
Figure 8.
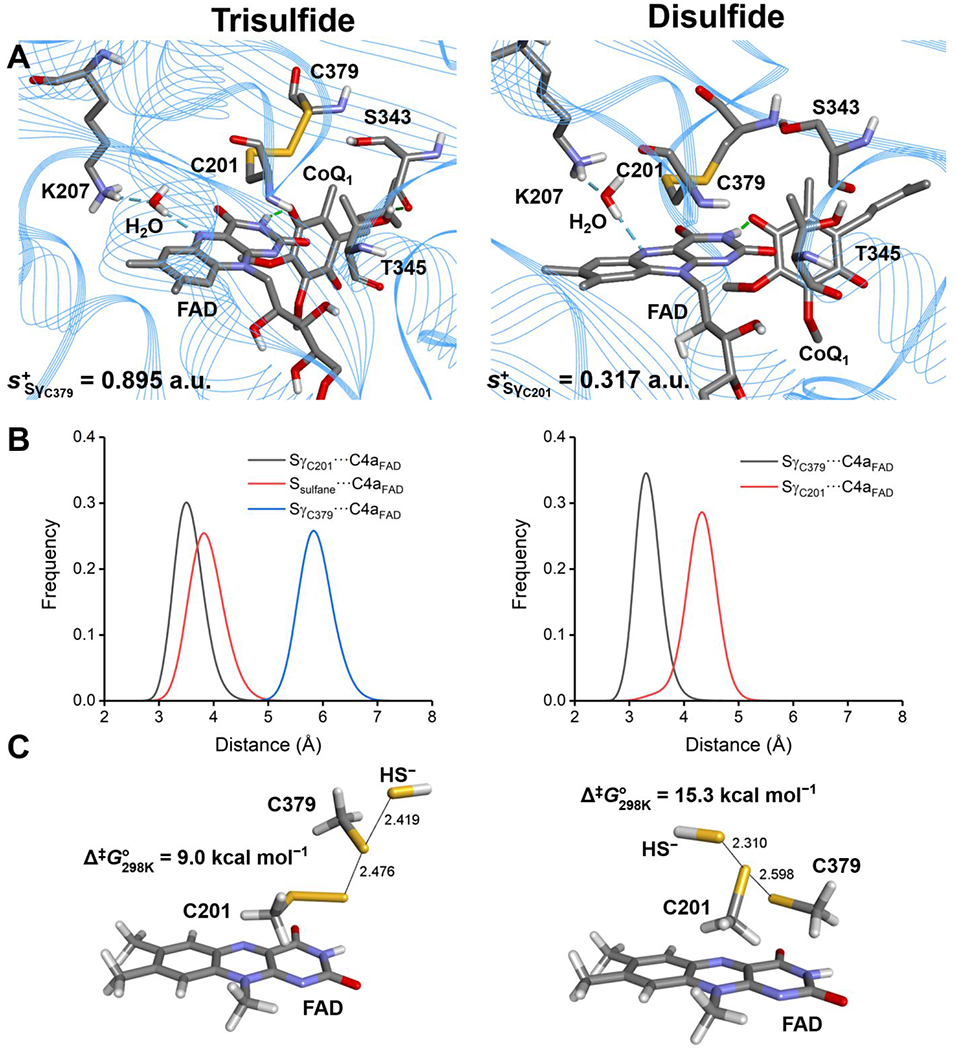
MD simulations and computational modeling of SQOR. A, Active site architecture in representative structures corresponding to the most populated cluster from 600 ns MD simulations of SQOR in the trisulfide (left panel) or disulfide state (right panel). Condensed local softness for the most electrophilic Sγ atom between Cys-201 and Cys-379 is reported for each system in atomic units (a.u.). B, Sulfur-to-C4a FAD distances for SγC201–C4aFAD/Ssulfane–C4aFAD/SγC379–C4aFAD (trisulfide, left panel) and SγC201–C4aFAD/SγC379–C4aFAD (disulfide, right panel) monitored along the corresponding MD trajectories. C, Structure of the transition states (TS) located for the sulfide anion attack on the trisulfide (left panel) or disulfide (right panel) using a reduced model of the active site of SQOR at the IEFPCM-DFT level of theory in a dielectric of ε = 10.125. Data correspond to interatomic distances in Å and Gibbs free-energy associated barriers at 298 K in kcal mol−1.