

Abstract
Purpose
Autophagy plays an important role in balancing the inflammatory response to restore homeostasis. The aim of this study was to explore the mechanism by which trehalose suppresses inflammatory cytokines via autophagy activation in primary human corneal epithelial cells (HCECs) exposed to hyperosmotic stress.
Methods
An in vitro dry eye model was used in which HCECs were cultured in hyperosmolar medium with the addition of sodium chloride (NaCl). Trehalose was applied in different concentrations. The levels of TNF-α, IL-1β, IL-6, and IL-8 were detected using RT-qPCR and ELISA. Cell viability assays, immunofluorescent staining of LC3B, and western blots of Beclin1, Atg5, Atg7, LC3B, and P62 were conducted. The key factors in upstream signaling pathways of autophagy activation were measured: P-Akt, Akt, and transcription factor EB (TFEB).
Results
Trehalose reduced the proinflammatory mediators TNF-α, IL-1β, IL-6, and IL-8 in primary HCECs at 450 mOsM. This effect was osmolarity dependent, and a level of 1.0% trehalose showed the most suppression. Trehalose promoted autophagosome formation and autophagic flux, as evidenced by increased production of Beclin1, Atg5, and Atg7, as well as higher LC3B I protein turnover to LC3B II, with decreased protein levels of P62/SQSTM1. The addition of 3-methyladenine blocked autophagy activation and increased the release of proinflammatory cytokines. Trehalose further activated TFEB, with translocation from cytoplasm to the nucleus, but diminished Akt activity.
Conclusions
Our findings demonstrate that trehalose, functioning as an autophagy enhancer, suppresses the inflammatory response by promoting autophagic flux via TFEB activation in primary HCECs exposed to hyperosmotic stress, a process that is beneficial to dry eye.
Keywords: corneal epithelial cells, trehalose, autophagy, hyperosmolarity, inflammation
Dry eye is a multifactorial disease of the ocular surface characterized by a loss of homeostasis of the tear film; it is accompanied by ocular symptoms in which tear film instability1 and hyperosmolarity, ocular surface inflammation and damage,2−4 and neurosensory abnormalities play etiological roles.5−7 The prevalence of dry eye involving symptoms with or without signs has been estimated to be as high as 50%,8 a public health burden that has had a serious impact on the lives, studies, and work of many people.9 Due to deficient tear production10 or tear over-evaporation,11 tear film hyperosmolarity has been shown to be an important factor in the initiation of ocular surface inflammation in dry eye patients,12,13 as well as in mouse models.14−16
Physiotherapy strategies, artificial tears as supplementation, anti-inflammatory eye-drop therapy (including non-steroidal anti-inflammatory drugs and glucocorticoid), and even surgeries are current therapies for various dry eye patients to improve their symptoms.17−19 However, many of these approaches are palliative rather than disease modifying, and they do not provide adequate symptom relief or prevent disease progression.20 Studies on the pathophysiology role of hyperosmolarity have led to new preventive and therapeutic approaches for patients with dry eye syndrome.12,21−23 Osmoprotectants are small organic molecules that are used by many types of cells to restore isotonic cell volume and stabilize protein function, thus allowing adaptation to hyperosmolarity.24,25 Our previous studies showed that osmoprotectants such as pterostilbene,26 l-carnitine,27 erythritol, and betaine28 can suppress the inflammatory response via their uptake, accompanied by a decreasing concentration of intracellular inorganic salts in human corneal epithelial cells (HCECs) exposed to hyperosmotic stress.
The main biological purpose of trehalose, a disaccharide comprised of two glucose molecules,29 is water regulation,30 as it seems to form a gel phase during cellular dehydration that protects the organelle and then allows rapid rehydration when a proper environment is reintroduced.31 It can also serve a hydration function in the eyes of dry eye patients.32 Trehalose protects corneal epithelial cells in culture from death by desiccation,33,34 and it has been used as an effective and safe eye drop for the treatment of moderate to severe dry eye syndrome since 2002.35 In addition, trehalose also appears to increase autophagy in a manner independent of mechanistic target of rapamycin complex 1 (mTORC1) inhibition.36−38 Autophagy is a well-conserved self-degradative pathway39 and is one of the main intracellular quality control systems in almost all eukaryotic cell types.40 The major physiological role of autophagy is ensuring the maintenance of cell and tissue homeostasis by recycling macromolecules, especially in response to many stressors, including starvation, and under several types of cell stresses such as hypoxia, infection, and inflammation.41 Moreover, autophagy also suppresses inflammation, which causes collateral cell and tissue damage.42 Conversely, a properly mounted, focused, and transient inflammation promotes cell and tissue repair and regeneration.43 Previous studies44,45 reported that autophagy ensures a well-balanced inflammatory response that is accompanied by restoration of homeostasis. The ability of autophagy to prevent excessive inflammation was first observed in mice rendered deficient in Atg16/1 in 2008.46
Catalytic inhibition of mTORC1 in cells leads to basic helix–loop–helix transcription factor EB (TFEB) activation; however, because rapamycin is quite ineffective at activating TFEB,47−49 it is important to explore an alternative approach to activating TFEB. Here, we suggest that the serine/threonine kinase Akt (protein kinase B) could serve as an actionable target that controls TFEB activity independently of mTORC1. We have found that trehalose, an mTOR-independent autophagy inducer, promotes cytoplasm-to-nucleus translocation of TFEB while inhibiting Akt and finally reducing inflammation to protect primary HCECs from hyperosmotic stress via activation of autophagy.
Materials and Methods
Primary Cultures of HCECs and In Vitro Hyperosmotic Stress Model
Fresh human corneoscleral tissues (<72 hours after death) not suitable for clinical use from donors 18 to 65 years of age were obtained from the Lions Eye Bank of Texas (Houston, TX). Based on our previous methods,50 primary HCECs were cultured in 12-well plates using explants from corneal limbal rims in a supplemented hormonal epidermal medium (SHEM) containing 5% fetal bovine serum (FBS). Confluent cultures at 14 to 18 days were switched to an equal volume (0.5 mL/well) of serum-free medium (SHEM without FBS) for 24 hours and then treated for 4 or 24 hours with isosmolar medium (312 mOsM) and hyperosmolar medium (450 mOsM), which was achieved by adding 69-mM sodium chloride (NaCl), with or without 1-hour prior incubation with trehalose (provided by Allergan, Dublin, Ireland). In some experiments, an autophagy inhibitor, 5-mM 3-methyladenine (3-MA; Sigma-Aldrich, St. Louis, MO, USA) was included. The osmolarity of the culture medium was measured by a vapor pressure osmometer located in the Body Fluids Clinical Chemistry Laboratory of the Houston Methodist Hospital–Texas Medical Center (Houston, TX).3 The cells treated for 4 hours were lysed in Buffer RLT Plus from the RNeasy Plus Mini Kit (Qiagen, Hilden, Germany) for total RNA extraction. The cells treated for 24 hours were used for immunostaining or were lysed in radioimmunoprecipitation assay (RIPA) buffer (Sigma-Aldrich) for western blot analysis. The supernatant of the conditioned medium was stored at –80°C before ELISA.
RNA Extraction, Reverse Transcription, and Quantitative Real-Time PCR
Total RNA was extracted with the Qiagen RNeasy Plus Mini Kit according to the manufacturer's instructions, quantified with a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA), and stored at –80°C before use. Quantitative real-time PCR (RT-qPCR) was performed as previously described.51 In brief, first-strand cDNA was synthesized by reverse transcription from 2.0 µg of total RNA using Ready-To-Go You-Prime First-Strand Beads (GE Healthcare, Piscataway, NJ, USA), and qPCR was performed in a StepOnePlus Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) with 10.0-µL reaction volume containing 4.5 µL of cDNA, 0.5 µL gene expression assay, and 5.0 µL TaqMan Fast Universal PCR Master Mix (Applied Biosystems). Taqman Gene Expression Assays (Applied Biosystems) used for this study included glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Hs99999905_m1), TNF-α (Hs00174128_m1), IL-1β (Hs01555413_m1), IL-6 (Hs00174131_m1), and IL-8 (Hs00174103_m1). The thermocycler parameters were 50°C for 2 minutes and 95°C for 10 minutes, followed by 40 cycles of 95°C for 15 seconds and 60°C for 1 minutes. A non-template control was included to evaluate DNA contamination. The results were analyzed by the comparative threshold cycle (Ct) method and normalized by GAPDH as an internal control.
Enzyme-Linked Immunosorbent Assay
Double-sandwich ELISA for human TNF-α, IL-1β, IL-6, and IL-8 (BioLegend, San Diego, CA, USA) was performed to determine the protein concentration of the pro-inflammatory cytokines (TNF-α, IL-1β, and IL-6) and chemokine (IL-8) in the conditioned medium from different treatments according to the manufacturer's protocol and our previous publication.52 Absorbance was read at a wavelength of 450 nm with a reference wavelength of 570 nm using Infinite M200 PRO multimode microplate readers (Tecan US, Morrisville, NC, USA).
Western Blot Analysis
Western blot was performed as previously described.3 In brief, equal amounts of protein measured using a Micro BCA Protein Assay Kit (Pierce Biotechnology, Rockford, IL, USA) were mixed with 6× SDS reducing sample buffer and boiled for 5 minutes before loading. The proteins (50 µg/lane) were separated on 4% to 15% Mini-PROTEAN TGX Stain-Free Precast Gels (Bio-Rad Laboratories, Hercules, CA, USA) and transferred electronically to 0.2-µm polyvinylidene difluoride membranes (Bio-Rad Laboratories). The membranes were blocked with 5% nonfat milk in TTBS (Tris-buffered saline, 0.1% Tween 20) for 1 hour at room temperature and incubated with primary antibodies against human Beclin1, ATG5, ATG7, LC3B, and P62/SQSTM1 (Novus Biologicals, Littleton, CO, USA); TFEB, Akt, Phospho-Akt (Ser473) (D9E) XP Rabbit mAb, and Histone H3 (96C10) Mouse mAb (Cell Signaling Technology, Danvers, MA, USA); or β-Actin (Boster Biological Technology, Pleasanton, CA, USA) overnight at 4°C. After five washes with TTBS for 5 minutes each, the membranes were incubated with HRP conjugated Goat anti-Rabbit IgG (H+L) Secondary Antibody (1:10,000; Thermo Fisher Scientific) or Goat anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody (1:10,000; Thermo Fisher Scientific) for 1 hour at room temperature. The signals were detected with a Clarity western ECL substrate (Bio-Rad Laboratories), and the images were acquired using a ChemiDoc MP Imaging System (Bio-Rad Laboratories).
Cytoplasmic and Nuclear Protein Extraction
Cytosolic and nuclear protein was fractionated with NE-PER Nuclear and Cytoplasmic Extraction Reagents (Pierce Biotechnology) according to the manufacturer's instructions.
Immunofluorescent Staining
HCEC cultures on eight-chamber slides were fixed with methanol at –20°C for 5 minutes. Indirect immunofluorescent staining was performed using our previous methods.53 Primary rabbit polyclonal antibody against human LC3B (Novus Biologicals) was used. Alexa Fluor 488-conjugated AffiniPure Goat anti-Rabbit IgG (H+L) secondary antibody (Jackson ImmunoResearch Laboratories, West Grove, PA) was applied, and 4′,6-diamidino-2-phenylindole (DAPI; Invitrogen, Carlsbad, CA, USA) was used for nuclear counterstaining. Secondary antibody was applied alone as a negative control and compared to isotype goat IgG. The staining was photographed with a Nikon A1 Confocal Laser Microscope System (Nikon Instruments, Melville, NY, USA) and processed with Image J software (National Institutes of Health, Bethesda, MD, USA).
MTT Assay
HCEC cultures were seeded at a density of 2 × 104 cells/mL onto 96-well plates with 200 µL of culture medium per well overnight and were treated for 24 hours with isosmolar medium (312 mOsM) or hyperosmolar medium (450 mOsM), with or without 1-hour prior incubation with trehalose. The proliferative activity of the cells was quantitatively determined by a Cell Growth Determination Kit, MTT based (Sigma-Aldrich). The optical density of absorbance at 570 nm was measured using Infinite M200 PRO multimode microplate readers (Tecan US).54
Statistical Analysis
Student's t-test was used to compare differences between two groups. One-way ANOVA was used to make comparisons among three or more groups, followed by Dunnett's post hoc test. P < 0.05 was considered statistically significant.
Results
Trehalose Suppresses Hyperosmolarity-Induced Inflammatory Reaction in an Osmolarity-Dependent Manner
Hyperosmotic stress at 450 mOsM stimulated the mRNA expression and protein production of pro-inflammatory cytokines (TNF-α, IL-1β, and IL-6) and chemokine (IL-8) in primary HCECs. As determined by RT-qPCR, the hyperosmolarity significantly stimulated TNF-α, IL-1β, IL-6, and IL-8 mRNA expression to 4.82 ± 1.89, 2.32 ± 0.98, 2.71 ± 0.63, and 2.37 ± 0.57 fold, respectively (n = 6) (Figs. 1A, 1C, 1E, 1G). These increased mRNA levels were confirmed at protein levels by ELISA, which showed that hyperosmolarity increased TNF-α, IL-1β, IL-6, and IL-8 protein levels in the medium supernatants from normal low levels (8.71 ± 1.29 pg/mL, 16.05 ± 6.21 pg/mL, 3.18 ± 0.37 ng/mL, and 4.30 ± 0.49 ng/mL) to 96.43 ± 12.64 pg/mL, 86.96 ± 10.79 pg/mL, 7.57±1.38 ng/mL, and 9.04 ± 1.07 ng/mL, respectively (n = 6) (Figs. 1B, 1D, 1F, 1H).
Figure 1.

Trehalose suppressed the hyperosmolarity-induced inflammatory reaction. The pro-inflammatory cytokines, TNF-α (A, B), IL-1β (C, D), and IL-6 (E, F) and the chemokine IL-8 (G, H) were significantly stimulated at mRNA and protein levels in primary HCECs exposed to 450-mOsM hyperosmotic medium. (A, C, E, G) RT-qPCR analyses. The relative fold differences in mRNA expression were determined using 312-mOsM isosmolar medium as an internal control. (B, D, F, H) ELISA analyses. These markers were largely suppressed by trehalose at 0.5% to 1.5%, but 1.0% showed the most inhibition. Data are mean ± SD of six independent samples. *P < 0.05, compared with 450-mOsM hyperosmotic medium without prior incubation with trehalose.
Trehalose at the concentration of 0.5% to 1.5 % displayed suppressive effects on the expression and production of these inflammatory markers at mRNA and protein levels induced by hyperosmolarity; 1.0% trehalose showed the most inhibition. TNF-α, IL-1β, IL-6, and IL-8 mRNA expression was significantly decreased to 1.81 ± 0.79, 1.09 ± 0.53, 1.81 ± 0.28, and 1.20 ± 0.17 fold, respectively (n = 6) (Figs. 1A, 1C, 1E, 1G) by 1% trehalose. These decreased mRNA levels were accompanied by significantly suppressed protein concentrations of TNF-α, IL-1β, IL-6, and IL-8 in the medium supernatants (26.06 ± 9.58 pg/mL, 27.05 ± 8.88 pg/mL, 4.46 ± 0.71 ng/mL, and 5.27 ± 0.75 ng/mL, respectively (n = 6) (Figs. 1B, 1D, 1F, 1H), as compared to concentrations in the hyperosmolar medium.
Trehalose Increases Osmolarity in a Concentration-Dependent Manner
Higher concentrations (2.0%–5.0%) of trehalose appeared to lose the protective effects in a concentration-dependent manner; in fact, 5.0% trehalose may have stimulated higher expression and production of these inflammatory-related markers. Further, we found that osmolarity-dependent inhibition of inflammatory-related markers by trehalose was due to its aggravated effect on SHEM osmolarity. Trehalose was observed to increase osmolarity in deionized water, SHEM, and hyperosmolar medium in a concentration-dependent manner. Osmolarity in deionized water increased with higher concentrations of trehalose, with an especially great increase being observed at the 5% concentration (Fig. 2A). Similarly, osmolarity increased in SHEM and hyperosmolar medium when different concentrations of trehalose were added (Figs. 2B, 2C); 1.0% trehalose slightly increased osmolarity in SHEM (from 312 mOsM to 335 mOsM) and in hyperosmolar medium (from 450 mOsM to 473 mOsM).
Figure 2.
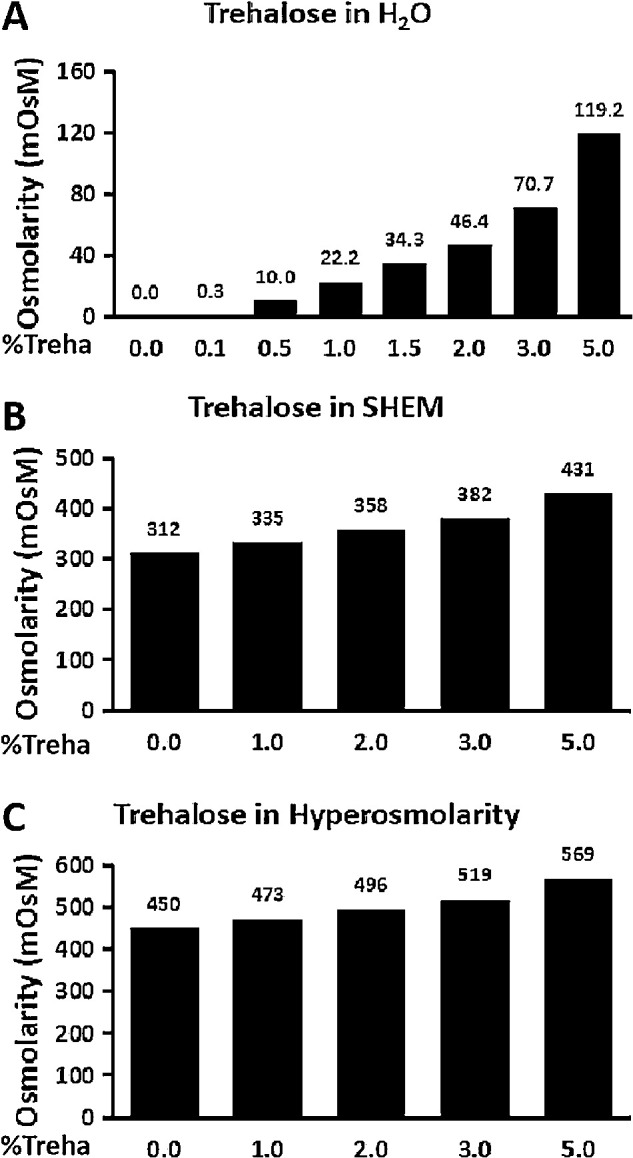
Trehalose increased osmolarity in the media. (A) Osmolarity of trehalose in H2O. (B) Osmolarity of trehalose in SHEM (312-mOsM isosmolar medium). (C) Osmolarity of trehalose in SHEM with the addition of 69-mM NaCl (450-mOsM hyperosmotic medium). Trehalose increased osmolarity in deionized water, SHEM, and hyperosmotic medium in a concentration-dependent manner.
Trehalose Enhances Autophagosome Formation and Autophagic Flux in HCECs Exposed to Hyperosmotic Stress
In the baseline condition, the production of autophagy-related proteins, such as Beclin1, Atg5, and Atg7, was not stimulated by 1.0% trehalose in primary HCECs cultured in SHEM (312 mOsM isosmolar medium) (Fig. 3A). However, when HCECs were exposed to hypertonic medium (450 mOsM), 1% trehalose significantly promoted autophagosome formation, as evidenced by the increased production of Beclin1, Atg5, Atg7, and LC3B. In addition to the higher turnover of LC3B I protein to form LC3B II, p62 protein (also known as SQSTM1) was observed to decrease significantly, indicating that trehalose enhances autophagic flux (Fig. 3B). This pattern of autophagy activation with the increased production and turnover of LC3B II, as well as the decreased p62 levels, became more pronounced in primary HCECs exposed to hyperosmotic stress with the addition of 1.0% trehalose when compared to HCECs without trehalose (n = 6) (Figs. 3C–3H).
Figure 3.

Trehalose promoted autophagosome formation and autophagic flux in HCECs exposed to hyperosmotic stress. (A) Western blot. The production of Beclin1, Atg5, and Atg7 was not stimulated by 1% trehalose in primary HCECs cultured in SHEM (312-mOsM isosmolar medium), with β-actin as the internal control. (B) Western blot. It was found that 1.0% trehalose enhanced the activation of autophagy, as evidenced by the increased protein levels of autophagy-related genes (Beclin1, Atg5, Atg7, and LC3B), the higher turnover of LC3B II, and the decreased protein levels of P62/SQSTM1 in primary HCECs exposed to hyperosmotic stress with β-actin as the internal control. (C–H) Quantitative analysis of relative expression levels of (C) Beclin1, (D) Atg5, (E) Atg7, (F) LC3B, (G) LC3B II/LC3B I, and (H) P62/SQSTM1. The data are presented as mean ± SD of six independent experiments. *P < 0.05, compared within groups.
The immunofluorescent staining showed that the percentage of cells containing LC3B puncta significantly increased with the 1.0% trehalose treatment in primary HCECs exposed to hyperosmotic stress. The number of LC3B puncta cells was greater compared to HCECs not receiving 1.0% trehalose and unexposed HCECs, which had the least: control versus hyperosmolarity, P < 0.05; control versus trehalose, P < 0.05; hyperosmolarity versus trehalose, P < 0.05 (n = 6) (Figs. 4A, 4B). Additionally, LC3B puncta were more abundant in primary HCECs with the 1.0% trehalose treatment compared to those without the 1.0% trehalose treatment and unexposed HCECs, which again had the least (n = 3) (Fig. 4C). As evaluated by the MTT cell survival assay, 1.0% trehalose effectively restored cell viability in conditions of hyperosmotic stress (hyperosmolarity vs. trehalose, P < 0.05) (Fig. 4D), which induced cell damage with lower survival rates.
Figure 4.

Trehalose induced autophagosome formation in HCECs exposed to hyperosmotic stress. (A) Representative immunofluorescence images of the punctate staining of LC3B in primary HCECs exposed to 450-mOsM hyperosmotic medium with or without prior incubation with 1.0% trehalose. (B) Percentage of autophagic cells. The fields were randomly selected, and at least 100 cells for each sample were analyzed (n = 3). (C) Average puncta per autophagic cell. Three cells were randomly chosen for analysis. (D) Cell viability analyzed by MTT assay. The cell viability was restored in primary HCECs exposed to 450-mOsM hyperosmotic medium with prior incubation with 1.0% trehalose. The data are presented as mean ± SD. *P < 0.05, compared within groups.
Trehalose-Induced TFEB Nuclear Translocation and Activation of the Autophagy–Lysosome Pathway Is Mediated by Akt Inhibition
To investigate the mechanism by which trehalose modulates autophagy, we investigated whether TFEB, a known autophagy activator, plays a role. We found that trehalose was able to induce TFEB translocation of cytoplasm to nucleus, as indicated by the upregulated level of nuclear TFEB after trehalose treatment as evaluated by western blot (Fig. 5A). Trehalose also enhanced autophagic flux, as indicated by P62/SQSTM1 and LC3B II turnover: control versus hyperosmolarity, P < 0.05; control versus trehalose, P < 0.05; hyperosmolarity versus trehalose, P < 0.05 (n = 6) (Fig. 5C). Finally, we sought to investigate the mechanism by which trehalose signals for TFEB nuclear translocation. Interestingly, we found that trehalose inhibited Akt substrates and pathways, and the phosphorylation of Akt was reduced in primary HCECs exposed to hyperosmotic stress (hyperosmolarity vs. trehalose, P < 0.05; n = 6) (Figs. 5A, 5B), as evaluated by western blot. The total level of Akt did not differ across these groups, suggesting that Akt signaling is inhibited with trehalose. Together, these results suggest that trehalose may lead to increased TFEB by reducing Akt signaling and further activation of autophagy.
Figure 5.

Trehalose-induced TFEB nuclear translocation and activation of the autophagy–lysosome pathway via Akt inhibition. (A) Western blot. Trehalose induced Akt inactivation, nuclear translocation of TFEB, and autophagosome formation and autophagic flux, as indicated by an increase of LC3B but a decrease of P62/SQSTM1 in primary HCECs exposed to hyperosmotic stress. (B, C) Quantitative analysis of relative expression levels of (B) LC3B II/LC3B I and (C) P-Akt/Akt. The data are presented as mean ± SD of six independent experiments. *P < 0.05, compared within groups.
Trehalose Induces Autophagy Against Inflammation in HCECs Exposed to Hyperosmotic Stress
To establish a direct link between autophagy induction by trehalose and a decrease in inflammatory cytokine release, 5-mM of 3-MA was applied as an autophagy inhibitor by blocking autophagosome formation. As evaluated by western blot, the activation of autophagy induced by trehalose was inhibited by 3-MA. LC3B induction and the high turnover of LC3B II were significantly suppressed, whereas the decreased levels of P62/SQSTM1 were largely restored by 3-MA in primary HCECs exposed to hyperosmotic stress (Figs. 6A–6D). As a consequence, the release of inflammatory cytokines suppressed by trehalose was also dramatically stimulated by 3-MA, evidenced by much higher protein levels of TNF-α (103.07 ± 30.64 pg/mL), IL-1β (90.10 ± 16.06 pg/mL), IL-6 (6.98 ± 1.84 ng/mL), and IL-8 (9.13 ± 0.72 ng/mL) in the medium supernatants of HCECs treated with NaCl, Trehalose and 3-MA, compared to without 3-MA (Figs. 6E–6H). The results further suggest that autophagy activation by trehalose suppresses the production of proinflammatory cytokines in HCECs under hyperosmotic stress.
Figure 6.

Trehalose induced autophagy against inflammation in HCECs exposed to hyperosmotic stress. (A) Western blot. The activation of autophagy induced by 1% trehalose was inhibited by 5-mM 3-MA, as evidenced by a decrease of LC3B and an increase of P62/SQSTM1 in primary HCECs exposed to hyperosmotic stress with β-actin as internal control. (B–D) Quantitative analysis of relative expression levels of (B) LC3B, (C) LC3B II/LC3B I, and (D) P62/SQSTM1. (E–H) ELISA analyses. The decreased release of (E) TNF-α, (F) IL-1β, (G) IL-6, and (H) IL-8 by trehalose was blocked by 5-mM 3-MA in HCECs exposed to 450-mOsM hyperosmotic medium. The data are presented as mean ± SD of four independent experiments. *P < 0.05, compared within groups.
Discussion
Trehalose as an organic osmoprotectant has been proven to play protective roles against the production of proinflammatory mediators in primary HCECs exposed to hyperosmotic stress, as well as in dry eye patients and animal models.33−35 Trehalose is a natural disaccharide used in food production that recently was found to be capable of enhancing the autophagic activity in various mammalian cells, including epithelial cells.37 However, the pathogenesis of trehalose-induced autophagy is not well understood in an in vitro dry eye model. Comprehensive results from the present study show that trehalose stimulates the cytoplasm-to-nucleus translocation of TFEB and the expression of genes regulating autophagy by inhibition of Akt. The activation of autophagy protects the HCECs from hyperosmolarity-induced cell damage, a potential mechanism underlying rises in pro-inflammatory cytokines and chemokine expression in dry eye disease and in an in vitro model of this condition.
Many studies, including ours, have reported that hyperosmotic stress can elicit an inflammatory response through different proinflammatory cytokines and chemokines.12,21−23 An increase in these molecules has been found in the HCEC culture model and the in vivo murine dry eye model, as well as in the tear fluid of dry eye patients.52,55 This study confirms previous findings and further reveals that the addition of trehalose was found to reduce secreted cytokine levels in primary HCECs exposed to hyperosmotic stress, suggesting that trehalose may be acting on the intracellular signaling pathways, thereby reducing the levels of inflammatory markers.34,56 However, higher concentrations (2.0%–5.0 %) of trehalose appeared to lose the protective effects in a concentration-dependent manner, and 5.0% trehalose may stimulate higher expression of these inflammatory markers, as the accumulation of trehalose increased osmolarity in deionized water, SHEM, and the hyperosmolar medium. The optimum concentration, then, was 1.0% trehalose.
Autophagy activity is linked to inflammation, and this interaction may be both inductive and suppressive and associated with the pathogenesis of several inflammatory diseases.43 It is established that Th1 cytokines, such as TNF-α, IL-1, and IL-6, exhibit effects of autophagy inducement,57 which could be related to the significant increase of autophagy activation in HCECs under hyperosmotic stress. Our data using primary HCECs exposed to hyperosmotic stress as an in vitro model of dry eye demonstrate that trehalose promoted autophagosome formation and autophagic flux, as evidenced by enhanced production of the autophagy-related protein markers Beclin1, Atg5, Atg7, and LC3B, as well as by LC3B I protein turnover to form LC3B II; protein levels of P62 protein and then inflammatory cytokine secretion were reduced. However, the addition of 3-MA blocked autophagosome formation and autophagic flux and increased the release of proinflammatory cytokines in HCECs exposed to hyperosmotic stress (Fig. 6). These findings establish a direct link between autophagy induction by trehalose and decreased inflammatory cytokine release.
Further, the increased average of LC3B puncta per primary HCECs and the greatly restored cell viability under hyperosmotic stress, both attributable to the addition of trehalose, indicate the protective role of trehalose. Compared to the 68.3 ± 11.1% cell survival rate in the hyperosmolarity condition, trehalose was able to effectively prevent cell death and restore cell viability at 89.7 ± 4.3%. The prevention of cell death by trehalose may be attributed to the enhanced autolysosome formation and autophagic flux. Autophagy is a pivotal intracellular process by which cellular macromolecules are degraded by various stimuli. A failure in the degradation of autophagic substrates such as impaired organelles and protein aggregates leads to their accumulation and the ultimately cell death characteristic of many stress conditions and degenerative diseases.37,43 In addition, as discussed above, autophagy activation reduced the production of inflammatory cytokines, another link to cell survival.
This study suggests that Akt control of TFEB activity is an actionable target that has potential relevance for the treatment of dry eye disease. Akt is a member of the AGC serine/threonine kinase family and plays a critical role in cell survival and the inhibition of apoptosis.58 Abnormal activation of Akt may occur through Akt mutation or dysregulation of upstream signaling pathways, and it is an important driving force of pathogen invasion.59 Interestingly, previous innovative studies have indicated that Akt regulates macroautophagy,60 a process whereby cellular material is enclosed into auophagosomes that fuse with lysosomes, creating autolysosomes. Although it is unclear how trehalose modulates Akt activation, the discovery that Akt regulates lysosomal function through TFEB is crucial to characterizing the role of Akt in autophagy pathways and offers a new perspective for understanding the cellular processes that are affected by the clinical use of Akt inhibitors.61−63 We found that trehalose induced inactivation of Akt and TFEB nuclear localization and increased activation of TFEB function in primary HCECs exposed to hyperosmotic stress in vitro. Subsequent induction of autophagy, perhaps to heal lysosomal damage, might lead to activation of the autophagy–lysosome pathway and further suppression of inflammation to protect HCECs.
Our findings are consistent with previous studies using primary HCECs but contrast somewhat with results by Panigrahi and colleagues,56 who reported autophagy induction by trehalose via increased phosphorylation of Akt with higher protein levels of P62 in corneal epithelial cells exposed to desiccation stress. This discrepancy regarding the mechanism of trehalose may be due to the utilization of different cell types and different stress models. Panigrahi et al.56 used a SV40 large T antigen immortalized HCE cell line (HCE-T), which may respond to trehalose differently than our primary HCECs. Their desiccation stress model was created by air-drying HCE-T cultures for 10 minutes at room temperature with 40% humidity after the medium completely aspirated, which is a seriously dry condition that could cause cell damage or even death. Our hyperosmotic stress model is relatively gentle, and the primary HCECs may respond to trehalose more physiologically.
In conclusion, our findings reveal that trehalose induces autophagy against inflammation by activation of TFEB via Akt inhibition in primary HCECs exposed to hyperosmotic stress. Follow-up studies are necessary to examine the clinical effect of trehalose application on the activation of autophagy or the consequences of its presence on the ocular surface.
Acknowledgments
Supported by grants from the National Institutes of Health (R01 EY023598 to DQL, EY011915 to SCP); by a NEI Center Core Grant for Vision Research (EY002520); by Allergan; by the Lions Foundation for Sight, Research to Prevent Blindness, Oshman Foundation, and William Stamps Farish Fund; by a Young Scientists Grant from the National Natural Science Foundation of China (81400380 to ZL); and by a grant from the Fundamental Research Funds for the Central Universities of China (XJJ2014076 to ZL).
Disclosure: Z. Liu, None; D. Chen, None; X. Chen, None; F. Bian, None; W. Qin, None; N. Gao, None; Y. Xiao, None; J. Li, None; S.C. Pflugfelder, None; D.-Q. Li, None
References
- 1. Gumus K, Crockett CH, Rao K, et al.. Noninvasive assessment of tear stability with the tear stability analysis system in tear dysfunction patients. Invest Ophthalmol Vis Sci. 2011; 52: 456–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhang L, Su Z, Zhang Z, Lin J, Li DQ, Pflugfelder SC. Effects of azithromycin on gene expression profiles of proinflammatory and anti-inflammatory mediators in the eyelid margin and conjunctiva of patients with meibomian gland disease. JAMA Ophthalmol. 2015; 133: 1117–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chi W, Hua X, Chen X, et al.. Mitochondrial DNA oxidation induces imbalanced activity of NLRP3/NLRP6 inflammasomes by activation of caspase-8 and BRCC36 in dry eye. J Autoimmun. 2017; 80: 65–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Li DQ, Pflugfelder SC. Matrix metalloproteinases in corneal inflammation. Ocul Surf. 2005; 3(suppl): S198–S202. [DOI] [PubMed] [Google Scholar]
- 5. Craig JP, Nichols KK, Akpek EK, et al.. TFOS DEWS II Definition and Classification report. Ocul Surf. 2017; 15: 276–283. [DOI] [PubMed] [Google Scholar]
- 6. Rahman EZ, Lam PK, Chu CK, Moore Q, Pflugfelder SC. Corneal sensitivity in tear dysfunction and its correlation with clinical parameters and blink rate. Am J Ophthalmol. 2015; 160: 858–866.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pflugfelder SC, Stern ME. Mucosal environmental sensors in the pathogenesis of dry eye. Expert Rev Clin Immunol. 2014; 10: 1137–1140. [DOI] [PubMed] [Google Scholar]
- 8. Stapleton F, Alves M, Bunya VY, et al.. TFOS DEWS II Epidemiology report. Ocul Surf. 2017; 15: 334–365. [DOI] [PubMed] [Google Scholar]
- 9. Tung CI, Perin AF, Gumus K, Pflugfelder SC. Tear meniscus dimensions in tear dysfunction and their correlation with clinical parameters. Am J Ophthalmol. 2014; 157: 301–310.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pflugfelder SC, De Paiva CS, Moore QL, et al.. Aqueous tear deficiency increases conjunctival interferon-γ (IFN-γ) expression and goblet cell loss. Invest Ophthalmol Vis Sci. 2015; 56: 7545–7550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pflugfelder SC. Tear dysfunction and the cornea: LXVIII Edward Jackson Memorial Lecture. Am J Ophthalmol. 2011; 152: 900–909.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Baudouin C, Aragona P, Messmer EM, et al.. Role of hyperosmolarity in the pathogenesis and management of dry eye disease: proceedings of the OCEAN group meeting. Ocul Surf. 2013; 11: 246–258. [DOI] [PubMed] [Google Scholar]
- 13. Pflugfelder SC, de Paiva CS. The pathophysiology of dry eye disease: what we know and future directions for research. Ophthalmology. 2017; 124: S4–S13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Luo L, Li DQ, Corrales RM, Pflugfelder SC. Hyperosmolar saline is a proinflammatory stress on the mouse ocular surface. Eye Contact Lens. 2005; 31: 186–193. [DOI] [PubMed] [Google Scholar]
- 15. Zhang X, De Paiva CS, Su Z, Volpe EA, Li DQ, Pflugfelder SC. Topical interferon-gamma neutralization prevents conjunctival goblet cell loss in experimental murine dry eye. Exp Eye Res. 2014; 118: 117–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen W, Zhang X, Li J, et al.. Efficacy of osmoprotectants on prevention and treatment of murine dry eye. Invest Ophthalmol Vis Sci. 2013; 54: 6287–6297. [DOI] [PubMed] [Google Scholar]
- 17. Jones L, Downie LE, Korb D, et al.. TFOS DEWS II Management and Therapy report. Ocul Surf. 2017; 15: 575–628. [DOI] [PubMed] [Google Scholar]
- 18. Pflugfelder SC, Stern M, Zhang S, Shojaei A. LFA-1/ICAM-1 interaction as a therapeutic target in dry eye disease. J Ocul Pharmacol Ther. 2017; 33: 5–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Moore QL, De Paiva CS, Pflugfelder SC. Effects of dry eye therapies on environmentally induced ocular surface disease. Am J Ophthalmol. 2015; 160: 135–142.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pflugfelder SC. Prevalence, burden, and pharmacoeconomics of dry eye disease. Am J Manag Care. 2008; 14(suppl): S102–S106. [PubMed] [Google Scholar]
- 21. Deng R, Hua X, Li J, et al.. Oxidative stress markers induced by hyperosmolarity in primary human corneal epithelial cells. PLoS One. 2015; 10: e0126561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Luo L, Li DQ, Pflugfelder SC. Hyperosmolarity-induced apoptosis in human corneal epithelial cells is mediated by cytochrome c and MAPK pathways. Cornea. 2007; 26: 452–460. [DOI] [PubMed] [Google Scholar]
- 23. Chen Z, Tong L, Li Z, et al.. Hyperosmolarity-induced cornification of human corneal epithelial cells is regulated by JNK MAPK. Invest Ophthalmol Vis Sci. 2008; 49: 539–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Deng R, Su Z, Hua X, Zhang Z, Li DQ, Pflugfelder SC. Osmoprotectants suppress the production and activity of matrix metalloproteinases induced by hyperosmolarity in primary human corneal epithelial cells. Mol Vis. 2014; 20: 1243–1252. [PMC free article] [PubMed] [Google Scholar]
- 25. Corrales RM, Luo L, Chang EY, Pflugfelder SC. Effects of osmoprotectants on hyperosmolar stress in cultured human corneal epithelial cells. Cornea. 2008; 27: 574–579. [DOI] [PubMed] [Google Scholar]
- 26. Li J, Ruzhi D, Hua X, et al.. Blueberry component pterostilbene protects corneal epithelial cells from inflammation via anti-oxidative pathway. Sci Rep. 2016; 6: 19408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hua X, Deng R, Li J, et al.. Protective effects of l-carnitine against oxidative injury by hyperosmolarity in human corneal epithelial cells. Invest Ophthalmol Vis Sci. 2015; 56: 5503–5511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hua X, Su Z, Deng R, Lin J, Li DQ, Pflugfelder SC. Effects of l-carnitine, erythritol and betaine on pro-inflammatory markers in primary human corneal epithelial cells exposed to hyperosmotic stress. Curr Eye Res. 2015; 40: 657–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Luyckx J, Baudouin C. Trehalose: an intriguing disaccharide with potential for medical application in ophthalmology. Clin Ophthalmol. 2011; 5: 577–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Elbein AD, Pan YT, Pastuszak I, Carroll D. New insights on trehalose: a multifunctional molecule. Glycobiology. 2003; 13: 17R–27R. [DOI] [PubMed] [Google Scholar]
- 31. Crowe JH. Trehalose and anhydrobiosis: the early work of J. S. Clegg. J Exp Biol. 2008; 211: 2899–2900. [DOI] [PubMed] [Google Scholar]
- 32. Teramoto N, Sachinvala ND, Shibata M. Trehalose and trehalose-based polymers for environmentally benign, biocompatible and bioactive materials. Molecules. 2008; 13: 1773–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Matsuo T. Trehalose protects corneal epithelial cells from death by drying. Br J Ophthalmol. 2001; 85: 610–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen W, Zhang X, Liu M, et al.. Trehalose protects against ocular surface disorders in experimental murine dry eye through suppression of apoptosis. Exp Eye Res. 2009; 89: 311–318. [DOI] [PubMed] [Google Scholar]
- 35. Matsuo T, Tsuchida Y, Morimoto N. Trehalose eye drops in the treatment of dry eye syndrome. Ophthalmology. 2002; 109: 2024–2029. [DOI] [PubMed] [Google Scholar]
- 36. Belzile JP, Sabalza M, Craig M, Clark E, Morello CS, Spector DH. Trehalose, an mTOR-independent inducer of autophagy, inhibits human cytomegalovirus infection in multiple cell types. J Virol. 2016; 90: 1259–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang X, Chen S, Song L, et al.. MTOR-independent, autophagic enhancer trehalose prolongs motor neuron survival and ameliorates the autophagic flux defect in a mouse model of amyotrophic lateral sclerosis. Autophagy. 2014; 10: 588–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lin X, Han L, Weng J, Wang K, Chen T. Rapamycin inhibits proliferation and induces autophagy in human neuroblastoma cells. Biosci Rep. 2018; 38: BSR20181822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Boya P, Esteban-Martinez L, Serrano-Puebla A, Gomez-Sintes R, Villarejo-Zori B. Autophagy in the eye: development, degeneration, and aging. Prog Retin Eye Res. 2016; 55: 206–245. [DOI] [PubMed] [Google Scholar]
- 40. Arias E, Cuervo AM. Chaperone-mediated autophagy in protein quality control. Curr Opin Cell Biol. 2011; 23: 184–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kaur J, Debnath J. Autophagy at the crossroads of catabolism and anabolism. Nat Rev Mol Cell Biol. 2015; 16: 461–472. [DOI] [PubMed] [Google Scholar]
- 42. Karin M, Clevers H. Reparative inflammation takes charge of tissue regeneration. Nature. 2016; 529: 307–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhong Z, Sanchez-Lopez E, Karin M. Autophagy, inflammation, and immunity: a troika governing cancer and its treatment. Cell. 2016; 166: 288–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Elliott EI, Sutterwala FS. Initiation and perpetuation of NLRP3 inflammasome activation and assembly. Immunol Rev. 2015; 265: 35–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhong Z, Umemura A, Sanchez-Lopez E, et al.. NF-κB restricts inflammasome activation via elimination of damaged mitochondria. Cell. 2016; 164: 896–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Saitoh T, Fujita N, Jang MH, et al.. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008; 456: 264–268. [DOI] [PubMed] [Google Scholar]
- 47. Martina JA, Chen Y, Gucek M, Puertollano R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy. 2012; 8: 903–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Roczniak-Ferguson A, Petit CS, Froehlich F, et al.. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci Signal. 2012; 5: ra42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Settembre C, Zoncu R, Medina DL, et al.. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012; 31: 1095–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kim HS, Jun Song X, de Paiva CS, Chen Z, Pflugfelder SC, Li DQ. Phenotypic characterization of human corneal epithelial cells expanded ex vivo from limbal explant and single cell cultures. Exp Eye Res. 2004; 79: 41–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Li DQ, Zhou N, Zhang L, Ma P, Pflugfelder SC. Suppressive effects of azithromycin on zymosan-induced production of proinflammatory mediators by human corneal epithelial cells. Invest Ophthalmol Vis Sci. 2010; 51: 5623–5629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Li DQ, Luo L, Chen Z, Kim HS, Song XJ, Pflugfelder SC. JNK and ERK MAP kinases mediate induction of IL-1beta, TNF-alpha and IL-8 following hyperosmolar stress in human limbal epithelial cells. Exp Eye Res. 2006; 82: 588–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chen Z, de Paiva CS, Luo L, Kretzer FL, Pflugfelder SC, Li DQ. Characterization of putative stem cell phenotype in human limbal epithelia. Stem Cells. 2004; 22: 355–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Liu Z, Zhou Q, Zhu J, et al.. Using genipin-crosslinked acellular porcine corneal stroma for cosmetic corneal lens implants. Biomaterials. 2012; 33: 7336–7346. [DOI] [PubMed] [Google Scholar]
- 55. Li DQ, Chen Z, Song XJ, Luo L, Pflugfelder SC. Stimulation of matrix metalloproteinases by hyperosmolarity via a JNK pathway in human corneal epithelial cells. Invest Ophthalmol Vis Sci. 2004; 45: 4302–4311. [DOI] [PubMed] [Google Scholar]
- 56. Panigrahi T, Shivakumar S, Shetty R, et al.. Trehalose augments autophagy to mitigate stress induced inflammation in human corneal cells. Ocul Surf. 2019; 17: 699–713. [DOI] [PubMed] [Google Scholar]
- 57. Qian M, Fang X, Wang X. Autophagy and inflammation. Clin Transl Med. 2017; 6: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Palmieri M, Pal R, Nelvagal HR, et al.. mTORC1-independent TFEB activation via Akt inhibition promotes cellular clearance in neurodegenerative storage diseases. Nat Commun. 2017; 8: 14338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009; 9: 550–562. [DOI] [PubMed] [Google Scholar]
- 60. Wang RC, Wei Y, An Z, et al.. Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science. 2012; 338: 956–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Rusmini P, Cortese K, Crippa V, et al.. Trehalose induces autophagy via lysosomal-mediated TFEB activation in models of motoneuron degeneration. Autophagy. 2019; 15: 631–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Palmieri M, Pal R, Sardiello M. AKT modulates the autophagy-lysosome pathway via TFEB. Cell Cycle. 2017; 16: 1237–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sardiello M. Transcription factor EB: from master coordinator of lysosomal pathways to candidate therapeutic target in degenerative storage diseases. Ann N Y Acad Sci. 2016; 1371: 3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]