischer D1306) for 10min in PBS. GCL volume was measured in 1:8 sections using the Cavalieri method by Stereoinvestigator. For zif268 cell counts, tissue was blocked in 1% BSA and incubated at 4°C for 48 h with primary antibody, rabbit antibody to zif268 (1:500, Cell Signaling, 4153). Tissue was then incubated in secondary rhodamine antibody to rabbit antibody (1:500, Thermo Fischer, # 31670) for two h, followed by DAPI staining (1:5000) for 30 min. Dorsal sections were identified as between bregma −1.58 and −2.18 and were imaged at 40x on a Zeiss LSM 880 with each overexpressing virus image matched to a control virus section. DAPI, zif268+ and GFP+ cells were counted manually using the cell counter plug in FIJI. Cells were counted in each channel separately and then combined to identify GFP+/zif268+ colabeled cells. 2-4 sections per mouse were analyzed and counts were averaged per animal.
Slice Preparation for recording
Mice were anesthetized with isoflurane and rapidly decapitated. Brains were removed and isolated in NMDG-HEPES artificial cerebral spinal fluid (aCSF) composed of (in mM): 93 NMDG, 2.5 KCl, 1.2 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 Glucose, 5 sodium ascorbate, 2 thiourea, 3 sodium pyruvate, 10 MgSO4.7H2O, 0.5 CaCl2.2H2O (pH adjusted with HCl to 7.35, mOsm between 300 and 310), saturated with 95% O2 and 5% CO2. Transverse hippocampal slices (300 μm) were prepared using a Leica VT1200 vibratome (Leica Microsystems Inc., Buffalo Grove, IL), and incubated at 34 C for 11-12 min in NMDG-HEPES aCSF. Slices were transferred to a modified HEPES aCSF containing (in μM): 92 NaCl, 2.5 KCl, 1.2 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 Glucose, 5 sodium ascorbate, 2 thiourea, 3 sodium pyruvate, 2 MgSO4.7H2O, 2 CaCl2.2H2O (Ph adjusted with NaOH to 7.35, mOsm between 300 and 310), and maintained at ambient temperature in oxygenated aCSF for at least forty-five mins before recordings began.
Electrophysiological Recordings
Slices were transferred to a recording chamber and superfused with aerated aCSF containing (in mM): 124 NaCl, 2.5 KCl, 1.2 NaH2PO4, 24 NaHCO3, 5 HEPES, 12.5 Glucose, 2 MgSO4.7H2O, 2 CaCl2.2H2O at a rate of 2 ml/min using a perfusion pump (Peri-Star Pro; World Precision Instruments, Sarasota, FL). For current clamp recordings of intrinsic membrane properties and current-injected firing, we used a potassium-gluconate–based intracellular recording solution containing (in mM): 135 K-Gluc, 5 NaCl, 2 MgCl2.6H2O, 10 HEPES, 0.6 EGTA, 4 Na2ATP, 0.4 Na2GTP, pH adjusted with KOH to 7.34, 280–290 mOsm. Spontaneous IPSCs and EPSCs (sIPSCs/sEPSCs) were recorded using a cesium-methanesulfonate-based internal solution containing (in mM): 135 Cs-Meth,10 KCL,10 HEPES, 1 MgCl26H2O, 0.2 EGTA, 4 MgATP, 0.3 Na2GTP, 20 phosphocreatine, pH adjusted with CsOH to 7.34, 280-290 mOsm. Recording electrodes were prepared from filamented borosilicate glass capillary tubes using a horizontal micropipette puller (P-1000; Sutter Instruments, Novato, CA). Whole-cell patch-clamp recordings were collected from neurons voltage-clamped at −55 mV for sEPSCs and +10 mV for sIPSCs; current clamp experiments were conducted at the cell’s resting potential and at −70 mV. A maximum of two cells per experimental condition were recorded from each animal, and all cells included in these analyses maintained a stable access resistance of 5–20 MΩ. Whole-cell currents were acquired using a MultiClamp 700B amplifier, digitized (Axon Digidata1550B, Axon Instruments, Union City, CA), and analyzed online and offline using an IBM-compatible personal computer and pClamp 10.3 software (Axon Instruments).
Flow cytometry
Nuclei were isolated as previously described (Lacar et al., 2016). Briefly, the whole brain was sectioned coronally using a mouse brain slicer matrix to isolate the dorsal hippocampus. The dorsal hippocampus was dissected out and placed in a nuclei isolation medium (sucrose 0.25 M, KCl 25 mM, MgCl2 5 mM, TrisCl 10 mM, dithiothreitol 100 mM, 0.1% Triton, protease inhibitors). Tissue was mechanically homogenized, allowing for separation of nuclei from cells. Samples were washed, resuspended in nuclei storage buffer (0.167 M sucrose, MgCl2 5 mM, and TrisCl 10 mM, dithiothreitol 100 mM, protease inhibitors) and filtered through 70 μM then 30 μM filters. Solutions and samples were kept cold throughout the protocol. Dissociated nuclei were then stained by 30 min incubation in anti-Prox1 Alexa Flour 647 (NBP-1-30045AF647, NovusBio, 1:1000) and anti-c-FOS Alexa Flour 488 (NBP2-50037AF488, NovusBio, 1:1000). Samples were gated and collected using a BD Influx sorter followed by analysis using the FlowJo software (Tree Star). Nuclei were gated first using forward and side scatter pulse area parameters (FSC-A and SSC-A) excluding debris, followed by exclusion of aggregates using pulse width (FSC-W and Trigger Pulse Width). The FOS+ population was gated using home cage control populations based on PROX1 and FOS fluorescence.
DNA Collection for bulk methylation sequencing
Brains from adult WT, transgenic, and virus injected mice were collected, quick frozen on dry ice and sectioned (200 μM) in cryostat. The dentate gyrus, CA1 and CA3 were microdissected from the sections. DNA was isolated using the QIAamp DNA Micro Kit according to manufacturer instructions (QIAGEN).
Reduced Representation Bisulfite Sequencing (RRBS)
In most experiments, we used RRBS (Akalin et al., 2012a) because of its high efficiency in sequencing CpGs and its higher coverage of sequenced reads relative to that of whole genome bisulfite sequencing (WGBS) (160X versus 30x) that allowed us to more accurately determine the extent of developmental methylation changes. Preparation for RRBS libraries, sequencing, and adaptor trimming were done by the Epigenomics Core at Weill Cornell Medicine. Single end 50 bp RRBS sequencing was performed as described, using Illumina HiSeq2000 and 2500 machines according to the manufacturer’s instructions. UCSC mm9 was prepared and indexed followed by alignment and methylation calling using Bismark(v17) (Akalin et al., 2012b). Differential methylation and statistical analysis were performed using the MethylKit package in Differential methylation and statistical analysis were performed using the MethylKit package (Akalin et al., 2012b) in R at default setting. Differentially methylated sites were defined as sites where the sliding linear model (SLIM)-corrected p values (q) were ≤ 0.01. Differentially methylated regions were defined as regions containing at least two differentially methylated CpG sites with < 1 kb distance between sites, and were referred to as an intermediate methylated region (IMR). Bed files for CpG islands were prepared using a publically available pipeline (https://www.r-bloggers.com/cpg-island-shelves/). Genomic coordinates for exons, introns, 5′UTR, 3′UTR were downloaded from the University of California Santa Cruz (UCSC) genome browser based on mm9. Promoters were defined as regions ± 500 bp from the transcription start site (TSS). To determine if IMRs are enriched in a genomic feature, we computed the Odds Ratio as: [(the number of IMR overlapping feature)/(the number of IMR not overlapping feature)] / [(the number of potential IMR overlapping feature)/(the number of potential IMR not overlapping feature)]. Odds Ratio > 1 reflects enrichment of the IMR to feature, while Odds Ratio < 1 reflects depletion. Statistically significant enrichments were computed using Chi-square test in R. Density plots were done using ggplot function (ggplot2, R).
Whole Genome Bisulfite Sequencing (WGBS)
Single-end 50 bp WGBS sequencing was performed using Illumina HiSeq2000 and 2500 machines according to the manufacturer’s instructions. The pipeline published by Lister et al. (2009) was used for alignment, methylation calls, and differential methylation.
Reduce Representation Oxidative Bisulfite Sequencing (RRoxBS)
To identify cytosines with hydroxymethylation (5hmC), RRoxBS, in combination with RRBS, was performed as previously described (Booth et al., 2012) with modifications for reduced representation sequencing. Library preparation, sequencing, and adaptor trimming were done by the Epigenomics Core at Weill Cornell Medicine as follows: Briefly, genomic DNA (100 ng/sample) was digested with 100U of MspI (New England Biolabs, Ipswich, MA), and cleaned up using QIAquick PCR purification columns (QIAGEN, Hilden, Germany). The digested DNA was spiked with 0.01% control DNA duplexes, provided by Cambridge Epigenetix (Cambridge, UK), containing C, 5-fC, 5-mC and 5-hmC bases at known positions. The control sequences were used to give a quantitative assessment of the efficiency of oxidation and bisulfite conversion. The rest of the library preparation was carried out using the Ovation Ultralow Methyl-Seq DR Multiplex with TrueMethyl oxBS kit and workflow (Tecan, Redwood, CA). The libraries were split into two aliquots after adaptor ligation: one aliquot was oxidized and bisulfite converted (oxBS), the other subjected to a mock oxidation before bisulfite conversion (BS). 2ul of the resulting material was assessed by qPCR to determine the optimal number of PCR amplification cycles. After amplification, the libraries were normalized and pooled according to the desired plexity, clustered at 6.5pM on single read flow cells and sequenced for 50 cycles on an Illumina HiSeq 2500. Illumina’s CASAVA 1.8.2 software was used to perform image capture, base calling and demultiplexing. Analysis of bisulfite treated sequence reads was carried out as described (Garrett-Bakelman et al., 2015), using CUTADAPT instead of FLEXBAR to trim the reads. Alignment to the mm10 genome and methylation calls were done using Bismark (v17) using the default options. Global CpG report was then used to compute percent CpG methylation and total coverage with base-pair resolution. To identify the level of 5hmC at a CpG site, we considered only the sites that are detected in the BS and oxBS experiments with delta methylation (BS – oxBS) being > 0. The BS experiment does not discriminate between 5mC and 5hmC, while the oxBS experiment detects 5mC. Therefore, the 5hmC methylation levels were inferred by subtracting oxBS methylation proportion from BS methylation proportion. Proportion test (prop.test, R) was then used to compute statistically hydroxyl-methylated (5hmC) cytosines. Therefore, CpGs with statistically significant differences between BS and oxBS experiments (p < 0.05, methylation proportion (BS – oxBS > 0)) were considered significantly hydroxy-methylated (PMID: 28428825).
Multiplexed single-nucleus RRBS (snRRBS)
Single nucleus RRBS were performed by first sorting PROX+/FOS+ and /FOS− neuronal nuclei in 96-well plates in 3 μL of 0.1 × CutSmart buffer (New England Biolabs) per well as described in the Flow Cytometry section of the Method Details. Plates were stored at −80°C. Processing of samples were as described for Multiplexed single-cell RRBS (Pastore et al., 2019) by the Epigenomic Core Facility of Weill Cornell Medicine. Briefly, nuclei were lysed, DNA was cut with Msp1 (Fermentas), and A-tailed DNA fragments were ligated with custom methylated adapters containing inline cell barcodes. Then, adaptor-ligated DNA from 24 nuclei were pooled together based on compatible cell barcodes, yielding 4 pools of 24 individual nuclei per 96-well plate. Each 24-nuclei pool was subjected to two cycles of bisulfite conversion (EpiTect Fast Bisulfite Kit, QIAGEN) according to manufacturer’s instructions. Converted DNA was then amplified using primers containing an Illumina i7 index. A different i7 index was used for every 24-nuclei pool, allowing multiplexing of 96 nuclei for paired-end sequencing on one Illumina HiSeq 2500 lane. Each pool of 96 nuclei was first demultiplexed by i7 barcodes, and then each pool of 24 nuclei was further demultiplexed by unique cell barcodes. Processing of the reads was adapted from Gaiti et al. (2019). Briefly, reads with at least 80% match for the template adapters were assigned to a given cell. Adapters were trimmed from the raw sequence reads. After adaptor removal, the first 5bp were trimmed from the 5’ end of the R1 and R2 fastq files. Reads were then aligned to the mm9 mouse genome using Bismark34 (v.0.14.5; parameters: -multicore 4 -X 1000 -un -ambiguous) using bowtie2-2.2.8 aligner35. Methylation states at each CpG were deduced using Bismark methylation extractor (-bedgraph -comprehensive). A total of 264/288 FOS+ and 233/288 FOS− cells passed the filtering criteria that included minimal coverage of 50,000 unique CpGs per cell, bisulfite conversion rates ≥ 99%, and number of reads below the 99th percentile of coverage in the cell. Collapsed methylation signals specific for the cell and site were saved as a matrix and inputted into R for statistical analysis and for data visualization. A total of 1,272,335 CpGs, which were also present in the bulk DGC methylome (≥ 3 cells in both FOS+ and FOS− populations), were detected across FOS+ and FOS− cells. Site differential methylation (DM) between FOS− and FOS+ cells was done using Barnard’s exact test (p < 0.05) (Barnard package, R) for all sites. Site coordinates were overlapped with other features using intersectBed tool. The methylation profiles across all FOS+ and FOS− cells was done using Rtsne package in R. For density plots, the average methylation per site was computed by dividing the cell counts for 100% methylation by the total number of cells within which a site was detected. The data was then used for density plots using ggplot2 in R. Odds Ratio for enrichment above chance was computed by (A = the number of sites overlapping feature/B = total number of sites)/(C = same number of random sites overlapping feature/D = total number of sites). This process was repeated 10 times. The average Odds Ratio was then computed and used for Chi-square analysis in R.
Single nuclei RNA-Seq on 10x Genomics platform
Single nucleus RNA-seq were performed by first sorting PROX1+/FOS+ and /FOS− neuronal nuclei in separate tubes in PBS as described in the Flow Cytometry section of the Method Details. Cell suspension were kept on ice and transferred to the Weill Cornell Epigenomics Core Facility for RNA-Seq. Standard 10x Genomics Chromium 3′ libraries (v3) were prepared according to the manufacturer’s recommendations. The FOS+ and FOS− 10x libraries were sequenced together on a HiSeq 2500 (Illumina) machine. 10x data were processed using CellRanger 3.1.0 with default parameters. Reads were aligned to the mouse reference genome mm10.
The CellRanger aggr pipeline was used to aggregate FOS+ and FOS− libraries into a single gene expression matrix consisting of 10,720 nuclei with 25,447 post-normalization reads. This matrix was loaded into R using the Seurat package (version 3.1.1) for downstream analysis (Butler et al., 2018). Default parameters were used unless specified. Nuclei were excluded from analysis if: 1) fewer than 200 genes were expressed 2) greater than 2500 genes were expressed and 3) more than 40% of UMIs were mapped to mitochondrial genes. Genes not expressed in at least three nuclei were also filtered out. Additionally, mitochondrial genes were filtered from the gene-nuclei matrix. The resulting expression matrix was composed of 13,382 genes (Table S2) and 5711 nuclei.
Gene expression was normalized to the total expression per nucleus, scaled by 10,000 and log-transformed. Following the identification of 2000 highly variable genes, the number of UMIs was regressed from each nucleus and gene expression was scaled. Principal component analysis (PCA) was performed on the highly variable genes. The top principal components (PCs) were selected using the JackStraw and Elbow methods and K-nearest neighbor (KNN) graph was constructed with the top 15 PCs. Nuclei were clustered with the Louvain algorithm (resolution = 0.6) and visualized in t-SNE space. Cluster markers were calculated through the Wilcoxon rank sum test. Cell types were identified based on expression of known markers. The top 5 markers per cluster was visualized on a heatmap based on average log fold change. Differential expression between FOS+ and FOS− nuclei were calculated with the Wilcoxon rank sum test. Genes were considered differentially expressed with a Bonferroni-corrected p value < 0.05
Computation of neighboring CpG methylation states
Methylated cytosine counts per reads were computed using an in-house pipeline (bam_to_meCount.py) provided in the Supplemental Information (Data S1). The output for bam_to_meCount.py script included the following columns in a table: Column1: read.-names, Column 2: The number of Cs methylated in CpG context, Column 3: The number of Cs unmethylated in CpG context, Column 4: The number of Cs methylated in non-CpG context, Column 5: The number of Cs unmethylated in non-CpG context, Column 6: number of bases that are not Cs, Column 7: read.length. From this table, the proportion of methylated CpGs was computed. To get the coordinates per read, bam files were converted to bed files using bamtobed function. The converted bed files were concatenated with the bam_to_meCount.py table output. To get the reads that overlap the IMRs, we used intersectBed function. Then reads with a minimum of 2 CpGs were extracted for plotting. To normalize the reads per IMR, the proportion of reads per IMR per methylation state was computed using the prop.table function (data.table package, R). The distribution of read proportion per IMR methylation state was plotted using boxplot function (R).
Ingenuity Gene Ontology Analysis
Lists of genes with IMRs and DM CpGs between FOS+ and FOS−cells, as well as with genes with DE were generated and used as input for Ingenuity Pathway Analysis (IPA). Top molecular/cellular functional categories, and functions within categories, were listed according to their corrected B-H value.
QUANTIFICATION AND STATISTICAL ANALYSIS
One-way, two-way, or repeated-measures ANOVAs with Sidak posthoc and paired and unpaired t tests were used. See statistics for DNA methylation and gene expression in corresponding sections.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| goat anti DCX | Santa Cruz | C18 |
| rabbit anti zif268 | Cell Signaling | 4153; RRID:AB_2097038 |
| anti-Prox1 Alexa Flour 647 | NovusBio | NBP-1-30045AF647 |
| anti-cFOS Alexa Flour 488 | NovusBio | NBP2-50037AF488 |
| Bacterial and Virus Strains | ||
| AAV1.Syn.GCaMP6s | Penn Vector Core | 100843-AAV1; RRID:Addgene_100843 |
| AAV-DJ-Syn-Dnmt3a-T2a-GFP | Stanford Vector Core | # 3097 |
| AAV-DJ-Syn-GFP | Standford Vector Core | # 3028 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Tamoxifen | Sigma-Aldrich | #T5648 |
| Isoflurane | Henry Schein | NDC 11695-6776-2 |
| Critical Commercial Assays | ||
| QIAamp DNA Micro Kit | QIAGEN | 56304 |
| TrueMethyl oxBS kit | Tecan | 0414-32 |
| EpiTect Fast Bisulfite Kit | QIAGEN | 59824 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6 | Jackson Laboratory | Stock No:000664 |
| Mouse: C57BL/6 | Taconic BioSciences | C57BL/6NTac |
| Mouse; Dnmt3af/f | Riken BioResource Center | Kaneda et al., 2004 |
| Mouse: Nestin-cre-ERT2 | Luis Parada of University of Texas Southwestern Medical Center, Dallas, TX | Chen et al., 2009 |
| Oligonucleotides | ||
| Nestin-cre Forward Primer: CAGCCTGTTACGTATAGCCG | Chen et al., 2009 | N/A |
| Nestin-cre Reverse Primer: GAGTCATCCTTAGCGCCGTA | Chen et al., 2009 | N/A |
| 16700 Primer: CCTAAGCACCAGGGTGTGAT | Chen et al., 2009 | N/A |
| 16701 Primer: TCACGGTTGGCCTTAGGGTT | Chen et al., 2009 | N/A |
| Dnmt3af/f Forward Primer CTGGTGATTGGAGGCAGTCCATGCA | Kaneda et al., 2004 | N/A |
| Dnmt3af/f Reverse Primer TAGCTGAGGCTGTCTGCATCGGACA | Kaneda et al., 2004 | N/A |
| Software and Algorithms | ||
| StereoInvestigator | MBF Bioscience | https://www.mbfbioscience.com/stereo-investigator |
| pClamp 10.3 | Molecular Devices | https://moleculardevices.app.box.com/s/rraipn0bep04avbr2tcufxww6at4ort5 |
| FlowJo | Tree Star | https://www.fiowjo.com/solutions/fiowjo/downloads |
| Bismark (v17) | Krueger and Andrews, 2011 | https://www.bioinformatics.babraham.ac.uk/projects/bismark/ |
| R | The R Foundation | https://www.r-project.org/ |
| Fiji | Schindelin et al., 2012 | https://fiji.sc |
| CASAVA 1.8.2 | Illumina | https://www.illumina.com/ |
| Other | ||
| RRBS sequencing data | This paper | IGV custom tracks/ uploaded to GEO: GSE72700 and GEO: GSE150551 |
| RRoxBS sequencing data | This paper | IGV custom tracks/ uploaded to GEO: GSE150551 |
| Single nuclei RNA-seq | This paper | uploaded to GEO: GSE150551 |
| Single nuclei RRBS data | This paper | IGV custom tracks/ uploaded to GEO: GSE150551 |
Highlights.
Numerous regions in DGCs show developmentally established methylation bistability
Increasing or reducing DNA methylation at regions alters neuron activation and excitability
Activated cells are enriched in one of the two states of bistable regions
Genes with bistable regions are associated with neuron excitability and plasticity
ACKNOWLEDGMENTS
We thank the following organizations for their support: the Epigenomics Core of Weill Cornell Medicine, the Applied Bioinformatics Core of Weill Cornell Medicine, the University of Pennsylvania Vector Core, and the Stanford Vector Core. We thank Paul Bergin for his guidance on behavior protocols and setup. We also thank Dr. Thu Huynh for her guidance on novel object interaction behavior. We thank Chloe Lopez-Lee for work on the hippocampal volume study. We are grateful for support by grants R01-MH103102 and R21-MH103715 from the NIH to M.T., a Medical Scientist Training Program grant from the National Institute of General Medical Sciences of the NIH under award T32GM007739 to the Weill Cornell/Rockefeller/Sloan Kettering Tri-Institutional MD-PhD Program, and a National Research Service Award from the NIMH under award F30MH115622 to R.N.F.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107789.
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPORTING CITATIONS
The following references appear in the Supplemental Information: Forrest et al. (2018); Helbig et al. (2019); Niday et al. (2017); Phelan et al. (2013); Zhou et al. (2009).
REFERENCES
- Abraham WC, and Tate WP (1997). Metaplasticity: a new vista across the field of synaptic plasticity. Prog. Neurobiol 52, 303–323. [DOI] [PubMed] [Google Scholar]
- Akalin A, Garrett-Bakelman FE, Kormaksson M, Busuttil J, Zhang L, Khrebtukova I, Milne TA, Huang Y, Biswas D, Hess JL, et al. (2012a). Base-pair resolution DNA methylation sequencing reveals profoundly divergent epigenetic landscapes in acute myeloid leukemia. PLoS Genet. 8, e1002781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akalin A, Kormaksson M, Li S, Garrett-Bakelman FE, Figueroa ME, Melnick A, and Mason CE (2012b). methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 13, R87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbosa FF, Santos JR, Meurer YS, Macêdo PT, Ferreira LM, Pontes IM, Ribeiro AM, and Silva RH (2013). Differential Cortical c-Fos and Zif-268 Expression after Object and Spatial Memory Processing in a Standard or Episodic-Like Object Recognition Task. Front. Behav. Neurosci 7, 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonthuis PJ, Huang WC, Stacher Horndli CN, Ferris E, Cheng T, and Gregg C (2015). Noncanonical Genomic Imprinting Effects in Offspring. Cell Rep. 72, 979–991. [DOI] [PubMed] [Google Scholar]
- Booth MJ, Branco MR, Ficz G, Oxley D, Krueger F, Reik W, and Balasubramanian S (2012). Quantitative sequencing of 5-methylcytosine and 5-hydroxymethylcytosine at single-base resolution. Science 336, 934–937. [DOI] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai DJ, Aharoni D, Shuman T, Shobe J, Biane J, Song W, Wei B, Veshkini M, La-Vu M, Lou J, et al. (2016). A shared neural ensemble links distinct contextual memories encoded close in time. Nature 534, 115–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron HA, and McKay RD (2001). Adult neurogenesis produces a large pool of new granule cells in the dentate gyrus. J. Comp. Neurol 435,406–417. [DOI] [PubMed] [Google Scholar]
- Chen J, Kwon CH, Lin L, Li Y, and Parada LF (2009). Inducible site-specific recombination in neural stem/progenitor cells. Genesis 47, 122–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen SJ, Munchow AH, Rios LM, Zhang G, Asgeirsdóttir HN, and Stackman RW Jr. (2013). The rodent hippocampus is essential for nonspatial object memory. Curr. Biol 23, 1685–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrest MP, Parnell E, and Penzes P (2018). Dendritic structural plasticity and neuropsychiatric disease. Nat. Rev. Neurosci 19, 215–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaidatzis D, Burger L, Murr R, Lerch A, Dessus-Babus S, Schübeler D, and Stadler MB (2014). DNA sequence explains seemingly disordered methylation levels in partially methylated domains of Mammalian genomes. PLoS Genet. 10, e1004143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaiti F, Chaligne R, Gu H, Brand RM, Kothen-Hill S, Schulman RC, Grigorev K, Risso D, Kim K-T, Pastore A, et al. (2019). Epigenetic evolution and lineage histories of chronic lymphocytic leukaemia. Nature 569 (7757), 576–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett-Bakelman FE, Sheridan CK, Kacmarczyk TJ, Ishii J, Betel D, Alonso A, Mason CE, Figueroa ME, and Melnick AM (2015). Enhanced reduced representation bisulfite sequencing for assessment of DNA methylation at base pair resolution. J. Vis. Exp 96, e52246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginno PA, Lott PL, Christensen HC, Korf I, and Chédin F (2012). R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Mol. Cell 45, 814–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good MA, Barnes P, Staal V, McGregor A, and Honey RC (2007). Context- but not familiarity-dependent forms of object recognition are impaired following excitotoxic hippocampal lesions in rats. Behav. Neurosci 121, 218–223. [DOI] [PubMed] [Google Scholar]
- Gunaydin LA, Grosenick L, Finkelstein JC, Kauvar IV, Fenno LE, Adhikari A, Lammel S, Mirzabekov JJ, Airan RD, Zalocusky KA, et al. (2014). Natural neural projection dynamics underlying social behavior. Cell 157, 1535–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JH, Kushner SA, Yiu AP, Cole CJ, Matynia A, Brown RA, Neve RL, Guzowski JF, Silva AJ, and Josselyn SA (2007). Neuronal competition and selection during memory formation. Science 316, 457–460. [DOI] [PubMed] [Google Scholar]
- Helbig KL, Lauerer RJ, Bahr JC, Souza IA, Myers CT, Uysal B, Schwarz N, Gandini MA, Huang S, Keren B, et al. ; Task Force for Neonatal Genomics; Deciphering Developmental Disorders Study (2019). De Novo Pathogenic Variants in CACNA1E Cause Developmental and Epileptic Encephalopathy with Contractures, Macrocephaly, and Dyskinesias. Am. J. Hum. Genet 104, 562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh TN, Shah M, Koo SY, Faraud KS, Santini E, and Klann E (2015). eIF4E/Fmr1 double mutant mice display cognitive impairment in addition to ASD-like behaviors. Neurobiol. Dis. S 3, 67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeger BN, Linker SB, Parylak SL, Barron JJ, Gallina IS, Saavedra CD, Fitzpatrick C, Lim CK, Schafer ST, Lacar B, et al. (2018). A novel environment-evoked transcriptional signature predicts reactivity in single dentate granule neurons. Nat. Commun 9, 3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkinson G, Pujadas E, Goutsias J, and Feinberg AP (2017). Potential energy landscapes identify the information-theoretic nature of the epigenome. Nat. Genet 49, 719–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneda M, Okano M, Hata K, Sado T, Tsujimoto N, Li E, and Sasaki H (2004). Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature 429, 900–903. [DOI] [PubMed] [Google Scholar]
- Kazachenka A, Bertozzi TM, Sjoberg-Herrera MK, Walker N, Gardner J, Gunning R, Pahita E, Adams S, Adams D, and Ferguson-Smith AC (2018). Identification, Characterization, and Heritability of Murine Metastable Epialleles: Implications for Non-genetic Inheritance. Cell 175,1259–1271.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlenkov A, Li J, Apontes P, Hurd YL, Byne WM, Koonin EV, Wegner M, Mukamel EA, and Dracheva S (2018). A unique role for DNA (hydroxy)methylation in epigenetic regulation of human inhibitory neurons. Sci. Adv 4, eaau6190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremerskothen J, Plaas C, Kindler S, Frotscher M, and Barnekow A (2005). Synaptopodin, a molecule involved in the formation of the dendritic spine apparatus, is a dual actin/alpha-actinin binding protein. J. Neurochem 92, 597–606. [DOI] [PubMed] [Google Scholar]
- Krueger F, and Andrews SR (2011). Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 27 (11), 1571–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacar B, Linker SB, Jaeger BN, Krishnaswami SR, Barron JJ, Kelder MJE, Parylak SL, Paquola ACM, Venepally P, Novotny M, et al. (2016). Nuclear RNA-seq of single neurons reveals molecular signatures of activation. Nat. Commun 7, 11022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landan G, Cohen NM, Mukamel Z, Bar A, Molchadsky A, Brosh R, Horn-Saban S, Zalcenstein DA, Goldfinger N, Zundelevich A, et al. (2012). Epigenetic polymorphism and the stochastic formation of differentially methylated regions in normal and cancerous tissues. Nat. Genet 44, 1207–1214. [DOI] [PubMed] [Google Scholar]
- Leutgeb JK, Leutgeb S, Moser MB, and Moser EI (2007). Pattern separation in the dentate gyrus and CA3 of the hippocampus. Science 315, 961–966. [DOI] [PubMed] [Google Scholar]
- Lev Maor G, Yearim A, and Ast G (2015). The alternative role of DNA methylation in splicing regulation. Trends Genet. 31, 274–280. [DOI] [PubMed] [Google Scholar]
- Levenson JM, Roth TL, Lubin FD, Miller CA, Huang IC, Desai P, Malone LM, and Sweatt JD (2006). Evidence that DNA (cytosine-5) methyltransferase regulates synaptic plasticity in the hippocampus. J. Biol. Chem 281,15763–15773. [DOI] [PubMed] [Google Scholar]
- Lisman J, Cooper K, Sehgal M, and Silva AJ (2018). Memory formation depends on both synapse-specific modifications of synaptic strength and cell-specific increases in excitability. Nat. Neurosci 21, 309–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo QM, et al. (2009). Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462, 315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XS, Wu H, Ji X, Stelzer Y, Wu X, Czauderna S, Shu J, Dadon D, Young RA, and Jaenisch R (2016). Editing DNA Methylation in the Mammalian Genome. Cell 167, 233–247.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinello K, Huang Z, Lujan R, Tran B, Watanabe M, Cooper EC, Brown DA, and Shah MM (2015). Cholinergic afferent stimulation induces axonal function plasticity in adult hippocampal granule cells. Neuron 85, 346–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maunakea AK, Chepelev I, Cui K, and Zhao K (2013). Intragenic DNA methylation modulates alternative splicing by recruiting MeCP2 to promote exon recognition. Cell Res. 23, 1256–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mundel P, Heid HW, Mundel TM, Krüger M, Reiser J, and Kriz W (1997). Synaptopodin: an actin-associated protein in telencephalic dendrites and renal podocytes. J. Cell Biol 139, 193–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niday Z, Hawkins VE, Soh H, Mulkey DK, and Tzingounis AV (2017). Epilepsy-Associated KCNQ2 Channels Regulate Multiple Intrinsic Properties of Layer 2/3 Pyramidal Neurons. J. Neurosci 37, 576–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onuchic V, Lurie E, Carrero I, Pawliczek P, Patel RY, Rozowsky J, Galeev T, Huang Z, Altshuler RC, Zhang Z, et al. (2018). Allele-specific epigenome maps reveal sequence-dependent stochastic switching at regulatory loci. Science 361, eaar3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S, Kramer EE, Mercaldo V, Rashid AJ, Insel N, Frankland PW, and Josselyn SA (2016). Neuronal Allocation to a Hippocampal Engram. Neuropsychopharmacology 41, 2987–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastore A, Gaiti F, Lu SX, Brand RM, Kulm S, Chaligne R, Gu H, Huang KY, Stamenova EK, Béguelin W, et al. (2019). Corrupted coordination of epigenetic modifications leads to diverging chromatin states and transcriptional heterogeneity in CLL. Nat. Commun 10, 1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelan KD, Shwe UT, Abramowitz J, Wu H, Rhee SW, Howell MD, Gottschall PE, Freichel M, Flockerzi V, Birnbaumer L, and Zheng F (2013). Canonical transient receptor channel 5 (TRPC5) and TRPC1/4 contribute to seizure and excitotoxicity by distinct cellular mechanisms. Mol. Pharmacol 83, 429–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picciotto MR, Higley MJ, and Mineur YS (2012). Acetylcholine as a neuromodulator: cholinergic signaling shapes nervous system function and behavior. Neuron 76, 116–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolls ET, and Treves A (2011). The neuronal encoding of information in the brain. Prog. Neurobiol 95, 448–490. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nature Methods 9 (7), 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Klein SS, Barboza L, Lohdi N, and Toth M (2016). Principles Governing DNA Methylation during Neuronal Lineage and Subtype Specification. J. Neurosci 36, 1711–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shipony Z, Mukamel Z, Cohen NM, Landan G, Chomsky E, Zeliger SR, Fried YC, Ainbinder E, Friedman N, and Tanay A (2014). Dynamic and static maintenance of epigenetic memory in pluripotent and somatic cells. Nature 513, 115–119. [DOI] [PubMed] [Google Scholar]
- Shukla S, Kavak E, Gregory M, Imashimizu M, Shutinoski B, Kashlev M, Oberdoerffer P, Sandberg R, and Oberdoerffer S (2011). CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature 479, 74–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer ZS, Yong J, Tischler J, Hackett JA, Altinok A, Surani MA, Cai L, and Elowitz MB (2014). Dynamic heterogeneity and DNA methylation in embryonic stem cells. Mol. Cell 55, 319–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skvortsova K, Zotenko E, Luu PL, Gould CM, Nair SS, Clark SJ, and Stirzaker C (2017). Comprehensive evaluation of genome-wide 5-hydroxymethylcytosine profiling approaches in human DNA. Epigenetics Chromatin 10, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler MB, Murr R, Burger L, Ivanek R, Lienert F, Schöler A, van Nimwegen E, Wirbelauer C, Oakeley EJ, Gaidatzis D, et al. (2011). DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature 480, 490–495. [DOI] [PubMed] [Google Scholar]
- Staley KJ, Otis TS, and Mody I (1992). Membrane properties of dentate gyrus granule cells: comparison of sharp microelectrode and whole-cell recordings. J. Neurophysiol 67, 1346–1358. [DOI] [PubMed] [Google Scholar]
- Temprana SG, Mongiat LA, Yang SM, Trinchero MF, Alvarez DD, Kropff E, Giacomini D, Beltramone N, Lanuza GM, and Schinder AF (2015). Delayed coupling to feedback inhibition during a critical period for the integration of adult-born granule cells. Neuron 85, 116–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winters BD, Forwood SE, Cowell RA, Saksida LM, and Bussey TJ (2004). Double dissociation between the effects of peri-postrhinal cortex and hippocampal lesions on tests of object recognition and spatial memory: heterogeneity of function within the temporal lobe. J. Neurosci 24, 5901–5908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie W, Barr CL, Kim A, Yue F, Lee AY, Eubanks J, Dempster EL, and Ren B (2012). Base-resolution analyses of sequence and parent-of-origin dependent DNA methylation in the mouse genome. Cell 148, 816–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Won J, Karlsson MG, Zhou M, Rogerson T, Balaji J, Neve R, Poirazi P, and Silva AJ (2009). CREB regulates excitability and the allocation of memory to subsets of neurons in the amygdala. Nat. Neurosci 12, 1438–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data reported in this study can be found in GEO under accession number GEO: GSE72700 and GEO: GSE150551. Most analyses were done using publicly available tools as listed in STAR Methods, with custom code available in the Supplemental Information.